

Abstract
We examined the mutagenic specificity of the widely used antibiotic ciprofloxacin (CPR), which displays weak to moderate mutagenic activity in several bacteria and generates short in-frame deletions in rpoB in Staphylococcus aureus. To determine the spectrum of mutations in a system where any gene knockout would result in a recovered mutant, including frameshifts and both short and long deletions, we examined CPR-induced mutations in the thymidylate synthase-encoding thyA gene. Here, any mutation resulting in loss of thymidylate synthase activity generates trimethoprim (Trm) resistance. We found that deletions and insertions in all three reading frames predominated in the spectrum. They tend to be short deletions and cluster in two regions, one being a GC-rich region with potential extensive secondary structures. We also exploited the well-characterized rpoB-Rifr system in Escherichia coli to determine that cells grown in the presence of sublethal doses of CPR not only induced short in-frame deletions in rpoB, but also generated base substitution mutations resulting from induction of the SOS system. Some of the specific point mutations prominent in the spectrum of a strain that overproduces the dinB-encoded Pol IV were also present after growth in CPR. However, these mutations disappeared in CPR-treated dinB mutants, whereas the deletions remained. Moreover, CPR-induced deletions also occurred in a strain lacking all three SOS-induced polymerases. We discuss the implications of these findings for the consequences of overuse of CPR and other antibiotics.
INTRODUCTION
Several bactericidal antibiotics have been reported to have low to moderate mutagenic activity when used at subinhibitory or sublethal concentrations (1–14), usually ranging from 3- to 8-fold over background levels of spontaneous mutations (with one exception [7]). In particular, ciprofloxacin (CPR) and its close derivative norfloxacin (NOR) display mutagenic activity in different detector systems (1–14). The mutagenic properties can result in an increase in the appearance of resistant mutants (1, 2, 4, 5). A previous study of CPR-induced mutations in the rpoB gene of Staphylococcus aureus detected short in-frame deletions (5). However, the rpoB system cannot detect out-of-frame deletions or insertions or in-frame deletions or insertions of more than 21 bp, since the integrity of the RNA polymerase must remain intact. Here, we have undertaken a study of the types of mutations resulting from treatment with CPR in Escherichia coli using the thyA-Trmr system, which detects trimethoprim (Trm)-resistant mutants that result from any mutations inactivating the thyA gene (15), including large or small deletions or additions, both in frame and out of frame. We also used the E. coli rpoB-Rifr system, which monitors mutations leading to rifampin (Rif) resistance (16–23), since the system is so extensively characterized that the spectra of different mutagenic pathways leave telltale fingerprints. This allows us to separate effects of CPR itself from those emanating from the CPR-induced processes. Treatment with CPR leads to complex changes in cellular metabolism and double-strand breaks (24) that result in the induction of the SOS system (1, 3, 10, 13, 25, 26). In fact, CPR can induce the SOS system to higher levels than UV irradiation (J. H. Miller, unpublished data). Using both the thyA-Trmr and rpoB-Rifr systems, we show here that treating E. coli with CPR results in an increase in both base substitution mutations and short deletions and insertions and that some of the base substitutions can be ascribed to the SOS system, while the deletions can be attributed to the action of CPR alone. An analysis of the deletions indicated that some cluster in a region that displays single-stranded DNA secondary-structure possibilities. CPR binds to a complex of DNA gyrase and DNA and also to a complex of topoisomerase IV and DNA, freezing it on the DNA (24). The resulting replication blockage may allow short single-strand regions to persist. We discuss the implications of these findings with regard to antibiotic overuse.
MATERIALS AND METHODS
E. coli strains.
The dinB-deficient strain used here is from the Keio collection described by Baba et al. (27), made from the starting strain BW25113 (lacIq rrnBT14 ΔlacZWJ16 hsdR514 ΔaraBADAH33 ΔrhaBADLD78) (28). The starting strain was used as the wild type (WT) in the experiments reported here unless otherwise stated. The dinB mutant carries a complete deletion of the dinB gene, with a kan insert in place of the gene. A second wild-type background was the CC101 to CC107 series of strains (29, 30). The background is ara (gpt-lac)5 thi-F′128 lacIZ proA+ proB+. Each strain carries a complete lac operon on the F′ plasmid with a frameshift mutation in lacI and a base substitution mutation in lacZ, except for CC107, which carries a frameshift mutation in lacZ. Thus, the CC strains are isogenic except for the base substitution or frameshift in lacZ. CC107 (30) was used principally in the experiments carried out here with this background. The wild-type strain used (see Table 2), ZK126 (31), is W3310 Δ lacU169 tna-2, and its derivative, SF2018 (32), deficient in all three SOS-induced DNA polymerases (Pol II, Pol IV, and Pol V), is ZK126 polB::Spcr dinB::Kanr umuDC::Camr. These two strains were a gift from Steven E. Finkel.
TABLE 2.
rpoB mutant frequencies in ZK126 and SF2018 strain backgrounds
| Strain background | Medium | f (10−8)a | No. (%) of deletions/insertions | No. (%) of base substitutions |
|---|---|---|---|---|
| ZK126 | LB | 3.1 (2.1–5.9) | 0 (0.0) | 10 (100.0) |
| LB + CPR | 12.5 (8.9–15.5) | 10 (50.0) | 10 (50.0) | |
| SF2018 | LB | 1.9 (0–3.4) | 0 (0.0) | 17 (100.0) |
| LB + CPR | 8.4 (8.3–10.2) | 13 (56.5) | 10 (43.5) |
The values in parentheses are 95% confidence limits.
Media.
The following media (33) were used: LB (10 g tryptone, 5 g yeast extract, 10 gm NaCl per liter) and minimal A [10.5 g K2HPO4, 4.5 g KH2PO4, 1 g (NH4)2SO4, 0.5 g sodium citrate · 2H2O per liter] supplemented with 10 ml of 20% glucose, 1 ml of 1 M MgSO4, and 0.5 ml of 1% thiamine hydrochloride (vitamin B1) per liter.
Growth conditions and mutant selection.
Unless otherwise stated, all genetic methods were as described by Miller (33). Overnight cultures containing different concentrations of a given mutagen were seeded with approximately 1 × 103 cells from a growing culture. For mutant frequency (f) determination, cultures were grown for 18 h at 37°C on a rotor at 50 rpm. For mutant collection, cultures were grown for 18 h at 37°C on a shaking platform at 250 rpm. The cells were plated on specific media, LB plus 100 μg/ml rifampin to select for Rifr mutants and minimal A glucose medium containing 50 μg/ml thymidine and 10 μg/ml trimethoprim to select for Trmr mutants. For the collection of mutants shown in Fig. 1, in the cases of spontaneous mutations in the CC107 background and CPR-induced mutations in the CC107 and dinB backgrounds, four mutants were sequenced per culture. Nonrepeating mutations within the same set of four are shown. Repeated occurrences within the same culture are represented by a plus sign, such as +1, or +4. None of the deletions or insertions detected in this study had repeated occurrences within the same culture, with a single exception. All the rest of the repeats were base substitutions. Since the total number of mutations sequenced, including the repeated occurrences, was used in each total given in Results below, the true percentage of deletions maybe somewhat underestimated, although these effects are relatively small (see below). The same is true for the CPR-induced deletions shown in Fig. 2, which shows the deletions resulting from CPR (10 ng/ml). All the other mutants were collected at only one per culture, including the wild type and the triple mutant lacking all SOS polymerases (see Table 2), the BW25113 controls in rpoB, the CC107 control in thyA, and the CPR (12 ng/ml and 14 ng/ml)-induced mutations in thyA. The last two sets combined yielded 18 of 31 mutants that carried deletions/insertions (58%), similar to what was seen for the other two sets of CPR-induced deletions in thyA.
FIG 1.

Sequence changes in ropB. The locations of spontaneous mutations in the two wild-type backgrounds are shown above the DNA sequence. The letters represent base substitutions. The locations of CPR-derived mutations in the wild-type CC107 background and a dinB-deficient strain are shown below. The solid lines are aligned with deleted base pairs; the arrows indicate locations of insertions. The numbers with plus signs in parentheses show the number of additional mutations at any given site that appeared more than once in a sample of four from the same culture. Mutants from three strains are shown in the following colors: green, BW25113 background; black, CC107 background; red, a dinB derivative of BW25113.
FIG 2.

Sequence changes in thyA. The locations of spontaneous mutations in the CC107 wild-type background are shown above the DNA sequence (black); the locations of CPR-derived mutations in the wild-type BW25113 background (red) and the CC107 background (black) are shown below. The letters represent base substitutions, the circles indicate the A·T→T·A base substitution at the hot spot at bp 131, the diamonds indicate deletions of a single base pair, the triangles indicate insertions of a single base pair, the solid lines are aligned with deleted base pairs, and the arrows indicate the locations of insertions. The numbers with plus signs in parentheses show the number of additional mutations at any given site that appeared more than once in a sample of four from the same culture.
Determination of mutant frequencies.
We inoculated 100 to 1,000 cells in a series of cultures in LB that were then grown for 18 h at 37°C with aeration prior to plating on the appropriate medium (LB plates with or without 100 μg/ml rifampin or glucose minimal medium plates containing 50 μg/ml thymidine plus 10 μg/ml trimethoprim). The frequencies of Rifr mutants were determined as described previously (19). Briefly, mutant frequency (f) was determined as the median frequency of a set of cultures (the number of cultures varied from 12 to 45) by dividing the number of Rifr mutants per milliliter by the titer of the culture as determined by plating on LB medium without rifampin; 95% confidence limits were determined according to the method of Dixon and Massey (34).
Chromosomal DNA isolation and sequencing.
For the rpoB gene, these steps were carried out exactly as described by Garibyan et al. (19). For the thyA gene, the PCR conditions were as follows: annealing temperature, 62°C; elongation time, 1 min 15 s per cycle; number of PCR cycles, 30. The primer sequences were as follows: thyA F/21mer, GGTGTGATCATGATGGTCTGG; thyA R/17mer, CACACTGGCGTCGGCTC; the sequencing primer was thyA F.
Secondary-structure predictions.
Secondary structures were determined by the Mfold program.
Antibiotics.
Ciprofloxacin and rifampin were purchased from Sigma (St. Louis, MO).
RESULTS
CPR mutagenesis in rpoB.
We grew cultures of two starting strains (see Materials and Methods) in LB with and without CPR at 10 ng/ml. The cultures were plated on LB plates containing 100 μg/ml Rif to look for resistant (Rifr) mutants. Table 1 shows that the mutation frequencies were increased in the presence of CPR. We purified mutants from different cultures for further analysis. We used two different starting strain backgrounds to allow comparisons with work carried out on mutants derived from each of the backgrounds.
TABLE 1.
rpoB mutant frequencies in BW25113 and CC107 strain backgrounds
| Strain background | Medium | f (10−8) in rpoBa |
|---|---|---|
| BW25113 | LB | 3.0 (1.8–4.7) |
| LB + CPR | 25.8 (15.2–33.1) | |
| CC107 | LB | 4.3 (3.8–5.7) |
| LB + CPR | 18.9 (14–21.8) |
The values in parentheses are 95% confidence limits.
DNA sequence analysis of rpoB mutations.
Rifr mutants result from mutations in rpoB that alter the respective binding sites for Rif while not greatly affecting the essential functions of RNA polymerase. Although most of these mutations described in E. coli are base substitutions, some short in-frame deletions and insertions have also been found (16, 23). So far, 92 different base substitution mutations have been characterized (16–23). We could detect 80 of the 92 mutations in rpoB by using a single primer pair. We analyzed this portion of rpoB in the study. There are 11 A·T→G·C, 17 G·C→A·T, 9 A·T→T·A, 10 A·T→CG, 18 G·C→T·A, and 15 G·C→C·G mutations included in the set. Close to 90% of the mutants did have mutational changes in this region; Fig. 1 shows the results. At the top of Fig. 1 are the 76 mutations found in LB without CPR in cultures from the CC107 background (black letters) from this study and 68 mutations from the BW25113 background (green letters), also from this study. These results adequately mirror our previous results from 444 sequenced spontaneous mutations in CC107 and its parent strain background (20). Note that all 588 spontaneous mutations from the latter study and from the present study are base substitutions. In contrast, we found that 32 of 103 mutations (31%) from the cultures grown in CPR were short deletions or insertions in rpoB. They are depicted at the bottom of Fig. 1 (black lines). All of the deletions or insertions involve a multiple of 3 bp, and they range in size from 3 bp to 21 bp, meaning that from 1 to 7 amino acids are deleted from the RNA polymerase β subunit. Deletions outnumber insertions by 28 to 4. Also apparent in the CPR spectrum is the large number, 10, of G·C→C·G changes at position 1576. A signature of the SOS-induced mutations in rpoB that we reported in the CC strain background is the transversion at position 1576, as shown by the shift in mutations at this site from the normal, spontaneous spectrum (G·C→A·T) to those found in a dinB expression plasmid-containing strain (G·C→T·A and G·C→C·G) (20). Therefore, we looked at CPR-induced mutations in a dinB strain that lacks Pol IV (Fig. 1, bottom, red). Here, it can be seen that the deletions still constitute 46% of the mutations (17 of 37), whereas the transversions at 1576 virtually disappear. (The percentage of CPR-induced deletions for both the wild-type and dinB strains even represents a slight underestimate [see Materials and Methods].)
CPR-induced deletions are not generated by SOS-induced polymerases.
We examined the spectra of mutations occurring in rpoB in a strain lacking all three SOS-inducible polymerases (Pol II, Pol IV, and Pol V). Table 2 shows that the deletions in rpoB that occur in CPR-treated cells appear in the starting strain background for the experiment (ZK126) and also in the triple mutant (SF2018; dnaB dinB umuC) lacking all three SOS polymerases. A previous study showed that deletions are very rare in the spectra of both of these strains in the absence of CPR, with no deletions and only one insertion found in ZK126 out of 125 sequenced and no deletions and one insertion out of 128 sequenced in SF2018 (23). The results shown in Table 2 indicate that the deletions result from the action of CPR itself. Thus, the spectrum of mutations in the wild-type strain grown in the presence of CPR seen at the bottom of Fig. 1 contains elements from the spontaneous background (notably, the G·C→A·T mutations at 1547); the induction of the SOS polymerases, including Pol IV (see above); and mutations directly induced by CPR itself.
DNA sequence analysis of thyA mutations.
We sequenced thyA mutations in the Trm-resistant mutants. In the absence of CPR, the thyA spectrum is dominated by a large hot spot that comprises nearly 60% of all the spontaneous mutations and results from an A·T→T·A change at position 131 (15) (see below). Viswanathan and coworkers have shown that it is caused by a quasipalindrome fold-back structure in the single-stranded regions of the DNA that induces repair enzymes to “correct” a “mispair” in the stem of the fold-back structure (15). We also found that this hot spot predominated when spontaneous mutations were monitored in the absence of added CPR. In the CC107 background, 39 of 52 (75%) sequenced mutations were at this hot spot (Fig. 2), and in BW25113, 104 of 145 (72%) sequenced mutations were at this site (Table 3 and data not shown). Deletions and insertions of more than 1 bp are virtually absent in these spectra but are well represented in the spectra of CPR-treated cells (Fig. 2). Even grouping the single-base-pair deletions and insertions with the larger deletions shows a significant difference. In the absence of CPR, they number 6 of 52 (11.5%) in CC107 and 14 of 145 (12.4%) in BW25113 (Table 3). However, in the presence of 10 ng/ml CPR, total deletions and insertions comprise 72% (30 of 42) and 52% (23 of 44) of the mutations in the two wild-type backgrounds, respectively, and 64% (21 of 33) in a dinB background (Table 3). Note that CPR-induced mutations occur at a 4.4-fold-higher rate in BW25113 than in the untreated control and at an 8.6-fold-higher rate in CC107. Multiplying the percentages of deletions and insertions by these factors showed that they occur at a 18.5-fold-higher rate in BW25113 and a 53.8-fold-higher rate in CC107. Smaller sample sizes were analyzed in the CC107 background for CPR (12 and 14 ng/ml), but together, they showed that 13 of 31 sequenced mutations were deletions/insertions (42%) (Table 3). Figure 2 shows the locations of the sequence changes in thyA. Here, one can see that even though all sizes of deletions are permitted, the deletions are still mainly short, and they tend to cluster in two regions of the gene. However, they are no longer exclusively in-frame deletions and insertions. Thus, out of 52 CPR-induced deletions in wild-type backgrounds shown in Fig. 2 and Table 3, 10 are in frame, 21 are in the +1 reading frame, and 21 are in the −1 frame. Of 14 insertions, 4 are in frame, 3 are in the +1 frame, and 6 are in the −1 frame.
TABLE 3.
thyA mutant frequencies in various strain backgrounds
| Strain background | Medium | f (10−8)a | No. (%) of deletions/insertions | No. (%) of base substitutions |
|---|---|---|---|---|
| BW25113 | LB | 67.5 (46–77) | 18 (12) | 127 (88) |
| LB + 10 ng/ml CPR | 474 (389–567) | 23 (52) | 21 (48) | |
| CC107 | LB | 38.0 (33.9–43.7) | 6 (11) | 46 (89) |
| LB + 10 ng/ml CPR | 110 (50.0–138) | 30 (71) | 12 (29) | |
| LB + 12 ng/ml CPR | 346 (199–659) | 5 (38) | 8 (62) | |
| LB + 14 ng/ml CPR | 730 (465–944) | 8 (44) | 10 (56) | |
| dinB | LB | 41.8 (37.1–42.8) | NDb | ND |
| LB + 10 ng/ml CPR | 327 (138–468) | 21 (64) | 12 (36) |
The values in parentheses are 95% confidence limits.
ND, not determined.
Possible elements involved in formation of short deletions/insertions.
As pointed out above, no frame or size restrictions exist for indels in thyA. The one requirement is that they result in an inactive protein. However, although they can appear anywhere in the gene/protein, these events do tend to cluster near bp 298 (14 of 53 deletions/insertions) (Fig. 2A) and near bp 511 (15 of 52 deletions) (Fig. 2B). Position 298 is centered in a localized GC-rich region (Fig. 2A), and a plausible secondary structure may form involving that region (Fig. 3). Given the replication-blocking consequences of CPR binding to gyrase (24) (see Discussion), one can envision such structures forming transiently, as blocked replication promotes the existence of single-stranded regions.
FIG 3.
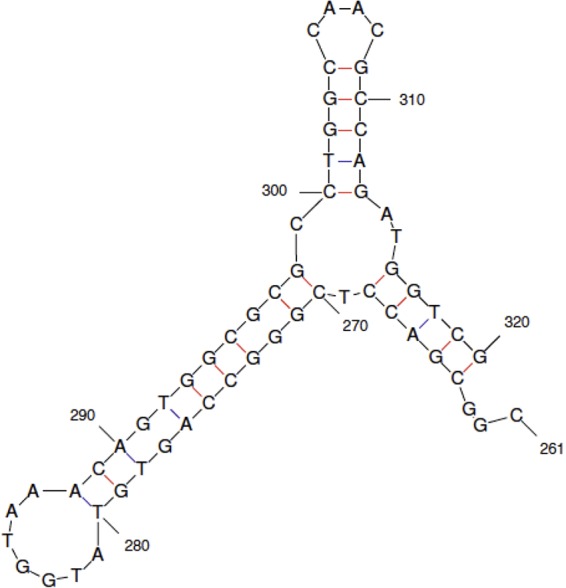
Potential single-strand secondary structure in thyA. Position 298 is centered within a region where a cluster of deletions of various lengths are observed. The Mfold program revealed a putative secondary structure between bp 261 and 320 (−16.4 kcal).
DISCUSSION
CPR and related fluoroquinolones, such as norfloxacin, induce mutations in a dose-dependent manner in several bacterial systems, including Salmonella enterica serovar Typhimurium (3, 6, 10, 11, 13, 14), E. coli (2, 4, 9), Streptococcus pneumoniae (8), S. aureus (5), Pseudomonas aeruginosa (12), and Mycobacterium fortuitum (7), although the level of mutagenesis (3- to 10-fold) is well below that for standard mutagens, with the exception of M. fortuitum (7). For example, Rifr in E. coli is increased 5,800-fold by alkylating agents, such as ethyl methanesulfonate (EMS) (19); 400-fold by UV light irradiation (19); and 150-fold and 4,800-fold by the base analogs 2-aminopurine and 5-bromodeoxyuridine, respectively (19, 35). However, the level of CPR-induced mutations is sufficient to promote increased evolution of CPR-resistant strains following exposure to CPR (1, 4, 5, 12), including in vivo studies using mice (4). Studies on the nature of CPR-induced mutations have been somewhat limited by the systems used or the small sample sizes analyzed. Cirz et al. (4) looked at mutations in E. coli after continuous growth in the presence of CPR and then selected for CPR resistance and found point mutations and a deletion of 3 bp at one position in the gyrA gene. The point mutations did not appear in very small samples of strains lacking any of the SOS-induced polymerases, Pol II, Pol IV, or Pol V (7, 6, and 4 CPR-resistant mutants, respectively), or lacking any pair of the polymerases (6, 2, and 3, respectively) grown with CPR. A single sequenced CPR-resistant mutant from a strain lacking all three polymerases resulted from the 3-bp deletion. Didier and coworkers (5) carried out the most comprehensive study, looking at mutations in the rpoB gene of S. aureus that resulted in Rifr mutants. They found that short in-frame deletions or insertions of 3, 6, 9, or 12 bp were well represented in the spectrum of mutations in a wild-type strain, but not in a recA strain. Previous work in S. aureus had found no reduction in CPR-induced mutation frequency in a lexA strain that cannot induce the SOS system (36), whereas the opposite result was reported for E. coli (4).
We used two systems to examine the nature of CPR-induced mutations. We employed the rpoB-Rifr system in E. coli because of the extensive characterization of mutational sites (19, 20). Here, we found a significant percentage of small deletions and insertions in rpoB resulting from CPR exposure (33 of 103 mutations sequenced in the wild-type strain [Fig. 1 and Table 2]), as has been detected in S. aureus rpoB (5) and very recently in a maximum-depth sequencing study of E. coli (37). We have gone further in this study by using a Pol IV-deficient (dinB) strain to pinpoint specific point mutations that result from the induction of the SOS system and by using a strain with the dinB, umuC, and polB genes deleted, and thus lacking all three SOS-induced polymerases, to show that the appearance of deletions in rpoB in response to CPR was unaffected (Table 2). Therefore, we could ascertain that even though recA strains are much more sensitive to CPR (38), the deletion mutations are not generated by any of the SOS polymerases. Viable rpoB mutants are restricted in the sense that any deletions or insertions must be small, confined to a specific region in rpoB, and in the correct reading frame, namely, a multiple of 3 bp added or deleted. The thyA-Trmr system has no such restrictions, as any mutation in thyA resulting in the inactivation of thymidylate synthase will yield a Trmr mutant. Figures 2 and 3 and Table 3 show that deletions and insertions do predominate in the spectrum of CPR-induced mutations, but now, out-of-frame deletions and insertions occur (25 in the +1 reading frame and 27 in the −1 reading frame versus 14 in the correct reading frame). Most importantly, even though the size restrictions are now removed, the deletions are predominantly relatively short (Fig. 2 and 3) and in fact tend to cluster in two regions. The largest cluster of deletions with different sizes contains a compelling secondary structure (−16.4 kcal) (Fig. 3). Viswanathan and coworkers (15) showed that a secondary structure emanating from a quasipalindrome earlier in the gene was responsible for the major hot spot among spontaneous mutations, an A·T→T·A change that accounts for more than half of all spontaneous mutations in thyA. This structure (−11.9 kcal) results in enzymatic (mismatch repair) correction of one of the “mispairs” in the stem. Why would we not see this type of mutation resulting from the same type of correction of what is a stronger secondary stem-and-loop structure? The most likely explanation is that the potential amino acid changes resulting from the correction of mispairs in the main stem (Fig. 3) probably do not result in an inactive enzyme. Likewise, in the stem for the hot spot at base 131 (15), correction of the mispair at base 136 would result in an observed A→G change, generating a Thr→Ala substitution, which is apparently not detrimental to the protein, as the mutation is not observed.
The fact that CPR strongly induces the SOS system (1, 3, 10, 13, 25, 26) yet has a relatively low mutation rate itself in E. coli (2, 4, 9) allows us to assess the levels of mutations resulting from the SOS polymerases. The rpoB-Rifr system is well characterized, allowing us to recognize the fingerprint of SOS induction, or at least the mutations resulting from increased expression of the dinB-encoded Pol IV, since they have been identified in rpoB by analyzing the spectrum of dinB overexpression (20). The fingerprint of Pol IV expression involves a shift at position 1576 from G·C→A·T mutations to the two transversions G·C→T·A and G·C→C·G (20). We can see this in Fig. 1, which shows a shift in the proportion of G·C→A·T versus G·C→C·G mutations from 21:2 without CPR to 7:10 with CPR. These mutations all but disappear in the dinB spectrum with CPR (Fig. 1). The true contribution of mutations caused by SOS-induced polymerases acting at undamaged bases after DNA damage-stimulated expression has proved elusive to measure because it is usually masked by the much higher frequency of mutations occurring opposite damaged bases and because simulations with mutants constitutively expressing the SOS system may not recreate levels of induction identical to those of the SOS system. Figure 1 argues that the levels of mutations occurring opposite undamaged bases, as reflected at the indicator position 1576, are indeed part of the CPR-induced mutation spectrum, yet this level represents a relatively low absolute mutation frequency.
What is the nature of the lesion that results in the CPR-induced mutations, the majority of which are deletions or insertions? Kohanski and coworkers and Foti and coworkers have presented extensive evidence that one pathway of killing for all bactericidal antibiotics involves the generation of hydroxyl radicals that damage DNA, leading to lethal double-strand breaks (39, 40). Additionally, it has been postulated that specifically 8-oxo-dG, particularly in the dGTP precursor, is the main DNA lesion responsible (40). Although we did not observe the mutational fingerprint of this lesion (A·T→C·G) as a major part of the CPR-induced spectra in the targets studied here (see also the recent study by Long and coworkers [41]), clearly reactive oxygen species (ROS) are involved in CPR mutagenesis (42). Are the mutational spectra from different bactericidal antibiotics similar? We briefly analyzed ampicillin (AMP)-induced mutations in rpoB, and although we found a mutation frequency 5 times over background, as seen in other work (2), we found only one deletion or insertion among 55 sequenced mutations with independent origins (data not shown). Thus, the spectra of AMP-induced mutations and CPR-induced mutations are not similar using this particular target gene. This can be explained by the findings of Gutierrez and coworkers (43), who showed that AMP promotes bacterial mutagenesis via an RpoS-mediated reduction in replication fidelity by depleting the cell of MutS. Kohanski and coworkers sequenced six norfloxacin-resistant mutants induced by ampicilliin and found that the four detected changes all resulted from base substitutions in either gyrA, gyrB, or the promoter for the acrAB operon (2). However, all three ampicillin-induced kanamycin-resistant mutants with detected sequence changes showed insertions in either the rpsL or arcA gene that resulted in truncation of the encoded protein (2). The CPR-gyrase complex, which freezes on DNA (24), may promote deletions/insertions by strongly inducing SOS recombination functions that incorrectly repair damaged DNA (9, 44, 45). Extensive secondary structures (Fig. 3) might increase incorrect recombinational repair or certain slippage mechanisms (46). There are several implications of the results reported here. As noted previously, the level of CPR-induced mutations is sufficient to accelerate the evolution of drug resistance in bacteria (1, 4, 5, 12). Because CPR is a widely used antibiotic, it should be appreciated that the surviving populations within the microbiome will have increased mutations and that the majority of them will be deletions within genes. A higher proportion of these mutations than of base substitutions have deleterious effects. Thus, although these effects would not be seen in terms of immediately killing bacteria, they can reduce the fitness of the commensal bacteria, rendering them more susceptible to being outgrown and replaced by unwanted strains. Also, the mutagenic effects of CPR are subject to being increased by synergy from other antibiotics used in combination treatments. Synergy of antibiotics with regard to killing has been widely studied (47), but the synergy of mutagens with regard to mutagenesis has not. Recently, we showed that certain pairs of widely used mutagens, some of which are employed in chemotherapy, display strong synergy with regard to mutagenesis (48). These studies should be expanded to examine certain antibiotic pairs.
ACKNOWLEDGMENT
Part of this research was funded by a Faculty Research Grant from the University of California.
REFERENCES
- 1.Blazquez J, Couce A, Rodriguez-Beltran J, Rodriguez-Rojas A. 2012. Antimicrobials as promoters of genetic variation. Curr Opin Microbiol 15:561–569. doi: 10.1016/j.mib.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 2.Kohanski MA, DePristo MA, Collins JJ. 2010. Sub-lethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol Cell 37:311–320. doi: 10.1016/j.molcel.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yim G, McClure J, Surette MG, Davies JE. 2011. Modulation of Salmonella gene expression by subinhibitory concentrations of quinolones. J Antibiot 64:73–78. doi: 10.1038/ja.2010.137. [DOI] [PubMed] [Google Scholar]
- 4.Cirz RT, Chin JK, Andes DR, Crecy-Lagard V, Craig WA, Romesberg FE. 2005. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol 3:e176. doi: 10.1371/journal.pbio.0030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Didier J-P, Villet R, Huggler E, Lew DP, Hooper DC, Kelley W, Vaudaux P. 2011. Impact of ciprofloxacin exposure on Staphylococcus aureus genomic alterations linked with emergence of rifampicin resistance. Antimicrob Agents Chemother 55:1946–1952. doi: 10.1128/AAC.01407-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gocke E. 1991. Mechanism of quinolone mutagenicity in bacteria. Mutat Res 248:135–143. doi: 10.1016/0027-5107(91)90095-6. [DOI] [PubMed] [Google Scholar]
- 7.Gillespie SH, Basu S, Dickens AL, O'Sullivan DM, McHugh TD. 2005. Effect of subinhibitory concentrations of ciprofloxacin on Mycobacterium fortuitum mutation rates. J Antimicrob Chemother 56:344–348. doi: 10.1093/jac/dki191. [DOI] [PubMed] [Google Scholar]
- 8.Henderson-Begg SK, Livermore DM, Hall LMC. 2006. Effect of subinhibitory concentrations of antibiotics on mutation frequency in Streptococcus pneumoniae. J Antimicrob Chemother 57:849–854. doi: 10.1093/jac/dkl064. [DOI] [PubMed] [Google Scholar]
- 9.Thi TD, Lopez E, Rodriguez-Rojas A, Rodriguez-Beltran J, Couce A, Guelfo JR, Castaneda-Garcia A, Blazquez J. 2011. Effect of recA inactivation on mutagenesis of Escherichia coli exposed to sublethal concentrations of antimicrobials. J Antimicrob Chemother 66:531–538. doi: 10.1093/jac/dkq496. [DOI] [PubMed] [Google Scholar]
- 10.Ysern P, Clerch B, Castario M, Gilbert I, Barbe J, Llagostera M. 1990. Induction of SOS genes in Escherichia coli and mutagenesis in Salmonella typhimurium by fluoroquinolones. Mutagenesis 5:63–66. doi: 10.1093/mutage/5.1.63. [DOI] [PubMed] [Google Scholar]
- 11.Levin DE, Marnett LJ, Ames BN. 1984. Spontaneous and mutagen-induced deletions: mechanistic studies in Salmonella tester strain TA102. Proc Natl Acad Sci U S A 81:4457–4461. doi: 10.1073/pnas.81.14.4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanimoto K, Tomita H, Fujimoto S, Okuzumi K, Ike Y. 2008. Fluoroquinolone enhances the mutation frequency for meropenem-selected carbapenem resistance in Pseudomonas aeruginosa, but use of the high-potency drug doripenem inhibits mutant formation. Antimicrob Agents Chemother 52:3795–3800. doi: 10.1128/AAC.00464-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Power EG, Phillips I. 1993. Correlation between umuC induction and Salmonella mutagenicity assay for quinolone antimicrobial agents. FEMS Microbiol Lett 112:251–254. doi: 10.1111/j.1574-6968.1993.tb06458.x. [DOI] [PubMed] [Google Scholar]
- 14.Mamber SW, Kolek B, Brookshire KW, Bonner DP, Fung-Tomc J. 1993. Activity of quinolones in the Ames Salmonella TA102 mutagenicity test and other bacterial genotoxicity assays. Antimicrob Agents Chemother 37:213–217. doi: 10.1128/AAC.37.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Viswanathan M, Lacirignola JJ, Hurley RL, Lovett ST. 2000. A novel mutational hotspot in a natural quasipalindrome in Escherichia coli. J Mol Biol 302:553–564. doi: 10.1006/jmbi.2000.4088. [DOI] [PubMed] [Google Scholar]
- 16.Jin DJ, Gross CA. 1988. Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J Mol Biol 202:45–58. doi: 10.1016/0022-2836(88)90517-7. [DOI] [PubMed] [Google Scholar]
- 17.Severinov K, Soushko M, Goldfarb A, Nikiforov V. 1994. RifR mutations in the beginning of the Escherichia coli rpoB gene. Mol Gen Genet 244:120–126. [DOI] [PubMed] [Google Scholar]
- 18.Reynolds MG. 2000. Compensatory evolution in rifampicin-resistant Escherichia coli. Genetics 156:1471–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garibyan L, Huang T, Kim TM, Wolff E, Nguyen A, Nguyen T, Diep A, Hu K, Iverson A, Yang H, Miller JH. 2003. Use of the rpoB gene to determine the specificity of base substitution mutations on the Escherichia coli chromosome. DNA Repair 2:593–608. doi: 10.1016/S1568-7864(03)00024-7. [DOI] [PubMed] [Google Scholar]
- 20.Wolff E, Kim M, Hu K, Yang H, Miller JH. 2004. Polymerases leave fingerprints: analysis of the mutational spectrum in Escherichia coli rpoB to assess the role of polymerase IV in spontaneous mutation. J Bacteriol 186:2900–2905. doi: 10.1128/JB.186.9.2900-2905.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wrande M, Roth JR, Hughes D. 2008. Accumulation of mutants in “aging” bacterial colonies is due to growth under selection, not stress-induced mutagenesis. Proc Natl Acad Sci U S A 105:11863–11868. doi: 10.1073/pnas.0804739105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makiela-Dzbenska K, Joncyzk P, Schaaper RM, Fijalkowska IJ. 2011. Proofreading deficiency of Pol I increases the levels of spontaneous rpoB mutations in E. coli. Mutat Res 712:28–32. doi: 10.1016/j.mrfmmm.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corzett CH, Goodman MF, Finkel SE. 2013. Competitive fitness during feast and famine: how SOS DNA polymerases influence physiology and evolution in Escherichia coli. Genetics 194:409–420. doi: 10.1534/genetics.113.151837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drlica K, Hisa H, Kerns R, Malik M, Mustaev A, Zhao X. 2009. Quinolones: action and resistance updated. Curr Top Med Chem 9:981–998. doi: 10.2174/156802609789630947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Theodore A, Lewis K, Vulic M. 2013. Tolerance of Escherichia coli to fluoroquinolone antibiotics depends on specific components of the SOS response pathway. Genetics 195:1265–1276. doi: 10.1534/genetics.113.152306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phillips I, Culebras E, Moreno F, Baquero F. 1987. Induction of the SOS response by new 4-quinolones. J Antimicrob Chemother 20:631–638. doi: 10.1093/jac/20.5.631. [DOI] [PubMed] [Google Scholar]
- 27.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cupples C, Miller JH. 1989. A set of lacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc Natl Acad Sci U S A 86:5345–5349. doi: 10.1073/pnas.86.14.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cupples CG, Cabrera M, Cruz C, Miller JH. 1990. A set of lacZ mutations in Escherichia coli that allow rapid detection of specific frameshift mutations. Genetics 125:275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zambrano MM, Siegele DA, Almiron M, Tormo A, Kolter R. 1993. Microbial competition: Escherichia coli mutants that take over stationary phase cultures. Science 259:1757–1760. doi: 10.1126/science.7681219. [DOI] [PubMed] [Google Scholar]
- 32.Yamanaka K, Minko IG, Finkel SE, Goodman MF, Lloyd RS. 2011. Role of high-fidelity Escherichia coli DNA polymerase I in replication bypass of a deoxyadenosine DNA-peptide cross-link. J Bacteriol 193:3815–3822. doi: 10.1128/JB.01550-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller JH. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria, p 194–195. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 34.Dixon WJ, Massey FJ Jr. 1969. Introduction to statistical analysis. McGraw-Hill, New York, NY. [Google Scholar]
- 35.Becket E, Tse L, Yung M, Cosico A, Miller JH. 2012. Polynucleotide phosphorylase plays an important role in the generation of spontaneous mutations in Escherichia coli. J Bacteriol 194:5613–5620. doi: 10.1128/JB.00962-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cirz RT, Jones MB, Gingles NA, Minogue TD, Jarrahi B, Peterson SN, Romesberg FE. 2007. Complete and SOS-mediated response of Staphylococcus aureus to the antibiotic ciprofloxacin. J Bacteriol 189:531–539. doi: 10.1128/JB.01464-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jee J, Rasouly A, Shamovsky I, Akivis Y, Steinman RS, Mishra B, Nudler E. 2016. Rates and mechanisms of bacterial mutagenesis from maximum-depth sequencing. Nature 534:693–696. doi: 10.1038/nature18313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu A, Tran L, Becket E, Lee K, Chinn L, Park E, Tran K, Miller JH. 2010. Antibiotic sensitivity profiles determined with an Escherichia coli gene knockout collection: generating an antibiotic bar code. Antimicrob Agents Chemother 54:1393–1403. doi: 10.1128/AAC.00906-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. 2007. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 40.Foti JJ, Devadoss B, Winkler JA, Collins JA, Walker GC. 2012. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 336:315–319. doi: 10.1126/science.1219192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Long H, Miller SF, Strauss C, Zhao C, Cheng L, Ye Z, Griffin K, Te R, Lee H, Chen C-C, Lynch M. 2016. Antibiotic treatment enhances the genome-wide mutation rate of target cells. Proc Natl Acad Sci U S A 113:E2496–E2505. doi: 10.1073/pnas.1601208113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goswami M, Mangoli SH, Jawali N. 2006. Involvement of reactive oxygen species in the action of ciprofloxacin against Escherichia coli. Antimicrob Agents Chemother 50:949–954. doi: 10.1128/AAC.50.3.949-954.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gutierrez A, Laureti L, Crussard S, Abida H, Rodríguez-Rojas A, Blázquez J, Baharoglu Z, Mazel D, Darfeuille F, Vogel J, Matic I. 2013. β-Lactam antibiotics promote bacterial mutagenesis via an RpoS-mediated reduction in replication fidelity. Nat Commun 4:1610. doi: 10.1038/ncomms2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.López E, Elez M, Matic I, Blázquez J. 2007. Antibiotic-mediated recombination: ciprofloxacin stimulates SOS-independent recombination of divergent sequences in Escherichia coli. Mol Microbiol 64:83–93. doi: 10.1111/j.1365-2958.2007.05642.x. [DOI] [PubMed] [Google Scholar]
- 45.Lopez E, Blazquez J. 2009. Effect of subinhibitory concentrations of antibiotics on intrachromosomal homologous recombination in Escherichia coli. Antimicrob Agents Chemother 53:3411–3415. doi: 10.1128/AAC.00358-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lovett ST. 2004. Encoded errors: mutations and rearrangements mediated by misalignment at repetitive DNA sequences. Mol Microbiol 52:1243–1253. doi: 10.1111/j.1365-2958.2004.04076.x. [DOI] [PubMed] [Google Scholar]
- 47.Yeh P, Tschumi AI, Kishony R. 2006. Functional classification of drugs by properties of their pairwise interactions. Nat Genet 38:489–494. doi: 10.1038/ng1755. [DOI] [PubMed] [Google Scholar]
- 48.Ang J, Song LY, D'Souza S, Hong IL, Luhar R, Yung M, Miller JH. 25 July 2016. Mutagen synergy: hypermutability generated by specific pairs of base analogs. J Bacteriol. doi: 10.1128/JB.00391-16. [DOI] [PMC free article] [PubMed] [Google Scholar]