

Abstract
Relebactam (REL [MK-7655]) is a novel class A/C β-lactamase inhibitor intended for use with imipenem for the treatment of Gram-negative bacterial infections. REL restores imipenem activity against some resistant strains of Klebsiella and Pseudomonas. In this multicenter, double-blind, controlled trial (NCT01506271), subjects who were ≥18 years of age with complicated intra-abdominal infection were randomly assigned (1:1:1) to receive 250 mg REL, 125 mg REL, or placebo, each given intravenously (i.v.) with 500 mg imipenem-cilastatin (IMI) every 6 h (q6h) for 4 to 14 days. The primary efficacy endpoint was the proportion of microbiologically evaluable (ME) subjects with a favorable clinical response at discontinuation of i.v. therapy (DCIV). A total of 351 subjects were randomized, 347 (99%) were treated, and 255 (73%) were ME at DCIV (55% male; mean age, 49 years). The most common diagnoses were complicated appendicitis (53%) and complicated cholecystitis (17%). Thirty-six subjects (13%) had imipenem-resistant Gram-negative infections at baseline. Both REL doses plus IMI were generally well tolerated and demonstrated safety profiles similar to that of IMI alone. Clinical response rates at DCIV were similar in subjects who received 250 mg REL plus IMI (96.3%) or 125 mg REL plus IMI (98.8%), and both were noninferior to IMI alone (95.2%; one-sided P < 0.001). The treatment groups were also similar with respect to clinical response at early and late follow-up and microbiological response at all visits. Pharmacokinetic/pharmacodynamic simulations show that imipenem exposure at the proposed dose of 500 mg IMI with 250 mg REL q6h provides coverage of >90% of carbapenem-resistant bacterial strains.
INTRODUCTION
The current epidemic of multidrug-resistant (MDR) bacterial infections is a critical challenge in health care today. The organisms known as “ESKAPE” pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) predominate in the resistance epidemic and have become a significant problem in hospital-acquired infections around the world (1–11). The emergence of several highly resistant Gram-negative pathogens over the past decade, namely, Acinetobacter species, MDR P. aeruginosa, and carbapenem-resistant Enterobacteriaceae (including Klebsiella species and Escherichia coli), poses a particularly troubling and escalating global health issue (2, 12).
A common mechanism of resistance is the production of β-lactamases (13), which can often be overcome by the coadministration of a β-lactamase inhibitor with a β-lactam antibiotic (7). However, the emergence of newly identified β-lactamase-mediated resistance, such as class A serine-containing β-lactamases (e.g., K. pneumoniae carbapenemases [KPCs]), class B metallo-β-lactamases, and the coupling of class A and C enzymes with β-lactam antibiotic entry porin mutations, has presented a tremendous challenge (14): these mechanisms can confer resistance to antibiotics previously used as a last resort, such as carbapenems. Without effective and tolerable antibiotics, infections caused by MDR Gram-negative organisms have a high mortality rate, particularly in vulnerable populations, such as immunocompromised patients, the elderly, and children. Thus, there is an urgent need for new, well-tolerated drugs with activity against these emerging antibiotic-resistant bacteria (2, 12, 15).
Relebactam (REL [MK-7655]) is a parenteral (intravenous [i.v.]), small-molecule β-lactamase inhibitor that is active against both class A and class C β-lactamases (16). In vitro susceptibility and hollow-fiber time-kill studies have shown that REL restores imipenem susceptibility to many imipenem-resistant isolates of AmpC-producing P. aeruginosa and Enterobacteriaceae expressing KPCs or combinations of impermeability and extended-spectrum β-lactamases (ESBLs)/AmpCs (17–19). In vivo infection models with imipenem-resistant P. aeruginosa and K. pneumoniae show that REL given with imipenem-cilastatin (IMI) has the potential to effectively treat severe Gram-negative bacterial infections (20).
In phase 1 trials, REL has been generally well tolerated when administered i.v. as single doses up to 1,150 mg, as multiple doses up to 625 mg (with 500 mg IMI) every 6 h (q6h) for 7 days, and as multiple doses of 500 mg (with 500 mg IMI) q6h for 14 days. No substantive changes in hematologic parameters, electrocardiograms, vital signs, or general well-being have occurred. Transient increases in liver transaminases were observed upon multiple dosing in a small number of subjects (unpublished data).
The target pharmacokinetic/pharmacodynamic (PK/PD) parameter for REL was determined in a delayed-treatment lung infection model of imipenem-resistant P. aeruginosa that has been validated through demonstrated correlation with clinical efficacy for other β-lactam antibacterials (unpublished data). Accordingly, the target PK/PD parameter for REL has been defined as a total plasma exposure (area under the concentration-time curve from 0 to 24 h [AUC0–24 h]) of 50.1 mg · h/liter (AUC for the free, unbound fraction of drug [fAUC] of 40 mg · h/liter) following q6h dosing (or AUC from 0 to infinity [AUC0–∞] of >12.5 mg · h/liter following single-dose administration). This is the plasma exposure of REL required to reach target efficacy (defined as a static effect on P. aeruginosa tissue burden). This exposure target has been further validated by in vitro hollow-fiber PK/PD time-kill studies (21) and is considered sufficient to inhibit class A and C β-lactamases so as to restore imipenem susceptibility to several resistant strains of Pseudomonas as well as carbapenem-resistant strains of K. pneumoniae and other Enterobacteriaceae expressing class A and/or C enzymes.
Extensive PK/PD information gathered from in vitro susceptibility testing, hollow-fiber experiments, and in vivo infection models, together with multiple-dose safety data from the phase 1 program, supports that REL doses at or above 125 mg q6h meet the identified PK/PD exposure target (19–23). However, simulations from a translational PK/PD model (24) suggest that higher concentrations of REL may be needed for efficacy against highly resistant strains of P. aeruginosa, which are identified at low frequency in surveillance studies of clinical isolates (25). Therefore, we evaluated the safety and efficacy of two REL dose levels, 125 mg and 250 mg, each given q6h with 500 mg IMI, for the treatment of complicated intra-abdominal infections (cIAI), an important area for new antimicrobial development because of their high prevalence, high morbidity and mortality, and the serious risk of clinical failure due to MDR Gram-negative bacilli (26, 27).
MATERIALS AND METHODS
Study design.
This was a prospective, multicenter, double-blind, randomized controlled trial (MK-7655 Protocol 004, NCT01506271) conducted from 16 November 2012 through 12 August 2014 at 45 sites in 20 countries. The original protocol and all amendments were approved by the relevant institutional review board or independent ethics committee at each study center. The study was conducted in accordance with the protocol, Good Clinical Practice guidelines, and the Declaration of Helsinki. Subjects provided written informed consent before any study procedures were performed.
Study participants were adults (≥18 years of age) with clinically suspected and/or bacteriologically documented cIAI requiring hospitalization and treatment with i.v. antibiotic therapy. Postoperative (or intraoperative) enrollment was encouraged. If preoperative data were available that strongly suggested an appropriate diagnosis for entry (see Appendix S1 in the supplemental material), then preoperative enrollment was allowed. Eligible diagnoses included the following: cholecystitis (including gangrenous) with rupture, perforation, or progression of infection beyond the gallbladder wall; diverticular abscess; appendiceal perforation and periappendiceal abscess; acute gastric and duodenal perforations, if operated on >24 h after occurrence; traumatic perforation of the intestines, if operated on >12 h after occurrence; peritonitis due to perforated viscus, surgical intervention, or other focus of infection (but not spontaneous bacterial peritonitis associated with cirrhosis and chronic ascites); and intra-abdominal abscess.
Exclusion criteria included an APACHE II score of >30, antibiotic therapy effective against the identified pathogen taken after collection of culture for admission and before initiation of study therapy, systemic antibiotic (for >24 h) effective against the presumed or documented etiologic pathogen(s) administered within the prior 72 h, and baseline renal dysfunction (creatinine clearance [CLCR] of <50 ml/min) or hepatic dysfunction (alanine aminotransferase [ALT] or aspartate aminotransferase [AST] of >3× the upper limit of normal [ULN]).
Randomization was stratified based on disease severity (APACHE II score of ≤15 or >15) at screening using a centralized system and a randomized allocation schedule. Subjects were assigned in a 1:1:1 ratio to receive i.v. study therapy of 250 mg REL plus IMI, 125 mg REL plus IMI, or placebo plus IMI, once every 6 h. REL (or placebo) and IMI were infused over 30 min from 2 separate i.v. bags or 2 separate infusion syringes but through a single cannula. Masking of study therapy was not required because the REL infusions were visually indistinguishable from the placebo infusion. At each study center, a pharmacist who was not involved in evaluation of the subjects prepared and accounted for the infusion bags of active i.v. study therapy and placebo. Treatment group assignments were not revealed to the subjects, study investigators and staff (other than the pharmacist), or sponsor personnel until all subjects completed the study and the database was locked.
The minimum duration of i.v. study therapy was 96 h, or 4 full days (16 doses for q6h dosing or 12 doses for q8h dosing [renal adjustment]). The total duration of i.v. study therapy generally did not exceed 7 days. If, in the investigator's judgment, additional treatment was needed, i.v. study therapy was allowed to continue up to 14 days. If i.v. therapy was required for >14 days, the subject was discontinued from i.v. study therapy so other medication could be administered and was monitored at subsequent time points for safety but was considered a failure for efficacy.
For pharmacokinetic analyses, whole blood (4 ml) to obtain plasma for determination of REL, imipenem, and cilastatin concentrations was collected from all subjects at screening (visit 1) and after the first dose of study therapy at visit 2 (at approximately 30 min and 4 h after the first i.v. drug infusion was started). The exact date and time of initiation of infusion and whole-blood collection were recorded for all plasma samples.
Data on adverse events (based on assessment of symptoms, vital signs, and/or physical examination findings) and laboratory safety measures were collected through 14 days after completion of i.v. study therapy. Data on serious adverse events were collected through the end of the study if considered by an investigator to be related to the investigational compound.
Subjects were evaluated for efficacy at discontinuation of i.v. therapy (DCIV), at 5 to 9 days after i.v. study therapy (early follow-up), and at 28 to 42 days after i.v. study therapy (late follow-up). Clinical response was determined by the investigator based on prespecified definitions. The evaluation was made on the basis of the resolution of presenting clinical signs and symptoms, including evidence of a systemic inflammatory response (i.e., fever, elevated white blood cell [WBC] count, decreased blood pressure, increased pulse/respiratory rate, hypoxemia, and/or altered mental status) and physical findings consistent with IAI (e.g., abdominal pain and/or tenderness, abdominal wall rigidity, abdominal mass, or ileus).
Favorable clinical response (cure or sustained cure) was defined as resolution of all or most presenting signs and symptoms of IAI infection without the need for additional antibiotic therapy. An unfavorable clinical response (failure) was defined as lack of response to study therapy as documented by persistence or worsening of presenting symptoms and/or signs of IAI infection. Failure could have also included the following: death related to the cIAI at any time, persisting or recurrent infection within the abdomen documented by findings from percutaneous or operative reintervention, postsurgical wound infection, relapse, or use of additional antibiotic for baseline or new cIAI.
Global response was also determined by the investigator at day 28 following randomization. To be considered a favorable global response (i.e., cure) the subject must have met all of the following prespecified criteria: resolution of presenting signs and symptoms of IAI, survival, no unplanned percutaneous or surgical procedures for IAI, no receipt of antibacterial therapy for index or emergent IAI, and no other event related to the index or emergent IAI that resulted in clinical instability or worsening. The global response was considered failure if the subject did not meet all of these criteria.
Microbiologic response was imputed from the clinical response if follow-up cultures from the site of infection were not available. Although adequacy of surgical source control was not formally adjudicated by an independent evaluator in this study, assessment of source control was performed by the study physicians as part of the evaluability assessments, as the study protocol directed investigators to follow Infectious Diseases Society of America/Surgical Infection Society (IDSA/SIS) guidelines for adequate surgical intervention (28). An additional study follow-up visit occurred at day 28 (+7 days) post-randomization (global follow-up) and included assessment of the global response as well as the microbiological response. Detailed response criteria are provided in Appendix S2 in the supplemental material.
Statistical analysis.
Safety and tolerability were assessed by clinical review of all relevant parameters (including adverse events and laboratory tests) in the all-patients-as-treated (APaT) population, which consisted of all randomized subjects who received at least one dose of i.v. study therapy. To gain further insight into the hepatic transaminase elevations observed in phase 1, two events of clinical interest (ECI) were prespecified: (i) a confirmed aspartate aminotransferase (AST) or ALT level of ≥5× ULN and (ii) an AST or ALT level of ≥3× ULN with a total bilirubin level of ≥2× ULN and alkaline phosphatase level of <2× ULN (Hy's law). Between-treatment differences and 95% confidence interval (CI) were calculated using the Miettinen and Nurminen method without stratification (29); P values also were calculated for the prespecified ECI.
Two interim analyses were conducted during the course of the trial, when ∼25% and ∼50% of planned subjects in the phase 2b program (which included this study and a similar study, Protocol 7655-003, in subjects with complicated urinary tract infection) had reached the early follow-up visit, to evaluate the safety and tolerability of each dose of REL plus IMI in comparison to the control regimen and to inform the dose selection for phase 3. The APaT population was used for both analyses and included only those subjects who had completed the early follow-up visit or discontinued the study before this visit. These analyses were conducted by a statistician who had no other responsibilities with respect to the study, and the results were reviewed by an internal data monitoring committee (DMC).
The primary efficacy endpoint was the proportion of subjects in the microbiologically evaluable (ME) population who achieved a favorable clinical response at DCIV. The ME population was defined as subjects who met the protocol definition of cIAI, had a prestudy/postoperative culture from the site of infection that grew at least one Gram-negative enteric and/or anaerobic pathogen, had no significant deviations from the protocol that could impact the efficacy assessment, and received ≥96 h of i.v. study therapy. A sensitivity analysis was performed for the primary endpoint using the microbiological intention-to-treat (MITT) population, which was defined as subjects who received at least one dose of i.v. study therapy and had a prestudy/postoperative culture from the site of infection that grew at least one Gram-negative enteric and/or anaerobic pathogen.
With ∼117 subjects per treatment group, the study was powered at 80% to demonstrate noninferiority of REL plus IMI versus the control regimen on the primary endpoint, at an overall one-sided 2.5% α level, if the underlying treatment difference is zero percentage points (assuming a noninferiority margin of −15 percentage points, a 90% clinical response rate for the control regimen, and a 60 to 65% microbiological evaluability rate). Because this was a phase 2 dose-ranging study, the selected noninferiority margin was larger than the 10% margin currently recommended in FDA guidance for cIAI (30).
For the primary efficacy endpoint, 95% CIs for between-treatment differences and associated P values were calculated by the unconditional asymptotic Miettinen and Nurminen method without stratification (29). Between-treatment differences and 95% CIs were also calculated for the secondary endpoints (clinical response at early and late follow-up, microbiologic response, and global response). Efficacy comparisons for imipenem-resistant infections used Fisher's exact test (one sided) due to the small number of subjects expected in this subgroup.
A closed testing procedure with a fixed testing sequence was used to adjust for multiplicity (two treatment comparisons in the primary hypothesis). REL at 250 mg plus IMI was compared to the control regimen first, at a one-sided confidence level of α = 0.025. If noninferiority was established for this dose, then 125 mg REL plus IMI was compared to the control regimen at a one-sided α level of 0.025. If noninferiority was established for either dose of REL relative to control, then superiority testing was performed for that dose of REL versus the control regimen.
RESULTS
Study population.
Of the 351 subjects randomized, 347 (98.9%) received at least one dose of i.v. study therapy, 337 (96%) completed the study, and 330 (94%) completed the study therapy regimen (Table 1). Overall, 277 (78.9%) of the randomized subjects were included in the MITT population, and 255 (72.6%) were included in the ME population (Fig. 1); reasons for exclusion from evaluation are provided in Appendix S3 in the supplemental material.
TABLE 1.
Disposition of subjects by treatment group in the randomized population
| Group |
n (%) in treatment groupa |
|||
|---|---|---|---|---|
| 250 mg REL + IMI | 125 mg REL + IMI | Placebo + IMI | Total | |
| Randomizedb | 118 | 116 | 117 | 351 |
| Treated with any study therapy | 117 (99.2) | 114 (98.3) | 116 (99.1) | 347 (98.9) |
| Completed study medication | 112 (94.9) | 107 (92.2) | 111 (94.9) | 330 (94.0) |
| Discontinued study medication because of: | 5 (4.2) | 7 (6.0) | 5 (4.3) | 17 (4.8) |
| Adverse event | 1 (0.8) | 4 (3.4) | 3 (2.6) | 8 (2.3) |
| Death | 0 (0.0) | 1 (0.9) | 0 (0.0) | 1 (0.3) |
| Lack of efficacy | 0 (0.0) | 0 (0.0) | 1 (0.9) | 1 (0.3) |
| Lost to follow-up | 0 (0.0) | 0 (0.0) | 1 (0.9) | 1 (0.3) |
| Physician decision | 0 (0.0) | 2 (1.7) | 0 (0.0) | 2 (0.6) |
| Protocol violation | 2 (1.7) | 0 (0.0) | 0 (0.0) | 2 (0.6) |
| Insufficient drug supply | 1 (0.8) | 0 (0.0) | 0 (0.0) | 1 (0.3) |
| Withdrawal by subject | 1 (0.8) | 0 (0.0) | 0 (0.0) | 1 (0.3) |
| Completed study | 114 (96.6) | 109 (94.0) | 114 (97.4) | 337 (96.0) |
| Discontinued from study because of: | 4 (3.4) | 7 (6.0) | 3 (2.6) | 14 (4.0) |
| Adverse event | 0 (0.0) | 2 (1.7) | 0 (0.0) | 2 (0.6) |
| Death | 0 (0.0) | 1 (0.9) | 0 (0.0) | 1 (0.3) |
| Lost to follow-up | 0 (0.0) | 1 (0.9) | 2 (1.7) | 3 (0.9) |
| Progressive disease | 0 (0.0) | 1 (0.9) | 0 (0.0) | 1 (0.3) |
| Protocol violation | 3 (2.5) | 1 (0.9) | 0 (0.0) | 4 (1.1) |
| Insufficient drug supply | 1 (0.8) | 0 (0.0) | 0 (0.0) | 1 (0.3) |
| Withdrawal by subject | 0 (0.0) | 1 (0.9) | 1 (0.9) | 2 (0.6) |
REL, relebactam; IMI, 500 mg imipenem-cilastatin.
Percentages are based on the number of subjects randomized as the denominator.
FIG 1.
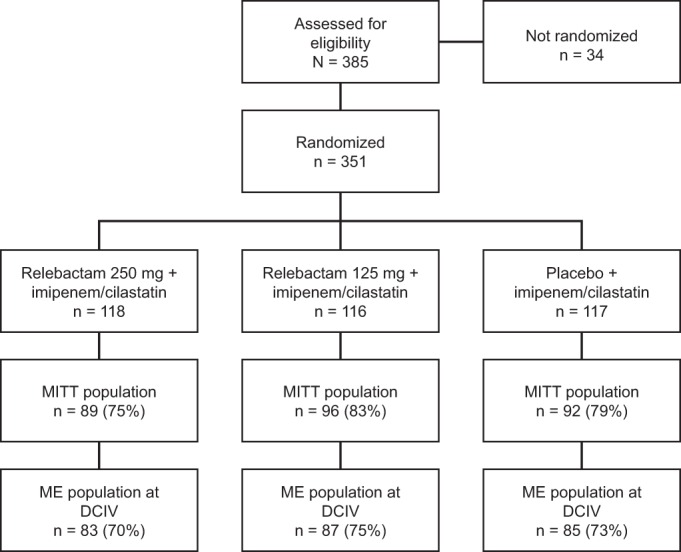
Profile of study enrollment and subject populations analyzed. MITT, microbiological intent to treat; ME, microbiologically evaluable; DCIV, discontinuation of i.v. study therapy.
In the ME population, 55.3% of the subjects were male, 92.5% were white, and 78.8% were <65 years of age (median age, 50.0 years; range, 18 to 88 years). Baseline characteristics, such as APACHE II score, time of enrollment, and presence of bacteremia, were balanced across the treatment groups (Table 2). The most common clinical diagnoses overall were complicated appendicitis (52.5%), complicated cholecystitis (16.5%), and perforated hollow viscus (11.4%). Most baseline diagnoses (94.5%) were not classified as postoperative infections. The treatment groups were generally similar with respect to the distribution of these characteristics (Table 3).
TABLE 2.
Baseline characteristics of the ME population
| Parameter | Result for treatment groupa |
|||
|---|---|---|---|---|
| 250 mg REL + IMI (n = 83) | 125 mg REL + IMI (n = 87) | Placebo + IMI (n = 85) | Total (n = 255) | |
| Gender, n (%) | ||||
| Female | 32 (38.6) | 45 (51.7) | 37 (43.5) | 114 (44.7) |
| Male | 51 (61.4) | 42 (48.3) | 48 (56.5) | 141 (55.3) |
| Race, n (%) | ||||
| Asian | 2 (2.4) | 3 (3.4) | 4 (4.7) | 9 (3.5) |
| Black | 1 (1.2) | 3 (3.4) | 0 (0.0) | 4 (1.6) |
| White | 77 (92.8) | 78 (89.7) | 81 (95.3) | 236 (92.5) |
| Multiracial | 3 (3.6) | 3 (3.4) | 0 (0.0) | 6 (2.4) |
| Hispanic or Latino | 5 (6.0) | 9 (10.3) | 1 (1.2) | 15 (5.9) |
| Age, yr | ||||
| Mean (SD) | 48.3 (18.4) | 49.7 (16.4) | 48.8 (17.7) | 49.0 (17.4) |
| <65 | 65 (78.3) | 66 (75.9) | 70 (82.4) | 201 (78.8) |
| ≥65 | 18 (21.7) | 21 (24.1) | 15 (17.6) | 54 (21.2) |
| Wt, mean (SD) kg | 79.3 (15.4) | 77.8 (15.5) | 79.4 (19.3) | 78.8 (16.8) |
| APACHE II score (stratum), n (%) | ||||
| ≤15 | 80 (96.4) | 84 (96.6) | 81 (95.3) | 245 (95.1) |
| >15 | 3 (3.6) | 3 (3.4) | 4 (4.7) | 10 (3.9) |
| Time of enrollment, n (%) | ||||
| Preoperative | 14 (16.9) | 17 (19.5) | 10 (11.8) | 41 (16.1) |
| Postoperative | 65 (78.3) | 68 (78.2) | 72 (84.7) | 205 (80.4) |
| Intraoperative | 4 (4.8) | 2 (2.3) | 3 (3.5) | 9 (3.5) |
| Bacteremia with cIAI, n (%) | 3 (3.6) | 4 (4.6) | 3 (3.5) | 10 (3.9) |
REL, relebactam; IMI, 500 mg imipenem-cilastatin.
TABLE 3.
Primary diagnoses for the ME population
| Clinical diagnosis |
n (%) with diagnosis by treatment groupa |
|||
|---|---|---|---|---|
| 250 mg REL + IMI (n = 83) | 125 mg REL + IMI (n = 87) | Placebo + IMI (n = 85) | Total (n = 255) | |
| Complicated appendicitis | 44 (53.0) | 47 (54.0) | 43 (50.6) | 134 (52.5) |
| Perforation | 22 (26.5) | 23 (26.4) | 20 (23.5) | 65 (25.5) |
| Periappendiceal abscess | 22 (26.5) | 24 (27.6) | 23 (27.1) | 69 (27.1) |
| Diverticular abscess | 5 (6.0) | 2 (2.3) | 5 (5.9) | 12 (4.7) |
| Complicated cholecystitis | 14 (16.9) | 15 (17.2) | 13 (15.3) | 42 (16.5) |
| Gangrenous | 9 (10.8) | 9 (10.3) | 8 (9.4) | 26 (10.2) |
| Perforation | 1 (1.2) | 2 (2.3) | 3 (3.5) | 6 (2.4) |
| Progression beyond gallbladder wall | 4 (4.8) | 4 (4.6) | 2 (2.4) | 10 (3.9) |
| Perforated hollow viscus | 8 (9.6) | 9 (10.3) | 12 (14.1) | 29 (11.4) |
| Stomach | 4 (4.8) | 1 (1.1) | 3 (3.5) | 8 (3.1) |
| Duodenum | 1 (1.2) | 5 (5.7) | 2 (2.4) | 8 (3.1) |
| Jejunum/ileum | 0 (0.0) | 1 (1.1) | 2 (2.4) | 3 (1.2) |
| Colon | 3 (3.6) | 2 (2.3) | 5 (5.9) | 10 (3.9) |
| Peritonitis | 3 (3.6) | 1 (1.1) | 0 (0.0) | 4 (1.6) |
| Posttraumatic | 1 (1.2) | 0 (0.0) | 0 (0.0) | 1 (0.4) |
| Postsurgical | 2 (2.4) | 1 (1.1) | 0 (0.0) | 3 (1.2) |
| Intra-abdominal abscess | 2 (2.4) | 3 (3.4) | 3 (3.5) | 8 (3.1) |
| Liver | 2 (2.4) | 2 (2.3) | 3 (3.5) | 7 (2.7) |
| Splenic | 0 (0.0) | 1 (1.1) | 0 (0.0) | 1 (0.4) |
| Multiple abscesses | 2 (2.4) | 2 (2.3) | 3 (3.5) | 7 (2.7) |
| Other | 5 (6.0) | 8 (9.2) | 6 (7.1) | 19 (7.5) |
| Postoperative infection | 3 (3.6) | 6 (6.9) | 5 (5.9) | 14 (5.5) |
REL, relebactam; IMI, 500 mg imipenem-cilastatin.
Safety endpoints.
At least one adverse event was reported by 45.8% of subjects overall (Table 4). Rates of drug-related adverse events and serious adverse events were generally similar across all treatment groups. One serious adverse event was considered drug related: severe thrombocytosis in a subject who received IMI alone and subsequently discontinued study therapy. An additional serious adverse event (paralytic ileus) was initially recorded as drug related but subsequently determined by the investigator not to be drug related. The rates of discontinuation due to an adverse event were low overall and similar across treatment groups. Adverse events leading to discontinuation were considered drug related in one subject who received 125 mg REL plus IMI (decreased creatinine clearance) and in three subjects who received IMI alone (thrombocytosis, nausea, and increased ALT). Three fatal adverse events occurred (septic shock, ventricular fibrillation, and intestinal infarction): all were in subjects receiving 125 mg REL plus IMI and were considered not drug related. The most common adverse events (incidence of >5% in any group) were diarrhea, nausea, and vomiting, which occurred at similar rates across the treatment groups.
TABLE 4.
Summary of adverse eventsa
| Parameter | Subjects with AE by treatment group, n (%) |
REL vs placebo comparison, % difference (95% CI)b |
|||
|---|---|---|---|---|---|
| 250 mg REL + IMI (n = 117) | 125 mg REL + IMI (n = 116) | Placebo + IMI (n = 114) | 250 mg REL + IMI | 125 mg REL + IMI | |
| At least 1 AE | 57 (48.7) | 55 (47.4) | 47 (41.2) | 7.5 (−5.4 to 20.1) | 6.2 (−6.7 to 18.8) |
| Drug-related AE | 16 (13.7) | 16 (13.8) | 11 (9.6) | 4.0 (−4.5 to 12.7) | 4.1 (−4.4 to 12.8) |
| Serious AE | 4 (3.4) | 11 (9.5) | 8 (7.0) | −3.6 (−10.3 to 2.4) | 2.5 (−5.0 to 10.1) |
| Serious and drug-related AE | 1 (0.9)c | 0 | 1 (0.9) | −0.0 (−4.0 to 3.9) | −0.9 (−4.8 to 2.4) |
| Death | 0 | 3 (2.6) | 0 | 0.0 (−3.3 to 3.2) | 2.6 (−0.7 to 7.3) |
| Discontinued due to AE | 1 (0.9) | 5 (4.3) | 3 (2.6) | −1.8 (−6.7 to 2.3) | 1.7 (−3.7 to 7.4) |
| Drug-related AE | 0 | 1 (0.9) | 3 (2.6) | −2.6 (−7.5 to 0.6) | −1.8 (−6.7 to 2.4) |
| Serious AE | 0 | 3 (2.6) | 1 (0.9) | −0.9 (−4.8 to 2.3) | 1.7 (−2.5 to 6.6) |
| Drug-related serious AE | 0 | 0 | 1 (0.9) | −0.9 (−4.8 to 2.3) | −0.9 (−4.8 to 2.4) |
| Most common AEs | |||||
| Diarrhea | 7 (6.0) | 7 (6.0) | 5 (4.4) | 1.6 (−4.7 to 8.1) | 1.6 (−4.6 to 8.2) |
| Nausea | 8 (6.8) | 9 (7.8) | 8 (7.0) | −0.2 (−7.3 to 6.9) | 0.7 (−6.5 to 8.0) |
| Vomiting | 7 (6.0) | 9 (7.8) | 3 (2.6) | 3.4 (−2.3 to 9.6) | 5.1 (−0.7 to 11.8) |
| Events of clinical interest | |||||
| AST or ALT of ≥5× ULN | 2 (1.7) | 0 | 2 (1.8) | −0.0 (−4.7 to 4.5) | −1.8 (−6.2 to 1.5) |
| AST or ALT of ≥3× ULN, total bilirubin level of ≥2× ULN, and alkaline phosphatase level of <2× ULN | 1 (0.9) | 0 | 0 | 0.9 (−2.4 to 4.7) | 0.0 (−3.3 to 3.2) |
AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CI, confidence interval; IMI, imipenem-cilastatin; ULN, upper limit of normal.
The difference in percentage and 95% CI are based on the unconditional and asymptotic Miettinen and Nurminen method without stratification.
Although subsequently determined by the investigator not to be drug related, this event appears in the table because the change in causality was not corrected in the clinical database prior to database lock.
The two prespecified ECIs ([i] confirmed AST or ALT level of ≥5× ULN and [ii] AST or ALT level of ≥3× ULN with total bilirubin level of ≥2× ULN and alkaline phosphatase level of <2× ULN) occurred in 5 subjects overall, with no significant difference between either dose of REL plus IMI and IMI alone (for all values, P > 0.05) (Table 4). For the subject meeting criteria for the second ECI (Hy's law), the event occurred on day 2 of the study and was not considered by the investigator to be related to study treatment. The ECI was instead thought to be related to septic shock and abdominal operative procedures, conditions that were present at randomization. Treatment with study drug was not interrupted during this event.
Favorable clinical response.
At the DCIV visit, the proportions of subjects in the ME population with a favorable clinical response were generally similar among the 3 treatment groups, ranging from 95.2% to 98.8% (Table 5). Both doses of REL plus IMI were noninferior to IMI alone with respect to the clinical response rate at DCIV (both at P < 0.001), although there was insufficient evidence to conclude that either dose of REL plus IMI is superior to IMI alone for this endpoint. Clinical response rates at the early and late follow-up visits were generally similar across the treatment groups (Table 5). Clinical response rates in the MITT population (Table 5) were consistent with the ME population.
TABLE 5.
Proportion of subjects with favorable clinical responsea
| Parameter | Result for treatment group, % (n/m) |
REL vs placebo comparison, % difference (95% CI)b |
|||
|---|---|---|---|---|---|
| 250 mg REL + IMI | 125 mg REL + IMI | Placebo + IMI | 250 mg REL + IMI | 125 mg REL + IMI | |
| ME population | |||||
| DCIV | 96.3 (78/81) | 98.8 (85/86) | 95.2 (79/83) | 1.1 (−6.2 to 8.6)* | 3.7 (−2.0 to 10.8)* |
| EFU | 94.9 (75/79) | 94.2 (81/86) | 96.3 (78/81) | −1.4 (−9.1 to 6.0) | −2.1 (−9.7 to 5.3) |
| LFU | 93.7 (74/79) | 95.3 (81/85) | 94.9 (75/79) | −1.3 (−9.6 to 6.9) | 0.4 (−7.2 to 8.2) |
| MITT population | |||||
| DCIV | 89.9 (80/89) | 91.7 (88/96) | 90.2 (83/92) | −0.3 (−9.6 to 8.9)§ | 1.4 (−7.2 to 10.3)* |
| EFU | 86.5 (77/89) | 88.5 (85/96) | 89.1 (82/92) | −2.6 (−12.7 to 7.2) | −0.6 (−10.0 to 8.9) |
| LFU | 87.6 (78/89) | 89.6 (86/96) | 85.9 (79/92) | 1.8 (−8.5 to 12.0) | 3.7 (−5.9 to 13.6) |
n/m, number of subjects with favorable clinical response/number of subjects with clinical response assessment; CI, confidence interval; DCIV, discontinuation of i.v. therapy; EFU, early follow-up (5 to 9 days after completion of i.v. study therapy); LFU, late follow-up (28 to 42 days after completion of i.v. study therapy).
The 95% CIs for between-treatment differences and associated P values are based on the unconditional asymptotic Miettinen and Nurminen method without stratification. *, P < 0.001; §, P = 0.002. (A P value of <0.025 indicates REL plus IMI is noninferior to control.)
The most common pathogens identified at baseline were E. coli (171 isolates), K. pneumoniae (38 isolates), and P. aeruginosa (37 isolates). The per-pathogen clinical response rates at DCIV were generally similar across treatment groups for nearly all pathogens identified (see Appendix S4 in the supplemental material), including the most common pathogens (Table 6).
TABLE 6.
Proportion of subjects in the ME population with favorable clinical response at DCIV by baseline pathogena
| Pathogen | Result for treatment group |
REL vs placebo comparison, % difference (95% CI)b |
||||||
|---|---|---|---|---|---|---|---|---|
| 250 mg REL + IMI (n = 81) |
125 mg REL + IMI (n = 86) |
Placebo + IMI (n = 83) |
250 mg REL + IMI | 125 mg REL + IMI | ||||
| n/m | % | n/m | % | n/m | % | |||
| Gram-positive aerobic cocci | 32/32 | 100 | 32/33 | 97.0 | 33/34 | 97.1 | 2.9 (−8.1 to 15.1) | −0.1 (−12.9 to 12.4) |
| Enterococcus faecalis | 7/7 | 100 | 5/5 | 100 | 5/5 | 100 | 0.0 (−37.4 to 45.6) | 0.0 (−46.1 to 46.1) |
| Streptococcus anginosus | 5/5 | 100 | 6/6 | 100 | 7/7 | 100 | 0.0 (−45.6 to 37.4) | 0.0 (−41.0 to 37.3) |
| Streptococcus constellatus | 2/2 | 100 | 5/6 | 83.3 | 6/6 | 100 | 0.0 | −16.7 (−57.9 to 28.5) |
| Gram-negative aerobic bacilli | 73/75 | 97.3 | 73/73 | 100 | 68/72 | 94.4 | 2.9 (−4.4 to 11.2) | 5.6 (0.4 to 13.5) |
| Enterobacter cloacae | 7/7 | 100 | 4/4 | 100 | 4/4 | 100 | 0.0 (−37.6 to 51.4) | 0.0 (−52.3 to 52.3) |
| Escherichia coli | 53/55 | 96.4 | 56/56 | 100 | 47/51 | 92.2 | 4.2 (−5.7 to 15.4) | 7.8 (1.1 to 18.6) |
| Klebsiella pneumoniae | 10/10 | 100 | 12/12 | 100 | 10/12 | 83.3 | 16.7 (−14.4 to 45.5) | 16.7 (−10.6 to 45.4) |
| Proteus mirabilis | 8/8 | 100 | 4/4 | 100 | 6/6 | 100 | 0.0 (−34.1 to 40.8) | 0.0 (−51.6 to 41.6) |
| Pseudomonas aeruginosa | 11/11 | 100 | 13/13 | 100 | 10/12 | 83.3 | 16.7 (−12.4 to 45.5) | 16.7 (−9.0 to 45.4) |
| Gram-negative anaerobic bacilli | 22/24 | 91.7 | 30/30 | 100 | 26/27 | 96.3 | −4.6 (−23.0 to 11.4) | 3.7 (−8.1 to 18.5) |
| Bacteroides fragilis | 11/11 | 100 | 8/8 | 100 | 12/12 | 100 | 0.0 (−26.7 to 25.1) | 0.0 (−33.6 to 25.2) |
| Bacteroides thetaiotaomicron | 6/6 | 100 | 6/6 | 100 | 6/7 | 85.7 | 14.3 (−29.9 to 52.8) | 14.3 (−29.9 to 52.8) |
The most common pathogens (those with at least 15 unique baseline isolates) are shown. CI, confidence interval; IMI, imipenem-cilastatin; n/m, number of subjects with pathogen and favorable clinical response/number of subjects with pathogen and clinical response assessment. Subjects with an indeterminate or missing response are excluded from the analysis.
The 95% confidence intervals are based on the unconditional asymptotic Miettinen and Nurminen method without stratification.
Imipenem-nonsusceptible infections.
At baseline, 36 subjects (13%) in the MITT population had Gram-negative infections that were imipenem nonsusceptible, including both complete and intermediate resistance, with comparable numbers in the 3 treatment groups. There were no apparent differences in subject characteristics between subjects with and those without IMI-nonsusceptible organisms. Thirty-four of these subjects were ME at the DCIV visit: all 34 had a favorable clinical response and a favorable microbiological response (14/14 receiving 250 mg REL plus IMI, 9/9 receiving 125 mg REL plus IMI, and 11/11 receiving IMI alone). Of the 40 pathogens isolated from these 34 subjects, 7 were nonsusceptible to imipenem alone but were susceptible to IMI plus REL; the remaining 33 organisms (5.3% of all isolates) were nonsusceptible to both imipenem alone and IMI plus REL (Table 7).
TABLE 7.
Baseline organisms nonsusceptible to imipenem alone
| Organism | No. of isolates |
||
|---|---|---|---|
| Totala | REL + IMI susceptible | REL + IMI nonsusceptible | |
| Total isolates | 40 | 7 | 33 |
| Acinetobacter baumannii complex | 3 | 3 | |
| Acinetobacter lwoffii | 1 | 1 | |
| Alcaligenes xylosoxidans | 1 | 1 | |
| Escherichia coli | 1 | 1 | |
| Morganella morganii | 5 | 5 | |
| Proteus mirabilis | 19 | 2 | 17 |
| Proteus penneri | 1 | 1 | |
| Proteus vulgaris | 1 | 1 | |
| Pseudomonas aeruginosa | 5 | 3 | 2 |
| Stenotrophomonas maltophilia | 3 | 3 | |
From 34 subjects, all 34 had a favorable clinical/global response and presumptive eradication at the end of study therapy.
Microbiologic response and global response.
Microbiological response rates in the ME population were similar for 250 mg REL plus IMI, 125 mg REL plus IMI, and IMI alone at all visits, including 28 days post-randomization (Table 8). Microbiological response was imputed in the majority of subjects with a favorable response rating since it is generally not possible nor medically acceptable to perform repeat intra-abdominal sampling on patients who are clinically improving or well. Global response was also assessed at 28 days post-randomization, in the MITT population. The proportions of subjects with a favorable global response were generally similar among the 3 treatment groups: 250 mg REL plus IMI, 86.5% (77/89); 125 mg REL plus IMI, 89.6% (86/96); IMI alone, 84.8% (78/92).
TABLE 8.
Proportion of subjects in the ME population with favorable microbiological responsea
| Parameter | Result for treatment group, % (n/m) |
REL vs placebo comparison, % difference (95% CI)b |
|||
|---|---|---|---|---|---|
| 250 mg REL + IMI | 125 mg REL + IMI | Placebo + IMI | 250 mg REL + IMI | 125 mg REL + IMI | |
| DCIV | 97.6 (81/83) | 100 (86/86) | 97.6 (82/84) | −0.0 (−6.3 to 6.2) | 2.4 (−2.0 to 8.3) |
| EFU | 97.4 (76/78) | 97.6 (80/82) | 97.5 (78/80) | −0.1 (−6.7 to 6.4) | 0.1 (−6.3 to 6.5) |
| LFU | 96.2 (75/78) | 97.6 (80/82) | 96.2 (75/78) | 0.0 (−7.4 to 7.4) | 1.4 (−5.1 to 8.6) |
| GFU | 96.2 (75/78) | 97.5 (79/81) | 96.2 (75/78) | 0.0 (−7.4 to 7.4) | 1.4 (−5.2 to 8.6) |
Subjects with indeterminate or missing response were excluded (2 at DCIV, 8 at EFU, 8 at GFU, and 8 at LFU). CI, confidence interval; DCIV, discontinuation of i.v. therapy; EFU, early follow-up; GFU, global follow-up; LFU, late follow-up; n/m, number of subjects with favorable microbiological response/number of ME subjects included in the analysis.
The 95% confidence intervals of between-treatment differences are based on the unconditional asymptotic Miettinen and Nurminen method without stratification.
Population PK modeling and probability of target attainment.
Population PK models of both REL and imipenem were constructed based on data from three adult phase 1 studies (a total of 149 subjects and 5,777 observed concentrations) and the current phase 2 study (326 patients and 1,436 observed concentrations) and were used to simulate the probability of target attainment for the 250-mg dose of REL.
Consistent with observed data from phase 1 studies, imipenem and REL were found to exhibit compatible PK, and a two-compartment model with first-order elimination provided the best fit to the data for both drugs. Between-subject variability for clearance (CL) and central and peripheral volumes (Vc and Vp) as well as proportional residual error were estimated for each drug. Cilastatin concentrations were not considered in the model, as any impact of changing cilastatin levels is captured in the imipenem PK. Creatinine clearance (CLCR) was the most influential covariate on the PK of both drugs since they are predominantly renally excreted. Body weight (BWT) was also found to be a significant covariate on CL for imipenem alone and on Vc and Vp for both drugs—all described by a power model. Imipenem and REL did not have a significant PK interaction; coadministration of these drugs did not alter the PK parameters of either entity compared with each drug being administered alone. Age, gender, and race did not significantly impact PK parameters for either imipenem or REL when tested in the population model, consistent with phase 1 findings. There was also a numerical finding that healthy volunteers had a lower estimated Vc for both imipenem and REL and a lower estimated CL for imipenem relative to cIAI patients, which may likely be a data artifact; the typical PK profiles appear similar for healthy volunteers and patients, implying this finding may not be clinically meaningful.
Using the final population PK models for both imipenem and REL, simulations were conducted to evaluate the probability of target attainment and recommend an optimal dosing regimen based on meeting the following criteria: (i) imipenem (lower bound), unbound time above MIC of ≥30%; (ii) REL (lower bound), median AUC0–24 h of ≥150 μM · h (equivalent to 50.1 mg · h/liter); (iii) REL (upper bound), median AUC0–24 h and Cmax of ≤800 μM · h and ≤122 μM, respectively (based on observed data at a 625-mg dose, the highest clinical dose administered in phase 1 multiple-dose studies in healthy adult volunteers).
Simulations showed that imipenem exposures at the proposed dose of 500 mg (with 250 mg REL) q6h provide coverage of MIC values that span the vast majority of distribution of P. aeruginosa and KPC-producing isolates from the SMART 2011 surveillance study (Fig. 2). Exposure projections of REL do not exceed the defined safety upper bound, while the efficacy PK target (AUC) for REL is exceeded across the range of body weights without the need for weight-based dosing adjustments (Fig. 3).
FIG 2.
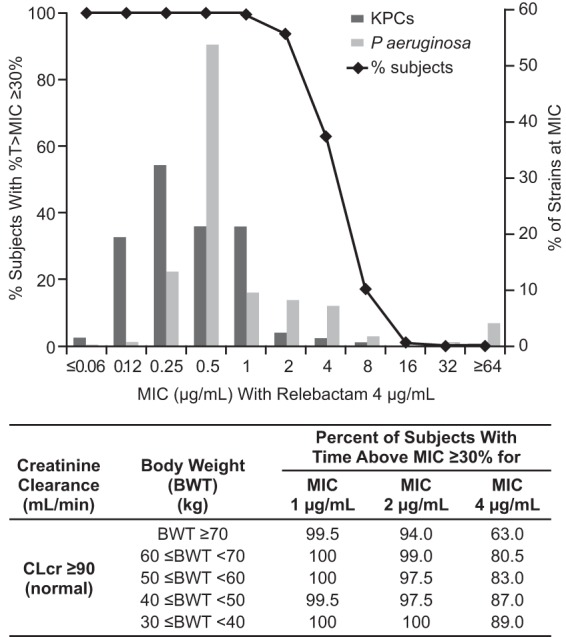
Probability of target attainment for imipenem at a dose of 500 mg q6h and distribution of KPC and P. aeruginosa strains as a function of imipenem MIC (as determined in the presence of 4 μg/ml REL). CLcr, creatinine clearance.
FIG 3.
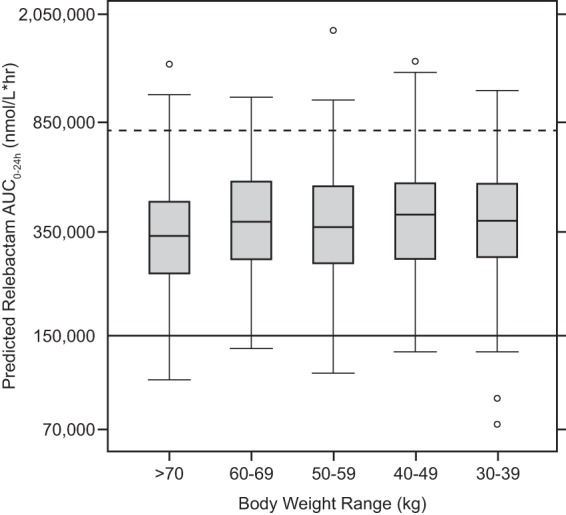
REL exposures (AUC0–24) in adults at a dose of 250 mg q6h across different body weight ranges. Boxes represent the median and 25th and 75th percentiles. Whiskers represent the 5th and 95th percentiles. Data points represent individual outlier values. The dashed horizontal line represents the upper bound of the therapeutic window for the median. The solid horizontal line represents the lower bound for the median.
DISCUSSION
This was a multicenter, double-blind, comparative clinical trial to evaluate the safety, tolerability, and efficacy of REL at 2 doses (250 mg and 125 mg) plus IMI (500 mg) compared to that of IMI alone in the treatment of cIAI. The comparative agent (imipenem-cilastatin) is licensed for the treatment of cIAI and is widely used in clinical practice to treat subjects with these complicated infections. This clinical trial studied serious bacterial infections requiring parenteral antibiotic therapy, including intraperitoneal infections associated most commonly with complicated appendicitis, complicated cholecystitis, and perforated hollow viscus, most of which were not postoperative infections.
Both doses of REL were generally well tolerated in this study. Incidence rates for adverse events overall, drug-related adverse events, serious adverse events, and discontinuation due to an adverse event in both REL-plus-IMI groups were comparable to those in the placebo-plus-IMI group. The most common adverse events and the prespecified events of clinical interest also occurred at similar rates across the treatment groups. An interim review of combined safety data from this study and another phase 2 study of REL plus IMI (Protocol 7655-003, in subjects with complicated urinary tract infection) confirmed that 250 mg REL plus IMI has a safety profile comparable to those of the other treatment arms.
In this phase 2 study, the primary efficacy objective was to evaluate the true impact of various doses of relebactam, in combination with imipenem-cilastatin, on clinical response at the time point at which antibacterial treatment was assessed as complete. Since transition to oral antibacterial therapy was not permitted in the trial, the completion of i.v. study therapy (DCIV) was equivalent to the completion of all antibacterial therapy and is consistent with an important decision point in clinical practice.
Rates of favorable clinical response at the DCIV visit in the ME population were generally similar across the 3 treatment groups and demonstrated that both doses of REL plus IMI are noninferior to IMI alone in the treatment of cIAI. There was insufficient evidence to conclude that either dose of REL plus IMI is superior to IMI alone. A supportive analysis in the MITT population demonstrated comparable clinical response rates for the 3 treatment groups at the DCIV visit, supporting the results obtained in the primary analysis. In addition, the REL-plus-IMI and control groups were generally similar in terms of the clinical response rates at early follow-up (5 to 9 days after the end of treatment) and late follow-up (28 to 42 days after end of treatment), the global response rates at 28 days after randomization, and the microbiological response rates at all visits.
Comparable clinical and microbiological response rates were observed among the 3 treatment groups for all major bacterial pathogens identified. A small group of subjects (n = 36 [13% of the MITT population]) had Gram-negative isolates at baseline that were nonsusceptible to imipenem; all 34 who were ME at the DCIV visit had a favorable clinical response, including 7 subjects with imipenem-nonsusceptible Gram-negative infections that were susceptible to REL plus imipenem. Most of the imipenem-nonsusceptible isolates did not have restoration of in vitro susceptibility to imipenem with the addition of REL; the mechanism of nonsusceptibility is not likely to be class A/C β-lactamase production in these bacteria. Also, about half of the isolates not susceptible to REL plus imipenem were Proteus mirabilis, which is inherently resistant to imipenem for reasons other than class A and/or C β-lactamases (31, 32); thus, it is not unexpected that REL did not impact imipenem susceptibility in these organisms.
Standard CLSI breakpoints were used to define nonsusceptibility to imipenem. The significance of the favorable observed clinical responses in the setting of imipenem nonsusceptibility is difficult to determine with certainty, given the variety of factors that impact clinical outcome. In cIAI, surgical control of the source is the key driver of clinical outcome. Thus, if a subject has adequate surgical debridement, then the relative contribution of antibiotics is small, and nonsusceptibility may not be a major factor in clinical outcome. Since the pool of nonsusceptible infections in this study was so small, we may not see a variety of surgical management or outcomes (i.e., poor outcomes resulting from inadequate source control). In addition, many IAIs are polymicrobial. In this trial, it is possible that the isolated nonsusceptible organisms were not the dominant pathogen for any given infection, and instead the dominant pathogen may have been a susceptible Gram-negative organism.
The most important limitation of this study is the small number of resistant pathogens that were identified, limiting the assessment of the clinical utility of REL in that specific infection subset. Related to this, in infections where source control plays an important role in treatment, such as IAI, clinical response may be favorable even where there is in vitro resistance, further limiting conclusions regarding IMI-resistant pathogens in this study. The efficacy of IMI plus REL in patients with IMI-resistant infections, the ultimate target for IMI plus REL, will be evaluated further in the ongoing phase 3 program. Second, the most severely ill patients (APACHE score of >30) and those with moderate to severe renal insufficiency (creatinine clearance of <50 ml/min) were excluded from this study, limiting the generalizability of our results; these patient populations will be included in the phase 3 program.
In conclusion, REL doses of 125 mg and 250 mg given with IMI once every 6 h for 4 to 14 days were generally well tolerated and demonstrated safety profiles similar to that of IMI alone. Treatment of cIAI with 125 or 250 mg REL plus IMI was associated with high rates of favorable clinical response and favorable microbiological response at the end of treatment and at follow-up visits conducted approximately 1 week and 4 to 6 weeks later. Both doses of REL plus IMI were noninferior to IMI alone on the primary endpoint (favorable clinical response at end of treatment). Simulations based on data from this study and three phase 1 studies led to the selection of a suitable dose for the fixed-dose combination for use in the phase 3 studies, 500 mg IMI plus 250 mg REL (with dose adjustment for patients with decreased renal function). This dose is expected to result in the majority of patients achieving exposures that lie within the defined therapeutic windows for both imipenem and REL and also to provide coverage of >90% of carbapenem-resistant bacterial strains, a significant step in combating the growing issue of antibacterial resistance.
Supplementary Material
ACKNOWLEDGMENTS
We thank the patients and families whose participation enabled the successful conduct of this study, as well as the study teams and study site personnel. Primary Investigators for MK-7655 Protocol 004 (by country) were as follows: Argentina, S. Attorri and M. Castelli; Brazil, A. Tarcísio de Faria Freire; Bulgaria, Y. Yordanov; Colombia, J. Prada Trujillo; Estonia, Ü. Kivistik; Germany, M. Winkler; Greece, G. Daikos and G. Nakos; Latvia, J. Gardovskis, M. Nalivaiko, and G. Pupelis; Lithuania, G. Barauskas, A. Razbadauskas, K. Strupas, and D. Venskutonis; Mexico, E. Rodriguez Noriega; Peru, C. Galvez Vasquez and M. Salas Perez; Poland, M. Ostrowski; Portugal, A. Vieira; Romania, C. Iancu, S. Neagu, D. Sandesc, C. Tecau, and L. Vasile; Russia, E. Matevosyan and S. Shlyapnikov; South Africa, J. Becker, H. Du Plessis, and C. Van Dyk; Taiwan, W.-J. Ko, T.-H. Lin, and Y.-S. Shan; Turkey, M. Kemal Celen and G. Yilmaz; Ukraine, B. H. Bezrodnyi, O. B. Datsenko, V. V. Ganzhyi, K. M. Mylytsya, O. V. Pyptyuk, V. H. Yareshko, and V. O. Syplyvyi; and United States, A. Castellanos, C. Lucasti, C. Schrock, and M. Sims.
Medical writing and editorial assistance were provided by Kim M. Strohmaier and Carol Zecca, employees of Merck & Co., Inc.
This study was designed by representatives of the study sponsor in collaboration with academic advisors. All data were collected by investigators and associated site personnel, analyzed by statisticians employed or contracted by the sponsor, and interpreted by the authors, including those from the sponsor. All authors participated in reviewing and editing the manuscript, approved the submitted draft, had full access to the data used to write the manuscript and vouch for their accuracy, and attest that the study was conducted in accordance with the protocol.
C. Lucasti has received grant support for clinical trials from Merck, Gilead, Allergan, Debio Pharmaceuticals, GSK, Cempra, Johnson & Johnson, Summit, The Medicines Company, and Theravance Biopharma and speaker fees from Allergan and The Medicines Company. L. Vasile, D. Sandesc, and D. Venskutonis received grant support from Merck for conducting this clinical trial. P. McLeroth is an employee of Covance, involved in the execution of this clinical trial. M. Brown, M. Losada, A. Pedley, N. Kartsonis, and A. Paschke are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., and may own stock and/or stock options in the company.
Funding Statement
Merck & Co., Inc., Kenilworth, NJ, USA, provided financial support and investigational drug supplies for the study.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00633-16.
REFERENCES
- 1.Rice LB. 2008. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J Infect Dis 197:1079–1081. doi: 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- 2.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. 2009. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 3.Curiao T, Morosini MI, Ruiz-Garbajosa P, Robustillo A, Baquero F, Coque TM, Canton R. 2010. Emergence of blaKPC-3-Tn4401a associated with a pKPN3/4-like plasmid within ST384 and ST388 Klebsiella pneumoniae clones in Spain. J Antimicrob Chemother 65:1608–1614. doi: 10.1093/jac/dkq174. [DOI] [PubMed] [Google Scholar]
- 4.Giani T, D'Andrea MM, Pecile P, Borgianni L, Nicoletti P, Tonelli F, Bartoloni A, Rossolini GM. 2009. Emergence in Italy of Klebsiella pneumoniae sequence type 258 producing KPC-3 carbapenemase. J Clin Microbiol 47:3793–3794. doi: 10.1128/JCM.01773-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gregory CJ, Llata E, Stine N, Gould C, Santiago LM, Vazquez GJ, Robledo IE, Srinivasas A, Tomashek KM. 2010. Outbreak of carbapenem-resistant Klebsiella pneumoniae in Puerto Rico associated with a novel carbapenemase variant. Infect Control Hosp Epidemiol 31:476–484. doi: 10.1086/651670. [DOI] [PubMed] [Google Scholar]
- 6.Kumarasamy KK, Toleman MA, Walsh TR, Bagaria J, Butt F, Balakrishnan R, Chaudhary U, Doumith M, Giske CG, Irfan S, Krishnan P, Kumar AV, Maharjan S, Mushtaq S, Noorie T, Paterson DL, Pearson A, Perry C, Pike R, Rao B, Ray U, Sarma JB, Sharma M, Sheridan E, Thirunarayan MA, Turton J, Upadhyay S, Warner M, Welfare W, Livermore DM, Woodford N. 2010. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infect Dis 10:597–602. doi: 10.1016/S1473-3099(10)70143-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nordmann P, Cuzon G, Naas T. 2009. The real threat of Klebsiella pneumoniae carbapenemase-producing bacteria. Lancet Infect Dis 9:228–236. doi: 10.1016/S1473-3099(09)70054-4. [DOI] [PubMed] [Google Scholar]
- 8.Österblad M, Kirveskari J, Koskela S, Tissari P, Vuorenoja K, Hakanen AJ, Vaara M, Jalava J. 2009. First isolations of KPC-2-carrying ST258 Klebsiella pneumoniae strains in Finland, June and August 2009. Euro Surveill 14:19349. [PubMed] [Google Scholar]
- 9.Roche C, Cotter M, O'Connell N, Crowley B. 2009. First identification of class A carbapenemase-producing Klebsiella pneumoniae in the Republic of Ireland. Euro Surveill 14:19163. [PubMed] [Google Scholar]
- 10.Tóth Á, Damjanova I, Puskás E, Jánvári L, Farkas M, Dobák A, Borocz K, Paszti J. 2010. Emergence of a colistin-resistant KPC-2-producing Klebsiella pneumoniae ST258 clone in Hungary. Eur J Clin Microbiol Infect Dis 29:765–769. doi: 10.1007/s10096-010-0921-3. [DOI] [PubMed] [Google Scholar]
- 11.Wendt C, Schütt S, Dalpke AH, Konrad M, Mieth M, Trierweiler-Hauke B, Weigand MA, Zimmermann S, Biegler K, Jonas D. 2010. First outbreak of Klebsiella pneumoniae carbapenemase (KPC)-producing K. pneumoniae in Germany. Eur J Clin Microbiol Infect Dis 29:563–570. doi: 10.1007/s10096-010-0896-0. [DOI] [PubMed] [Google Scholar]
- 12.Chopra I, Schofield C, Everett M, O'Neill A, Miller K, Wilcox M, Frere JM, Dawson M, Czaplewski L, Urleb U, Courvalin P. 2008. Treatment of health-care-associated infections caused by Gram-negative bacteria: a consensus statement. Lancet Infect Dis 8:133–139. doi: 10.1016/S1473-3099(08)70018-5. [DOI] [PubMed] [Google Scholar]
- 13.Lefevre S, Debat H, Thomas D, Friboulet A, Avalle B. 2001. A suicide-substrate mechanism for hydrolysis of β-lactams by an anti-idiotypic catalytic antibody. FEBS Lett 489:25–28. doi: 10.1016/S0014-5793(01)02075-0. [DOI] [PubMed] [Google Scholar]
- 14.Overturf GD. 2010. Carbapenemases: a brief review for pediatric infectious disease specialists. Pediatr Infect Dis J 29:68–70. doi: 10.1097/INF.0b013e3181c9c118. [DOI] [PubMed] [Google Scholar]
- 15.Thomson JM, Bonomo RA. 2005. The threat of antibiotic resistance in Gram-negative pathogenic bacteria: B-lactams in peril! Curr Opin Microbiol 8:518–524. [DOI] [PubMed] [Google Scholar]
- 16.Young K, Raghoobar SL, Hairston NN, Painter RE, Racine F, Dorso KL, Park Y-W, Ogawa AM, Wisniewski D, Hermes J, Blizzard TA, Hammond ML, Motyl MR. 2010. In vitro activity of the class A and C β-lactamase inhibitor MK-7655, abstr F1-2139. Abstr 50th Intersci Conf Antimicrob Agents Chemother. [Google Scholar]
- 17.Livermore DM, Warner M, Mushtaq S. 2013. Activity of MK-7655 combined with imipenem against Enterobacteriaceae and Pseudomonas aeruginosa. J Antimicrob Chemother 68:2286–2290. doi: 10.1093/jac/dkt178. [DOI] [PubMed] [Google Scholar]
- 18.Young K, Motyl M. 2015. Retrospective analysis of the effect of MK-7655 (relebactam) on imipenem non-susceptible Pseudomonas aeruginosa and KPC-expressing Enterobacteriaceae, abstr C-156. Abstr 55th Intersci Conf Antimicrob Agents Chemother. [Google Scholar]
- 19.Hirsch EB, Ledesma KR, Chang KT, Schwartz MS, Motyl MR, Tam VH. 2012. In vitro activity of MK-7655, a novel β-lactamase inhibitor, in combination with imipenem against carbapenem-resistant Gram-negative bacteria. Antimicrob Agents Chemother 56:3753–3757. doi: 10.1128/AAC.05927-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Powles MA, Galgoci A, Misura A, Liberator P, Hammond M. 2010. In vivo efficacy of the beta-lactamase inhibitor, MK-7655, in combination with imipenem in murine models of infection, abstr F1-2140. Abstr 50th Intersci Conf Antimicrob Agents Chemother. [Google Scholar]
- 21.Wu J, Racine F, Rizk ML, Wismer MK, Harradine P, Young K, Motyl M. 2014. Exploring PK/PD relationship of a novel beta-lactamase inhibitor MK-7655 in combination with imipenem in a hollow fiber infection model, abstr P1740. Abstr 24th Annu Eur Congress Clin Microbiol Infect Dis. [Google Scholar]
- 22.Tang W, Dingley K, Blizzard L, Young K, Motyl M. 2010. MK-7655 human dose projection based on its pharmacokinetics in preclinical species, abstr F1-2141. Abstr 50th Intersci Conf Antimicrob Agents Chemother. [Google Scholar]
- 23.Mavridou E, Melchers RJ, van Mil AC, Mangin E, Motyl MR, Mouton JW. 2015. Pharmacodynamics of imipenem in combination with β-lactamase inhibitor MK7655 in a murine thigh model. Antimicrob Agents Chemother 59:790–795. doi: 10.1128/AAC.03706-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rizk ML, Ahmed GF, Young K, Motyl M, Butterton J, Racine F, Wu J, Li C, Wenning L. 2012. A semi-mechanistic pharmacokinetic/pharmacodynamic (PK/PD) model for MK-7655, a novel beta-lactamase inhibitor (BLI) for use in combination with imipenem/cilastatin, abstr A-1763. Abstr 52nd Intersci Conf Antimicrob Agents Chemother. [Google Scholar]
- 25.Young K, Hackel M, Lascols C, Bouchillon S, Badal R. 2012. Response to imipenem plus MK-7655, a novel β-lactamase inhibitor, in a surveillance study population of P. aeruginosa from SMART 2009, abstr C2-724. Abstr 52nd Intersci Conf Antimicrob Agents Chemother. [Google Scholar]
- 26.Falagas ME, Barefoot L, Griffith J, Ruthazar R, Snydman DR. 1996. Risk factors leading to clinical failure in the treatment of intra-abdominal or skin/soft tissue infections. Eur J Clin Microbiol Infect Dis 15:913–921. doi: 10.1007/BF01690508. [DOI] [PubMed] [Google Scholar]
- 27.Lee YR, McMahan D, McCall C, Perry GK. 2015. Complicated intra-abdominal infections: the old antimicrobials and the new players. Drugs 75:2097–2117. doi: 10.1007/s40265-015-0506-7. [DOI] [PubMed] [Google Scholar]
- 28.Solomkin JS, Mazuski JE, Bradley JS, Rodvold KA, Goldstein EJC, Baron EJ, O'Neill PJ, Chow AW, Dellinger EP, Eachempati SR, Gorbach S, Hilfiker M, May AK, Nathens AB, Sawyer RG, Bartlett JG. 2010. Diagnosis and management of complicated intra-abdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Clin Infect Dis 50:133–164. doi: 10.1086/649554. [DOI] [PubMed] [Google Scholar]
- 29.Miettinen O, Nurminen M. 1985. Comparative analysis of two rates. Stat Med 4:213–226. doi: 10.1002/sim.4780040211. [DOI] [PubMed] [Google Scholar]
- 30.Center for Drug Evaluation and Research. 2015. Guidance for industry: complicated intra-abdominal infections: developing drugs for treatment. Department of Health and Human Services, Food and Drug Administration, Rockville, MD: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm321390.pdf. [Google Scholar]
- 31.Neuwirth C, Siebor E, Duez JM, Pechinot A, Kazmierczak A. 1995. Imipenem resistance in clinical isolates of Proteus mirabilis associated with alterations in penicillin-binding proteins. J Antimicrob Chemother 36:335–342. doi: 10.1093/jac/36.2.335. [DOI] [PubMed] [Google Scholar]
- 32.Villar HE, Danel F, Livermore DM. 1997. Permeability to carbapenems of Proteus mirabilis mutants selected for resistance to imipenem or other beta-lactams. J Antimicrob Chemother 40:365–370. doi: 10.1093/jac/40.3.365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.