

Abstract
Outer membrane proteins (OMPs) of microorganisms play important roles in directly interacting with host cells. Brucella species inhibit the apoptosis of host cells to benefit their own intracellular survival and replication. However, the association between OMP31 of Brucella and host cell apoptosis, and the underlying mechanism are unclear. In this study, an OMP31 gene deletion mutant based on B. melitensis 16M was constructed. Following the infection of RAW264.7 cells with B. melitensis 16M or the mutant strain, colony formation, apoptosis, tumor necrosis factor (TNF)-α levels and the levels of key downstream factors of the apoptosis pathways triggered by TNF-α, namely caspase-3, −8 and −9, cytochrome c, B-cell lymphoma 2 (Bcl-2) and Bcl-2-associated X protein (Bax) were detected. The mutant strain was shown to have the same phenotype as the parent strain using traditional microbiological tests. However, the mutant strain had impaired intracellular survival, with higher levels of apoptosis and TNF-α expression in infected RAW164.7 macrophages than the parent strain. The downstream factors of apoptosis triggered by TNF-α, including increased caspase-8, −3 and −9, cytochrome c and Bax, and decreased Bcl-2, indicated that the classical and mitochondrial cell death pathways were involved. It may be concluded that OMP31 from Brucella inhibited apoptosis and benefitted the intracellular survival of this microorganism. Furthermore, TNF-α may have served as a switch triggering classical death and mitochondrial cell death pathways.
Keywords: Brucella melitensis 16M, OMP31, apoptosis, tumor necrosis factor-α
Introduction
Brucellae are facultative intracellular bacterial pathogens (1–3). Although Brucella species lack most of the classical virulence factors such as invasive proteases, exotoxins, capsules fimbriae, virulence plasmids and lysogenic phages (4), they are able to grow in phagocytes. Similar to other intracellular bacterial pathogens, including Shigella, Salmonella, Yersinia and Mycobacterium, Brucella species are able to modulate the apoptosis of host cells to benefit intracellular survival and replication (5). To replicate in macrophage cells, Brucella have evolved mechanisms such as inhibition of apoptosis to modulate cell death machinery (6,7), which in turn favors pathogen survival and replication (7,8); cell death by apoptosis is a common response of mammalian cells to a wide variety of bacterial infections (9).
Tumor necrosis factor (TNF)-α is important for the autocrine stimulation of macrophage functions, which is important for triggering specific immunity against a number of intracellular pathogens. TNF-α is one of many physiological factors that are able to trigger apoptosis (10). Furthermore, TNF-α-induced apoptosis has been indicated to play a role in a variety of pathologies linked to chronic inflammation and auto-immune diseases (11,12). Analysis of Brucella infection in mice indicates a crucial and central role for TNF-α in the control of Brucella invasion and mouse recovery (13). Furthermore, TNF-α contributes to resistance to intracellular Brucella at different stages of infection, at the levels of innate and specific immunity (13).
Outer membrane protein (OMP)25 from Brucella suis has been reported to inhibit the production of TNF-α by human macrophages (14). Furthermore, B. suis-infected macrophages secreted significantly lower amounts of TNF-α than did macrophages infected with a ΔOMP25 B. suis mutant (14). OMP31 has a significant homology (34% identity) with Brucella OMP25 (15). In a previous study, OMP31 was reported to be an important immunogenic major outer membrane protein and antigen (16), which is present in all Brucella species, with the exception of B. abortus (17).
However, whether the inhibition of cellular apoptosis is associated with OMP31 from Brucella melitensis is unknown. It may be hypothesized that OMP31 from B. melitensis benefits survival and replication in macrophages by inhibiting apoptosis through TNF-α signaling following Brucella infection. In the present study, to test this hypothesis, an OMP31 gene deletion mutant based on B. melitensis 16M was constructed and the apoptosis of RAW264.7 macrophages induced by the mutant or parent strain was assessed using flow cytometry. To determine the mechanism of apoptosis associated with OMP31 of B. melitensis, enzyme-linked immunosorbent assay (ELISA) and reverse transcription-quantitative polymerase chain reaction (RT-qPCR) assays for the detection of the levels of B-cell lymphoma 2 (Bcl-2), Bcl-2-associated X protein (Bax), caspase-2, −3, −6 and −9, TNF-α and cytochrome c (Cyt c) were carried out.
Materials and methods
Bacterial strains and macrophage cell line
The strains used in these experiments were B. melitensis strain 16M (Library for Brucellosis of Chinese Centre for Disease Control and Prevention) and Bacillus subtilis strain BAA12545 (gifted by Professor Wenge Hu; College of Life Sciences, Shihezi University, Shihezi, Xinjiang, China), which was engineered for its sacB gene and used for the screening of mutant strains by counter-selection. B. melitensis was grown on Brucella agar (BD Biosciences, Franklin Lakes, NJ, USA). Sucrose medium was utilized for sacB counter-selection as previously described (18).
The RAW264.7 macrophage cell line was purchased from the China Academy Typical Culture Preservation Committee cell library (Shanghai, China). The cells were cultured at 37°C with 5% CO2 in complete tissue culture medium (c-DMEM) consisting of Dulbecco's modified Eagle's medium (American Type Culture Collection, Manassas, VA, USA) supplemented with 10% heat-inactivated fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Construction of OMP31 gene deletion mutant
Genomic DNA of B. melitensis strain 16M was used as the template for amplification of OMP31 upstream and downstream segments by PCR. The primer sequences used are shown in Table I. The flanking sequences were separately amplified, and overlap extension PCR (19) was then employed to combine the flanking sequences to generate contiguous DNA sequences of the OMP31 fragment. The fragment was cloned into a pMD18-T vector (D101A; Takara Biotechnology Co., Ltd., Dalian, China). Following SphI/XhoI digestion of the recombinants, the target fragments were introduced into a pGEM-7zf (+) suicide vector (P2251; Promega Corp., Beijing, China). The sacB gene of Bacillus subtilis was amplified by PCR and inserted into the recombinant pGEM-7zf (+) vector. Thus, the suicide plasmid pGB-OMP31 was generated. The pGB-OMP31 plasmid was introduced into B. melitensis strain 16M by electroporation as described previously (20). Ampicillin-resistant and sucrose-sensitive integrants were selected according to the study of Campos et al (20). The mutant, designated 16MΔOMP31, was identified by western blot assay and PCR tests (21) following subculture for 15 generations. Conventional microbiological tests, including assays of lipopolysaccharide antigens, phage typing, dye sensitivities, CO2 requirements, H2S production, and metabolic properties (21), were carried out to identify its phenotype.
Table I.
Primer sequences for the construction of the mutant Brucella melitensis strain 16MΔomp31.
| Target | Primer | Primer sequences (5′→3′) | Restriction site |
|---|---|---|---|
| Upstream | OMP31-N | F: GCATGCCCCATCCTGTCGCTTTGTGT | SphI |
| R: CTCGAGAATCACATTCGGCGAAAAAG | XhoI | ||
| Downstream | OMP31-C | F: CTCGAGTTATCAGGCGGCGGGAATTG | XhoI |
| R: GGATCCAGGCTTCGCTCGGTCACAGG | BamHI | ||
| SacB gene | sacB | F: GGATCCGGGCTGGAAGAAGCAGACCGCTA | BamHI |
| R: GAGCTCGCTTATTGTTAACTGTTAATTGTCC | SacI | ||
| Detection for Brucella | L7/L12 | F: ATGGCTGATCTCGCAAAGA | – |
| R: TTACTTGAGTTCAACCTTG | – | ||
| Detection for mutant | DC | F: CGTACATATTGGCGAGGGTG | – |
| R: CCGTCAGGAAGGGTTCAGTG | – |
omp31, outer membrane protein 31.
Macrophage infection and survival assay
RAW264.7 macrophages were plated in 12-well plates in DMEM without antibiotics at a concentration of 5×105 cells per well and incubated overnight at 37°C with 5% (vol/vol) CO2. The RAW264.7 cells were then infected with B. melitensis 16M or the mutant strain 16MΔOMP31 in triplicate wells of a 24-well plate at a multiplicity of infection (MOI) of 100:1. Following 4 h of incubation at 37°C in an atmosphere containing 5% CO2, the cells were washed three times with phosphate-buffered saline (PBS, pH 7.0) and treated with 50 µg/ml gentamicin to kill extracellular Brucella. The infected cells were then lysed with 0.1% Triton X-100 in PBS at 2, 4, 8, 12 and 24 h post infection. The number of viable colony forming units (CFUs) was determined by plating a series of 1:10 dilutions on tryptic soy agar plates.
Determination of cell apoptosis following infection by B. melitensis 16M or mutant
RAW264.7 cells cultured in 12-well plates were infected with B. melitensis 16M or mutant strain at a MOI of 100:1 in triplicate wells as described above. At 2, 4, 8, 12 and 24 h post infection, the RAW264.7 cells were washed with PBS and harvested. Apoptosis was then investigated. Briefly, B. melitensis 16M or mutant strain-infected RAW264.7 cells were incubated with Annexin V and propidium iodide (PI) using an Annexin V-FLUOS staining kit (Roche Diagnostics Corporation, Indianapolis, IN, USA) at room temperature for 20 min. Immediately afterwards, apoptotic RAW264.7 cells were detected using flow cytometry.
Detection of TNF-α by ELISA
Cells cultured in 12-well plates were infected with B. melitensis 16M or mutant strain in triplicate wells as described above. At 2, 4, 8, 12 and 24 h post infection, TNF-α production in the culture supernatants was tested using an ELISA kit (BD Biosciences).
Total RNA isolation and RT-qPCR analysis of TNF-α, Bax and Bcl-2
Cells cultured in 12-well plates were infected with B. melitensis 16M or mutant strain in triplicate wells as described above. Total RNA of the RAW264.7 cells was isolated using TRIzol reagent (CW0580Sl; CWBio, Beijing, China) and was further purified using a Qiagen RNeasy mini kit (Qiagen, Inc., Valencia, CA, USA), used according to the manufacturers' protocols. A Nanodrop 2100 instrument (NanoDrop Technologies; Thermo Fisher Scientific, Inc.) was used to assess the concentrations and quality of the RNA samples. Reverse transcription, cDNA quantification and cDNA amplification were performed exactly as described in the instructions of the Super RT One Step RT-PCR kit (CW0742S; CWBio). Specific primers used are listed in Table II. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a control. Relative mRNA levels of a particular gene in mutant strains were normalized using the 2−ΔΔCq method with GAPDH as a reference gene (22).
Table II.
Primer sequences for detection of Bax, Bcl-2 and TNF-α by reverse transcription-quantitative polymerase chain reaction.
| Primers | Primer sequences (5′→3′) |
|---|---|
| GAPDH | F: GCAGTGGCAAAGTGGAGATT |
| R: CCTTGACTGTGCCGTTGAAT | |
| TNF-α | F: GCCTCCCTCTCATCAGTTCTAT |
| R: CACTTGGTGGTTTGCTACGAC | |
| Bax | F: GCCTTTTTGCTACAGGGTTT |
| R: TGCTGTCCAGTTCATCTCCA | |
| Bcl-2 | F: GACTTCTCTCGTCGCTACCG |
| R: ACAATCCTCCCCCAGTTCAC |
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; TNF, tumor necrosis factor; Bax,; Bcl-2,; F, forward; R, reverse.
Detection of Cyt c by ELISA
RAW264.7 cells cultured in 12-well plates were infected with B. melitensis 16M or mutant strain at a MOI of 100:1 in triplicate wells as described above. At 12 h post infection, Cyt c from the cells was detected using a Cyt c ELISA kit (Elabscience, Wuhan, China) according to the manufacturer's protocol.
Detection of caspase-3, −8 and −9 by ELISA
RAW264.7 cells cultured in 12-well plates were infected with B. melitensis 16M or mutant strain at a MOI of 100:1 in triplicate wells as described above. At 4 h post infection, the concentrations of caspase-3, −8 and −9 were measured using caspase-3, −8 and −9 ELISA kits (BioVision, Inc., Milpitas, CA, USA) according to the manufacturer's instructions.
Statistical analysis
To determine the statistical significance of the immune response to the parent and mutant, differences were analyzed using analysis of variance tests and least significant difference post-hoc tests, with SPSS statistical analysis software (version 13.0; SPSS Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Mutant strain 16MΔOMP31 has the same phenotype as the parent strain
The OMP31 gene deletion mutant based on B. melitensis 16M was successfully constructed. To assess the mutant strain, conventional microbiology tests were carried out to identify its phenotype. These results showed that the lipopolysaccharide of the mutant strain remained smooth and the mutant exhibited the same traits as Brucella melitensis 16M (data not shown), including lipopolysaccharide antigens, phage typing, dye sensitivities, CO2 requirements, H2S production, and metabolic properties.
Deletion of OMP31 impairs the intracellular survival ability of Brucella following infection of RAW264.7 cells
Brucella has developed the ability to inhibit apoptosis to benefit intracellular replication. To assess the intracellular replication of the bacteria in RAW264.7 macrophages, the number of CFUs was counted following infection with Brucella melitensis 16M or the mutant strain. The results demonstrated that at 2–10 h post-infection, particularly at 4 h post-infection, parent stain impaired the ability of the bacteria to replicate in RAW264.7 cells. However, at 10–24 h post infection, the OMP31 mutant strain impaired the ability of the bacteria to replicate in RAW264.7 cells.
Mutant strain 16MΔOMP31 increases the apoptosis of RAW264.7 cells compared with the parent strain
Whether the deletion of OMP31, in addition to impairing the intracellular colony formation of Brucella in macrophages, also affected the apoptosis level was investigated. To evaluate the apoptosis of RAW264.7 cells induced by B. melitensis 16M or the mutant, flow cytometric analysis was conducted. The results demonstrated that the deletion of OMP31 increased the proportion of apoptotic RAW264.7 cells from 2 h post infection (Fig. 1). At 2–24 h post-infection, the mutant strain triggered more apoptosis cells than the parent strain. However, at 2–4 h post-infection, the mutant strain promoted RAW264.7 apoptosis and the parent strain inhibited apoptosis slightly. At 4–24 h post-infection, both the parent and mutant strains promoted RAW264.7 apoptosis.
Figure 1.
Percentage apoptosis of RAW 264.7 macrophages infected by Brucella melitensis 16MΔOMP31 mutant and parent strain. *P<0.05 vs. medium.
Mutant strain 16MΔOMP31 increases the TNF-α levels of RAW264.7 cells compared with the parent strain
The cytokine TNF-α plays a key role in immunity and inflammation by inducing cellular responses such as apoptosis (23). Therefore, following the detection of apoptosis, the TNF-α levels (RNA and secretion) induced by B. melitensis 16M or the mutant strain were detected. The results showed that the deletion of OMP31 increased the levels of TNF-α for the mutant strain in comparison with those for B. melitensis 16M from 4 h after infection for RNA (Fig. 2A), and 8 h post infection for protein (Fig. 2B). At 2–12 h post-infection, the concentration and transcriptional level of TNF-α increased rapidly, triggered by both mutant stain and parent strain, although the mutant stain induced greater TNF-α expression.
Figure 2.

Tumor necrosis factor (TNF)-α levels for RAW264.7 macrophages infected with Brucella melitensis 16MΔOMP31 mutant or parent strain. (A) Transcriptional levels and (B) secretion levels. *P<0.05.
Mutant strain 16MΔOMP31 increases caspase-3 and −8 levels in RAW264.7 cells compared with the parent strain
Triggering of the TNF receptor-1 (TNFR-1) by TNF-α may induce classical apoptosis by activating the initiator protease caspase-8 in the death receptor pathway (24). TNF-α induced apoptosis is mediated via the cell surface receptor TNFR-1 and involves the formation of two signaling complexes that sequentially activate NF-κB and caspases (25). Other studies have suggested that TNF-α may trigger apoptosis via an additional mechanism, involving constituents of acidic vesicles that can generate ceramides as second messengers such as Bcl-2 and activation of caspase-3 (26,27). Therefore, caspase-3 and 8 were detected by ELISA. The levels of caspase-3 and −8 were increased after 4 h post infection with the mutant strain 16MΔOMP31 (Fig. 3), to a greater extent than with the parent strain.
Figure 3.
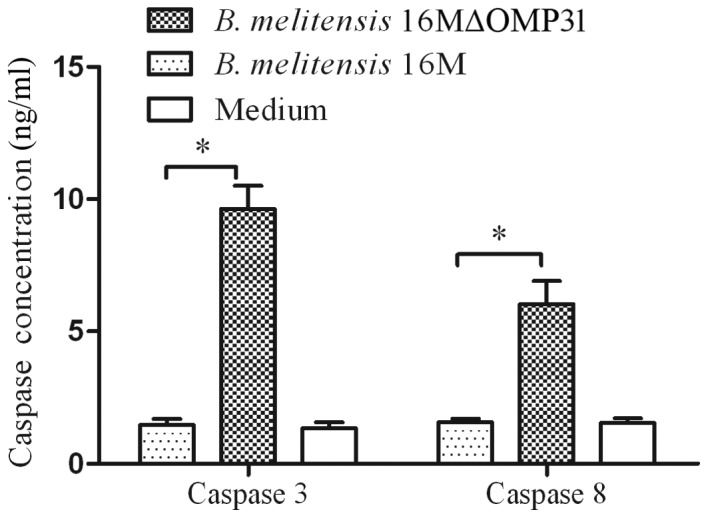
Classical cell death pathway is targeted by OMP31 from Brucella melitensis 16M. Caspase 3 and 8 concentrations of RAW264.7 macrophages infected with B. melitensis 16MΔOMP31 mutant or parent strain. *P<0.05.
Mutant strain 16MΔOMP31 increases Bax expression and reduces Bcl-2 levels of RAW264.7 cells compared with the parent strain
Bax, a member of the Bcl-2 protein family, is involved in the control of apoptotic events, and may accelerate apoptosis (28). Bcl-2 prolongs cell survival following the Bax-induced release of Cyt c (29). High levels of Bcl-2 inhibit apoptosis by attenuating the release of Cyt c while high levels of Bax induce apoptosis by binding to mitochondrial membranes and increasing the permeability of the membranes, facilitating the release of Cyt c (30). Cyt c activates caspase cascades in the cytosol, ultimately leading to cell death (31). To identify the possible pathway underlying the effects of OMP31, following the increase of TNF-α expression and apoptosis percentage, the transcriptional levels of Bcl-2 and Bax were detected. The present results for Bcl-2 (Fig. 4A) showed that the deletion of OMP31 reduced the bcl-2 level from 8 to 24 h post infection, although not at 4 h post infection. The results for Bax (Fig. 4B) revealed that the deletion of OMP31 raised the Bax level from 2 to 24 h post infection, but not at 4 h post infection. At 4–24 h post-infection, the mutant strain and parent strain both impaired Bcl-2 transcription and induced Bax transcription. Similarly, the mutant strain induced a greater change than the parent strain.
Figure. 4.

Mitochondria death pathway involvement of OMP31 in Brucella melitensis 16M. Transcriptional levels of (A) Bcl-2 and (B) Bax in RAW264.7 cells infected by B. melitensis 16MΔomp31 mutant or parent strain. (C) Cytochrome c (Cyt c) release from RAW264.7 cells infected by B. melitensis 16MΔomp31 mutant or parent strain. (D) Caspase-3 and −5 levels of RAW264.7 cells infected by B. melitensis 16MΔOMP31 mutant or parent strain. *P<0.05.
Mutant strain 16MΔOMP31 increases the Cyt c level of RAW264.7 cells compared with the parent strain
Cyt c is an essential component of the mitochondrial respiratory chain. It is a soluble protein, localized in the intermembrane space, and is loosely attached to the surface of the inner mitochondrial membrane. Cyt c release from mitochondria is a key event that has an important role in initiating the process of apoptosis in mammalian cells (32). To further investigate the pathway involved in the apoptosis induced by the deletion of OMP31, Cyt c release from infected cells was detected in the present study. The results showed that the deletion of OMP31 raised the concentration of Cyt c released after infection for 12 h (Fig. 4C). At 2–12 h post-infection, the concentration of Cyt c rapidly increased, triggered by both the mutant and parent strains.
Mutant strain 16MΔOMP31 increases caspase-9 levels in RAW264.7 cells compared with the parent strain
Caspases are a family of proteases with major roles in apoptosis. They are considered as downstream effectors of apoptosis, with caspase activation being an irreversible step in apoptotic signaling. Caspase-3, −6 and −7, known as executioner caspases, are downstream of the caspase-8 or −9 activation cascade (33,34). In a previous experiment of the present study, caspase-3 and −8 levels were found to be raised by the mutant strain. To further define the pathway induced by the deletion of OMP31, following the observation of a high level of Cyt c release, the levels of caspase-9 were detected (Fig. 4D). The results showed that caspase-9 levels were increased more than caspase-8 levels at 4 h post infection with the mutant strain. The mutant strain triggered more caspase-3 and −9 than the parent strain, and the concentration of caspase-9 was greater than that of caspase-3.
Discussion
In 1999, Caron et al reported that Brucella species released a specific, protease-sensitive inhibitor of TNF-α expression (35). It has since been reported that OMP25 from B. suis inhibits the production of TNF-α by human macrophages (14). Furthermore, B. suis-infected macrophages were shown to secrete significantly less TNF-α than did macrophages infected with a ΔOMP25 B. suis mutant (14). TNF-α is important for the autocrine stimulation of macrophage functions, which is important in the triggering of specific immunity against intracellular pathogens such as Brucella. TNF-α contributes to resistance to intracellular Brucella infection, via both innate and specific immunity (13). In the present study, the number of CFUs and TNF-α levels in macrophages infected with mutant and parent strains of B. melitensis were detected and the results showed that the deletion of OMP31 increased the secretion of TNF-α and impaired intracellular colony formation.
TNF-α is able to trigger apoptosis (10). Triggering of TNF receptor-1 (TNFR-1) by TNF-α induces classical apoptosis by activating the initiator protease caspase-8 in the death receptor pathway (24). TNF-α-induced apoptosis is mediated via the cell surface receptor TNFR-1, and involves the formation of two signaling complexes that sequentially activate NF-κB and caspases (25). Other studies have suggested that TNF-α may trigger apoptosis via an additional mechanism, involving constituents of acidic vesicles that generate ceramides as second messengers for Bcl-2 and activation of caspase-3 (26,27). In the present study, the flow cytometry results indicated that the mutant strain induced higher levels of apoptosis than did the parent strain. The results of the colony formation, TNF-α and flow cytometry experiments indicate that OMP31 contributes to the inhibition of apoptosis triggered by TNF-α and benefits the survival and replication of B. melitensis in RAW264.7 cells.
Caspase are cysteine proteases that cleave protein substrates at the sites of aspartic acid residues, and play a central role in the regulation and execution of apoptosis (36). To investigate the classical apoptosis route that is triggered by TNF-α (24), caspase-3 and −8 were detected and the results indicated that the classic pathway was triggered by TNF-α and involved OMP31 from Brucella.
Cyt c release from mitochondria has been observed in cells undergoing apoptosis following induction by signals including Fas and TNF activation, growth factors and exposure to chemotherapeutic drugs (37). The overexpression of Bcl-2 or Bcl-xL blocks the release of Cyt c and inhibits the apoptotic response (38,39). Bax is a member of the Bcl-2 protein family, which can accelerate apoptosis (28). Bax mostly exists in the cytosol and translocates to mitochondria following an apoptotic stimulus (29). The mitochondrial apoptosis pathway is regulated by members of the Bcl-2 protein family. Bcl-2 prolongs the survival of cells following the Bax-induced release of Cyt c (29). Notably, the present study also detected another pathway, the mitochondrial pathway (31,40), that is induced by TNF-α and is influenced by OMP31 from Brucella. All the results obtained for Cyt c, Bcl-2, Bax and caspase-9 demonstrated that apoptosis was triggered by TNF-α, and was more dependent upon the mitochondrial death pathway than the classical apoptosis pathway.
Notably, the results of the present study suggest that TNF-α induces caspases directly and also indirectly via mitochondria, with both pathways contributing to the apoptosis of RAW264.7 cells following infection by B. melitensis. In all, the present study indicated that OMP31 promoted apoptosis mediated by TNF-α-mitochondria-apoptosis pathway, not only TNF-α-caspase8-caspase3-pathway.
In summary, the cellular death response triggered by cytotoxic agents depends on the type and dose of chemotherapeutic stress within the cellular context and may involve classic apoptosis. In common with most intracellular bacteria, Brucella uses apoptosis inhibition as a strategy to replicate and survive in host cells. By constructing an OMP31 deletion mutant based on B. melitensis and analyzing the apoptosis induced by mutant and parent, the present study indicated that OMP31 from B. melitensis contributes to the inhibition of apoptosis to benefit survival and replication in RAW264.7 cells. Significantly, the results suggest that the pathway inhibited by OMP31 was triggered by TNF-α, and TNF-α may trigger downstream pathways, specifically the classical or mitochondrial death pathways. Further studies are required to investigate how and when the downstream pathway is triggered.
Acknowledgements
This study was supported by grants from the National Science & Technology Pillar Program (grant no. 2013BAI05B05), the National Nature Science Foundation of China (grant nos. 31060334 and 31260596) and the Key Discipline of Pathogenic Biology and Scientific Research Start-up Funding (grant no. PXY-BSQD-2015006), both from Pingdingshan University.
References
- 1.Schurig GG, Sriranganathan N, Corbel MJ. Brucellosis vaccines: Past, present and future. Vet Microbiol. 2002;90:479–496. doi: 10.1016/S0378-1135(02)00255-9. [DOI] [PubMed] [Google Scholar]
- 2.Boschiroli ML, Foulongne V, O'Callaghan D. Brucellosis: A worldwide zoonosis. Curr Opin Microbiol. 2001;4:58–64. doi: 10.1016/S1369-5274(00)00165-X. [DOI] [PubMed] [Google Scholar]
- 3.Franco MP, Mulder M, Gilman RH, Smits HL. Human brucellosis. Lancet Infect Dis. 2007;7:775–786. doi: 10.1016/S1473-3099(07)70286-4. [DOI] [PubMed] [Google Scholar]
- 4.Roop RM, II, Gee JM, Robertson GT, Richardson JM, Ng WL, Winkler ME. Brucella stationary-phase gene expression and virulence. Annu Rev Microbiol. 2003;57:57–76. doi: 10.1146/annurev.micro.57.030502.090803. [DOI] [PubMed] [Google Scholar]
- 5.Gao LY, Kwaik YA. The modulation of host cell apoptosis by intracellular bacterial pathogens. Trends In Microbiol. 2000;8:306–313. doi: 10.1016/S0966-842X(00)01784-4. [DOI] [PubMed] [Google Scholar]
- 6.Häcker G, Fischer SF. Bacterial anti-apoptotic activities. FEMS Microbiol Lett. 2002;21:1–6. doi: 10.1111/j.1574-6968.2002.tb11194.x. [DOI] [PubMed] [Google Scholar]
- 7.Fernandez-Prada CM, Zelazowska EB, Nikolich M, Hadfield TL, Roop RM, II, Robertson GL, Hoover DL. Interactions between Brucella melitensis and human phagocytes: Bacterial surface O-polysaccharide inhibits phagocytosis, bacterial killing and subsequent host cell apoptosis. Infect Immun. 2003;71:2110–2119. doi: 10.1128/IAI.71.4.2110-2119.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gross A, Terraza A, Ouahrani-Bettache S, Liautard JP, Dornand J. In vitro Brucella suis infection prevents the programmed cell death of human monocytic cells. Infect Immun. 2000;68:342–351. doi: 10.1128/IAI.68.1.342-351.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zychlinsky A. Apoptosis in bacterial pathogenesis. Biochem Soc Trans. 1996;24(Suppl):591S. [Google Scholar]
- 10.Aggarwal BB. Signalling pathways of the TNF superfamily: A double-edged sword. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 11.Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, Thurman RG. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999;117:942–952. doi: 10.1016/S0016-5085(99)70354-9. [DOI] [PubMed] [Google Scholar]
- 12.Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. IKKbeta is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFalpha. Immunity. 2003;19:725–737. doi: 10.1016/S1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 13.Dornand J, Gross A, Lafont V, Liautard J, Oliaro J, Liautard JP. The innate immune response against Brucella in humans. Vet Microbiol. 2002;90:383–394. doi: 10.1016/S0378-1135(02)00223-7. [DOI] [PubMed] [Google Scholar]
- 14.Jubier-Maurin V, Boigegrain RA, Cloeckaert A, Gross A, Alvarez-Martinez MT, Terraza A, Liautard J, Köhler S, Rouot B, Dornand J, Liautard JP. Major outer membrane protein Omp25 of Brucella suis is involved in inhibition of tumor necrosis factor alpha production during infection of human macrophages. Infect Immun. 2001;69:4823–4830. doi: 10.1128/IAI.69.8.4823-4830.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vaíno N, Cloeckaert A, Zygmunt MS, Dubray G. Cloning, nucleotide sequence, and expression of the Brucella melitensis omp31 gene coding for an immunogenic major outer membrane protein. Infec Immun. 1996;64:3744–3751. doi: 10.1128/iai.64.9.3744-3751.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghasemi A, Salari MH, Zarnani AH, Pourmand MR, Ahmadi H, Mirshafiey A, Jeddi-Tehrani M. Immune reactivity of Brucella melitensis-vaccinated rabbit serum with recombinant Omp31 and DnaK proteins. Iranian J Microbiol. 2013;5:19–23. [PMC free article] [PubMed] [Google Scholar]
- 17.Cloeckaert A, Debbarh HS, Vizcaíno N, Saman E, Dubray G, Zygmunt MS. Cloning, nucleotide sequence, and expression of the Brucella melitensis bp26 gene coding for a protein immunogenic in infected sheep. FEMS Microbiol Lett. 1996;140:139–144. doi: 10.1111/j.1574-6968.1996.tb08327.x. [DOI] [PubMed] [Google Scholar]
- 18.Pelicic V, Reyrat JM, Gicquel B. Generation of unmarked directed mutations in mycobacteria, using sucrose counter-selectable suicide vectors. Mol Microbiol. 1996;20:919–925. doi: 10.1111/j.1365-2958.1996.tb02533.x. [DOI] [PubMed] [Google Scholar]
- 19.Pelicic V, Reyrat JM, Gicquel B. A general method of in vitro preparation and specific mutagenesis of DNA fragments: Study of protein and DNA interactions. Nucleic Acids Res. 1988;16:7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campos E, Cravero SL, Delgui L, Mora I, Kahn N, Arese AI, Rossetti OL. Brucella abortus INTA2, a novel strain 19 (Delta)bp26: luc (Delta)bmp18 double mutant lacking drug resistance markers. Vet Microbiol. 2002;87:1–13. doi: 10.1016/S0378-1135(02)00016-0. [DOI] [PubMed] [Google Scholar]
- 21.Alton GG, Jones LM, Pietz DE. Laboratory techniques in brucellosis. Monogr Ser World Health Organ. 1975;55:1–163. [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCt method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Wang P, Qiu W, Dudgeon C, Zambetti GP, Yu J, Zhang L. PUMA is directly activated by NF-kappaB and contributes to TNF-alpha-induced apoptosis. Cell Death differ. 2009;16:1192–1202. doi: 10.1038/cdd.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ashkenazi A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat Rev Cancer. 2002;2:420–430. doi: 10.1038/nrc821. [DOI] [PubMed] [Google Scholar]
- 25.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/S0092-8674(03)00521-X. [DOI] [PubMed] [Google Scholar]
- 26.Dbaibo GS, Perry DK, Gamard CJ, Platt R, Poirier GG, Obeid LM, Hannun YA. Cytokine response modifier A (CrmA) inhibits ceramide formation in response to tumor necrosis factor (TNF)-alpha: CrmA and Bcl-2 target distinct components in the apoptotic pathway. J Exp Med. 1997;185:481–490. doi: 10.1084/jem.185.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Monney L, Olivier R, Otter I, Jansen B, Poirier GG, Borner C. Role of an acidic compartment in tumor-necrosis-factor-alpha-induced production of ceramide, activation of caspase-3 and apoptosis. Eur J Biochem. 1998;251:295–303. doi: 10.1046/j.1432-1327.1998.2510295.x. [DOI] [PubMed] [Google Scholar]
- 28.Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rossé T, Olivier R, Monney L, Rager M, Conus S, Fellay I, Jansen B, Borner C. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature. 1998;391:496–499. doi: 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- 30.Parone PA, James D, Martinou JC. Mitochondria: Regulating the inevitable. Biochimie. 2002;84:105–111. doi: 10.1016/S0300-9084(02)01380-9. [DOI] [PubMed] [Google Scholar]
- 31.Antonsson B. Bax and other pro-apoptotic Bcl-2 family ‘killer-proteins’ and their victim the mitochondrion. Cell Tissue Research. 2001;306:347–361. doi: 10.1007/s00441-001-0472-0. [DOI] [PubMed] [Google Scholar]
- 32.Liu K, Shu D, Song N, Gai Z, Yuan Y, Li J, Li M, Guo S, Peng J, Hong H. The role of cytochrome c on apoptosis induced by Anagrapha falcifera multiple nuclear polyhedrosis virus in insect Spodoptera litura cells. PloS One. 2012;7:e40877. doi: 10.1371/journal.pone.0040877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sadowski-Debbing K, Coy JF, Mier W, Hug H, Los MJ. Caspases - their role in apoptosis and other physiological processes as revealed by knock-out studies. Archivum Immunologiae Et Therapiae Experimentalis. 2002;50:19–34. [PubMed] [Google Scholar]
- 34.Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases−2, −3, −6, −7, −8, and −10 in a caspase−9−dependent manner. J Cell Biol. 1999;144:281–292. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caron E, Gross A, Liautard JP, Dornand J. Brucella species release a specific, protease-sensitive, inhibitor of TNF-alpha expression, active on human macrophage-like cells. J Immunol. 1996;156:2885–2893. [PubMed] [Google Scholar]
- 36.Cryns V, Yuan J. Proteases to die for. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- 37.Reed JC. Cytochrome c: Can't live with it - can't live without it. Cell. 1997;91:559–562. doi: 10.1016/S0092-8674(00)80442-0. [DOI] [PubMed] [Google Scholar]
- 38.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 39.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 40.Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21:92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]