

Abstract
Primary glioblastoma (GBM) is the most prevalent brain cancer, with fast progression and a poor prognosis. Current treatment options are unable to fully manage GBM since it is highly resistant to radiation and chemotherapy, and it cannot be completely removed by surgery. Thus, immunotherapeutic strategies utilizing tumor-infiltrating T cells have been investigated. In the present study, the T-cell response in GBM patients was examined in resected tumor samples and peripheral blood samples by flow cytometry. It was found that tumor-infiltrating T cells represented a rare population in all tumor cells, and were more refractory to anti-cluster of differentiation 3 (CD3) stimulation than their peripheral blood counterparts. A number of strategies were then assessed to boost tumor-infiltrating T-cell proliferation, and it was found that pre-incubation with 20 U/ml interleukin (IL)-2, as well as sequestration of IL-10 in culture, improved tumor T-cell proliferation following anti-CD3 stimulation. The stimulation of blood antigen-presenting cells by lipopolysaccharide, however, did not improve tumor T-cell proliferation. Overall, the present results provided a viable strategy for improving tumor-infiltrating CD3+ T-cell responses in GBM patients.
Keywords: glioblastoma, tumor-infiltrating T-cell, IL-2
Introduction
Primary glioblastoma (GBM) is the de novo presentation of World Health Organization (WHO) grade IV glioma without a preexisting lower grade tumor, and is the most frequent glioma subtype, with aggressive progression and a poor prognosis (1–3). Genetic alterations, including mutations in epidermal growth factor receptor (EGFR), altered AKT and mammalian target of rapamycin signaling pathways, and environmental factors such as cytomegalovirus infections, have been associated with an increased risk of glioblastoma, but little is known about the induction of the tumor (4,5). As GBM is one of the most resistant tumors to radiation and chemotherapy (6–8), the most effective treatment option is currently limited to surgical resection; however, complete surgical removal of the tumor is extremely difficult, since tumor cells invade the surrounding brain (9). Better therapeutic approaches are therefore urgently required.
To overcome the limitations of conventional treatments, the emerging field of immunotherapy has been investigated in GBM as a treatment option. Since mutant versions of EGFR are frequently found in GBM tumors, specific peptides contained in EGFR mutations have been investigated for use in vaccines (10,11). However, a number of significant challenges remain. GBM patients are profoundly immunosuppressive, both locally within the tumor and systemically (12). Multiple mechanisms exist to suppress effective immune responses targeted toward the tumor, including the blood-brain barrier that restricts lymphocyte trafficking (13), the local production of immunosuppressive cytokine transforming growth factor β (14,15), the increased expression of B7-H1 in glioma and the induction of T-cell apoptosis (16,17), the increased frequency of circulating regulatory T cells (18) and defective T-cell priming by microglial cells (19). On the other hand, stronger immune responses are associated with lower glioma incidences and/or increased survival. The incidence of glioma is inversely associated with allergies (20–22). Patient survival has been positively associated with the infiltration of effector T cells (23,24). Coexpression network analysis has also linked a series of immune-associated genes with GBM pathology and/or patient survival (25). Together, these studies suggest that an effective immune response mediated by tumor-infiltrating cells in the immune system can positively impact GBM outcome, but the manner in which to overcome the immunosuppressive microenvironment in GBM tumors remains a significant challenge for developing T-cell-based immunotherapies.
The present study examined the functions of tumor-infiltrating cluster of differentiation 3 (CD3)+ T cells in resected GBM tumors. It was found that compared with autologous peripheral blood CD3+ T cells, tumor-infiltrating CD3+ T cells were highly refractory to direct T-cell receptor stimulation by anti-CD3 antibodies, and that using autologous blood antigen-presenting cells (APCs) did not rescue the tumor T-cell responses. The study then investigated a number of strategies to improve the tumor T-cell response in vitro, and found that long-term interleukin (IL)-2 stimulation, as well as depletion of IL-10 in culture, can rescue tumor-infiltrating CD3+ T-cell proliferation in a subset of GBM patients. The study also examined whether stimulating APCs with Toll-like receptor (TLR) 4 ligand lipopolysaccharide (LPS) can improve APC stimulation, and found that LPS stimulation on autologous blood APCs did not improve the proliferation of the tumor-infiltrating T cells. These results provided a viable strategy for improving tumor-infiltrating CD3+ T-cell responses in GBM patients.
Materials and methods
Subjects
A total of 23 GBM patients, graded according to the WHO classification (2), were recruited for the study, including 16 males between 33 and 52 years of age, and 7 females between 35 and 55 years of age. No patients were taking any other forms of medication at the time. All patients provided written informed consent. The study was approved by the Ethical Board of the General Hospital of Shenyang Military Area Command of the Chinese People's Liberation Army (Shenyang, Liaoning, China). Not all patients were included in all experiments due to the low numbers of tumor-infiltrating T cells. Peripheral blood samples were obtained on the day of diagnosis prior to any treatment, and resected tumors were obtained by surgery. Efforts were made by the neurosurgeon to preserve healthy brain tissues during surgery, so small amounts of normal brain tissues were available in only 5 patients. Peritumoral tissues were not characterized as normal brain. Tumor and brain tissues were inspected intraoperatively by experienced neurosurgeons to confirm their identity prior to laboratory submission.
Sample preparation
Freshly isolated tumor tissue was mechanically dissociated and passed through a 70-mm pore size mesh, followed by collagenase digestion to obtain a single-cell suspension (19). For harvesting the brain/tumor mononuclear cells, homogenized brain/tumor cells were centrifuged across a 70–30% Percoll gradient at 500 × g for 30 min at 18°C, as previously described (26). Efficacy was confirmed by flow cytometry (Fig. 1A). For peripheral blood mononuclear cell (PBMC) isolation, heparinized blood was subjected to Ficoll-Hypaque gradient centrifugation. Purified peripheral blood APCs were obtained using a Pan Monocyte Isolation kit following the manufacturer's instructions (Miltenyi Biotec Inc., Cambridge, MA, USA) (27). Only fresh samples were used. The culture medium used was RPMI 1640 supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin and 2mM L-glutamine, unless otherwise specified.
Figure 1.

T-cell composition in GBM patients. (A) Gating of CD3+ T cells and their CD4 vs. CD8 expression in resected tumors. (A) Tumor leukocytes. (B) Percentages of CD3+ T cells in all undifferentiated cells in resected tumor and normal brain tissues in GBM subjects (unequal variances t-test; ***P<0.001). (C) Correlation between the percentages of T cells in tumor and brain tissues in the 5 patients with normal brain tissue attached to the tumor resection. Pearson's correlation test. (D) CD8-to-CD4 ratio in TI and PB T cells in GBM patients, calculated by dividing the percentage of CD8+ T cells by the percentage of CD4+ T cells (unequal variances t-test; ***P<0.001). (E) Correlation between the CD8-to-CD4 ratios in PB or TI T cells (Pearson's correlation test). GBM, glioblastoma; CD, cluster of differentiation; SSC, side scatter; FSC, forward scatter; TI, tumor-infiltrating; PB, peripheral blood.
Antibodies
For flow cytometry, cells were first labeled with Fixable Violet Dead Cell Stain (Thermo Fisher Scientific Inc., Waltham, MA, USA), and then stained with fluorescently-labeled anti-human CD3 [clone HIT3a; mouse immunoglobulin (Ig)G2aκ; catalog no. 555339], CD4 (clone RPA-T4; mouse IgG1κ; catalog no. 560650) and/or CD8 (clone RPA-T8; mouse IgG1κ; catalog no. 555369) monoclonal antibodies obtained from BD Biosciences (Franklin Lakes, NJ, USA), at 2 µg per 1 million cells. Forward scatter, side scatter and surface antigen expression was assessed by FACS Calibur (BD Biosciences). For stimulation, unlabeled anti-human CD3 monoclonal antibody (clone OKT3; mouse IgG2aκ; catalog no. 555329), anti-human CD28 monoclonal antibody (clone CD28.2; mouse IgG1κ; catalog no. 556620), anti-human IL-10 monoclonal antibody (clone JES3-9D7; rat IgG1, catalog no. 554495) and isotype control (clone R3-34; rat IgG1κ; catalog no. 554682) were added accordingly.
Proliferation assay
PBMCs or tumor leukocytes were labeled with carboxyfluorescein succinimidyl ester (CFSE) according to the manufacturer's protocol (Thermo Fisher Scientific Inc.), and then placed in a 96-well plate at 105 cells per well in 200 µl culture medium. The cells were incubated for 6 days at 37°C in 5% CO2. Upon harvesting, the cells were stained with surface antibodies, and the percentage of proliferating cells, a.k.a., CFSElo cells, in all CD3+ T cells was measured using FlowJo software (version 10.0.7r2; FlowJo LLC, Ashland, OR, USA). For IL-2 pre-incubation, recombinant human IL-2 protein (Thermo Fisher Scientific Inc.) was added accordingly in culture medium prior to CFSE-labeling. IL-2-containing medium was replaced every 3 days for time course experiments for a maximum of 9 days. For LPS stimulation of APCs, purified peripheral blood APCs were first incubated in culture medium with 2 µg/ml LPS for 3 days, after which the APCs were washed and added to the CFSE-labeled tumor cells.
Statistical analysis
Unequal variance t-tests were used for comparison between two groups. One-way analysis of variance was used for comparisons between multiple groups, and if the groups were significantly different, Tukey's multiple comparisons test or Dunnett's test was used. Paired tests were used for comparisons between different conditions on the same sample from the same subject. All statistical analyses were performed using Prism 6.0 software (GraphPad Software Inc., La Jolla, CA, USA). Bars represent the mean ± standard deviation. P<0.05 was considered to indicate a statistically significant difference.
Results
Tumor-infiltrating CD3+ T cells are rare in GBM tumors and exhibit a lower CD8-to-CD4 ratio than their peripheral blood counterparts
First, the lymphocyte composition was examined in resected tumors from 17 GBM patients by flow cytometry. Surrounding normal brain tissues attached to the tumor resection were also obtained in 5 out of 17 patients. It was found that CD3+ T cells represent a rare population in tumors and normal brain tissues, accounting for 0.005–0.23% of all tumor cells and 0.0009–0.005% of brain cells (Fig. 1A and B). In the 5 GBM patients with tumor and brain samples, the frequencies of tumor-infiltrating CD3+ T cells were directly correlated with that in the normal brain (Fig. 1C). CD3+ T cells can be categorized into CD8+ T cells, which are primarily recognized as cytotoxic T cells and are assessed in various immunotherapeutic strategies to eliminate cancerous cells, and CD4+ T cells, which are primarily known as helper T cells. Notably, within the CD3+ T-cell compartment, the CD8-to-CD4 ratio was significantly downregulated in the tumors compared with that in the autologous blood of GBM patients (Fig. 1D). The CD8-to-CD4 ratios in the tumor and blood samples of the same patient were not correlated (Fig. 1E).
Tumor-infiltrating CD3+ T cells in GBM are highly refractory to proliferation by anti-CD3 stimulation, independent of APC stimulation
Due to the low numbers of T cells observed in the resected tumors, proliferation responses in tumor T cells and peripheral blood T cells were then examined by CFSE staining. It was found that peripheral blood T cells and tumor T cells have significantly increased proportions of CFSElo cells following 6 days of stimulation by anti-CD3 monoclonal antibodies (Fig. 2A and B). Although no significant background differences were observed between tumor T cells and peripheral blood T cells when incubated in medium, the peripheral blood T cells exhibited significantly higher proportions of CFSElo cells than the tumor T cells following anti-CD3 stimulation.
Figure 2.

Proliferation of T cells in PB or TI cells, as measured by CFSE staining. (A and B) Percentages of CFSElo cells in PB or TI CD3+ T cells in unstimulated medium or after stimulation with anti-CD3 antibodies. PB mononuclear cells or tumor leukocytes were labeled with CFSE and then incubated in medium without or with 2 µg/ml anti-human CD3 monoclonal antibody at 105 cells per well in a 96-well plate. The cells were harvested at day 6 and the percentages of CFSElo cells among the CD3+ cells were examined by flow cytometry (one-way analysis of variance, followed by Tukey's multiple comparisons test; ***P<0.001 and **P<0.01). (C) Percentages of CFSElo cells in PB or TI T cells in unstimulated medium or after stimulation with anti-CD3 antibodies, with addition of purified APCs from autologous PB samples. Purified autologous APCs were added prior to CFSE labeling (one-way analysis of variance, followed by Tukey's multiple comparisons test; ***P<0.001). (D) Percentages of CFSElo cells in PB or TI T cells after stimulation with anti-CD3 and anti-CD28 monoclonal antibodies. The anti-human CD28 monoclonal antibodies were added together with anti-CD3 antibodies (unequal variances t-test; **P<0.01). GBM, glioblastoma; CD, cluster of differentiation; TI, tumor-infiltrating; PB, peripheral blood; CFSE, carboxyfluorescein succinimidyl ester; APC, antigen-presenting cell.
It was hypothesized that the lower proliferation of tumor-infiltrating T cells in GBM patients may be due to ineffective stimulation by tumor APCs in the tumor cell culture, since previously, glioma-infiltrating microglia cells were shown to be defective at stimulating allogeneic T cells, possibly due to low expression levels of costimulatory molecules CD80/CD86 and CD40 (19). To test this hypothesis, APCs were first isolated from 13 peripheral blood samples, and then autologous APCs were added into tumor leukocyte or PBMC cultures at 1-to-1 APC-to-tumor leukocyte or PBMC ratios. Again, it was found that the tumor-infiltrating T cells underwent significantly less proliferation following anti-CD3 stimulation compared with the autologous peripheral blood T cells in the same patient (Fig. 2C). Furthermore, since CD80/CD86 signaling acts by clustering CD28 molecules expressed on the T-cell surface, in a separate experiment, anti-CD28 antibodies were added together with anti-CD3 antibodies during incubation, and it was found that tumor T cells still underwent less proliferation than blood T cells from the same patient (Fig. 2D).
In conclusion, a defect in proliferative responses was observed following anti-CD3 stimulation in tumor-infiltrating T cells in GBM patients. This defect was independent of APCs and of stimulation by anti-CD28 antibodies.
Tumor-infiltrating T-cell proliferation can be rescued by long-term IL-2 stimulation and with IL-10 neutralization
Next, an attempt was made to rescue tumor-infiltrating T-cell proliferation in GBM patients. IL-2 provides survival and proliferation signals to activated T cells through IL-2 receptor signaling (28). Resected tumor leukocytes were obtained from 6 newly recruited GBM patients and were cultured in medium supplemented with different concentrations (from 0–20 U/ml) of recombinant IL-2 for 3 days, after which the cells were stained with CFSE and stimulated with anti-CD3 antibody for 6 days. It was found that tumor-infiltrating T-cell proliferation was significantly higher when the T cells were pre-incubated with 20 U/ml IL-2 compared with 0 U/ml (Fig. 3A). The study also examined the effect of pre-incubation time in rescuing tumor-infiltrating T-cell responses, and found that the longest pre-incubation period with 20 U/ml IL-2 (9 days) most effectively increased the proliferation of the tumor-infiltrating T cells, although pre-incubation for 3 or 6 days also enhanced proliferation (Fig. 3B).
Figure 3.

Proliferation of tumor-infiltrating T cells in various conditions. Six patients were included and each bar represents one subject. (A) Effect of pre-incubating with various concentrations of IL-2 on the proliferation of tumor T cells. Tumor leukocytes were pre-incubated with 0, 2, 5 or 20 U/ml IL-2 for 3 days in culture medium, and then the cells were washed, stained with CFSE, washed again and stimulated with 2 µg/ml anti-CD3 antibody. After 6 days, the cells were harvested and the percentages of CFSElo cells in CD3+ cells were examined by flow cytometry. Data were normalized against the results with 0 U/ml IL-2 in each patient (repeated measures one-way analysis of variance, followed by Dunnett's test; **P<0.01). (B) Effect of the lengths of pre-incubation with IL-2 on the proliferation of tumor T cells. Tumor leukocytes were pre-incubated with 20 U/ml IL-2 for 0, 3, 6, or 9 days in culture medium, and then the cells were stained with CFSE and stimulated with 2 µg/ml anti-CD3 antibody. The percentages of CFSElo cells were examined after 6 days. Data were normalized against the results without IL-2 pre-incubation (0 day) in each patient (repeated measures one-way analysis of variance, followed by Dunnett's test; **P<0.01 and *P<0.05). (C) Proliferation of tumor-infiltrating T cells without or with IL-10 depletion. Tumor leukocytes were stained with CFSE and stimulated with 2 µg/ml anti-CD3 antibody for 6 days. Concurrent with the addition of anti-CD3 antibodies, 2 µg/ml anti-human IL-10 monoclonal antibody or isotype control antibody were added (paired t-test). NS, not significant; IL, interleukin; CD, cluster of differentiation; CFSE, carboxyfluorescein succinimidyl ester.
Previous studies found that IL-10 production by tumor-infiltrating microglia cells was elevated in the serum and microglia cells of glioma patients compared with healthy controls (29,30). In the present study, in order to reduce potential inhibition by IL-10, an attempt was made to sequester the IL-10 in culture by adding anti-IL-10 monoclonal antibodies. It was found that in 5 out of 6 patients, proliferation was enhanced with the addition of anti-IL-10 antibodies (Fig. 3C), but this result was not statistically significant.
Treatment with LPS does not improve APC-stimulation of tumor-infiltrating T cells
It has been shown that LPS stimulation of blood APCs, but not glioma-infiltrating microglia cells, could enhance the proliferation of allogeneic T cells (19). The present study examined whether the LPS stimulation of peripheral blood APCs could enhance the proliferation of autologous tumor-infiltrating T cells under anti-CD3 stimulation. Unexpectedly, in 4 out of 6 subjects, pre-treatment with LPS on peripheral blood APCs reduced the proliferation of the tumor-infiltrating T cells (Fig. 4). It was therefore not found that LPS could enhance peripheral blood APC-mediated stimulation of tumor-infiltrating T-cell proliferation.
Figure 4.
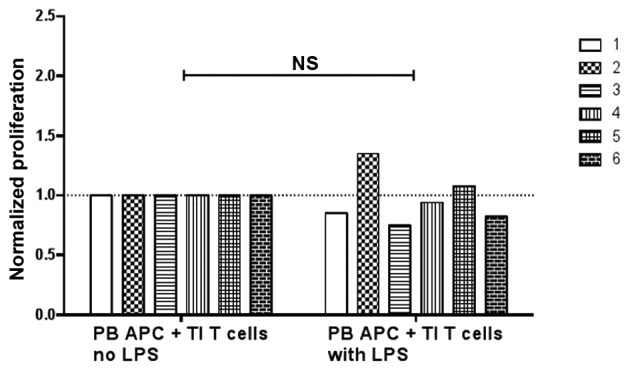
Proliferation of TI T cells when incubated with PB APCs. Six patients were included and each bar represents one subject. Purified blood APCs were pre-incubated without or with 2 µg/ml LPS for 3 days and then added to autologous tumor leukocyte culture. The cells were then labeled with CFSE and stimulated with anti-CD3 antibodies for 6 days. Data were normalized against the results without LPS in each patient (paired t-test). NS, not significant; TI, tumor-infiltrating; PB, peripheral blood; APC, antigen-presenting cell; LPS, lipopolysaccharide.
Discussion
Current treatment of GBM is limited by the resistance of the tumor to radiation and chemotherapy. Moreover, surgical removal of the tumor often fails to completely remove tumor tissues, since tumor cells invade the surrounding normal brain tissues (6–9). To resolve these difficulties, therapeutic options using tumor antigen-specific T cells have been investigated, but their application has been a daunting challenge due to the existence of the blood-brain barrier and the immunosuppressive microenvironment inside the tumor. The main goal of the present study was to examine various options to enhance tumor-infiltrating T-cell responses in GBM. It was first observed that the number of tumor-infiltrating T cells was extremely low in the resected tumor and the surrounding normal brain tissues. Also, although peripheral blood T cells were previously described as exhibiting immunosuppression with increased percentages of regulatory T cells (18), the present found that the tumor-infiltrating T cells were more suppressed than their peripheral counterparts in terms of proliferation following anti-CD3 stimulation. Local activation of infiltrating T cells by tumor antigens and their subsequent proliferation are considered beneficial for anti-tumor responses; therefore, the present study examined a number of strategies to enhance the proliferation by tumor-infiltrating T cells. It was found that by manipulating IL-2 concentrations and their pre-incubation time in the cell culture, tumor-infiltrating T-cell proliferation could be effectively enhanced. Sequestering IL-10 by anti-IL-10 antibodies also improved tumor-infiltrating T-cell proliferation in 5 out of 6 patients. In conclusion, the present data showed that although tumor-infiltrating T cells were more refractory to anti-CD3 stimulation, strategies exist to rescue their responses, such as the addition of IL-2 and the sequestration of IL-10.
Previous studies have shown that the capacity of glioma-infiltrating microglia cells to be stimulated by TLRs, to secrete cytokines and to upregulate costimulatory molecules are impaired; thus, these cells are less effective at stimulating and supporting T-cell responses (19). The present study found that the lower proliferation did not appear to be solely due to the ineffective stimulation by tumor microglia, since the addition of blood APCs in the cell culture did not resolve the difference in the proliferative response between tumor T cells and autologous blood T cells. A previous study has also shown that LPS-stimulated peripheral blood APCs can induce allogeneic T-cell proliferation more effectively than unstimulated APCs (19). In the present study, however, it was found that tumor T cells from 4 out of 6 subjects actually underwent less proliferation when the autologous blood APCs were stimulated with LPS. This discrepancy may have resulted from the fact that in the present assay, LPS were added for 3 days and were washed away prior to the APC T-cell coculture, while in the previous study (19), LPS were added concurrently during the coculture, and therefore, direct stimulation on the allogeneic T cells by LPS cannot be ruled out. More importantly, since LPS stimulation of APCs was found to increase IL-10 production under certain conditions (31), we speculate that in GBM patients, where systemic immune suppression has been observed (12), LPS may have further enhanced the regulatory phenotype of patient APCs.
Further studies are required to examine the activity of APCs in GBM patients under various stimulatory conditions and to assess their efficacy in stimulating autologous T-cell responses for potential T-cell-mediated immunotherapy.
Acknowledgements
This study was supported by the Natural Science Foundation of Liaoning Province (grant no. 2013020195).
References
- 1.Gladson CL, Prayson RA, Liu WM. The pathobiology of glioma tumors. Annu Rev Pathol. 2010;5:33–50. doi: 10.1146/annurev-pathol-121808-102109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, Burger PC, Cavenee WK. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61:215–229. doi: 10.1093/jnen/61.3.215. [DOI] [PubMed] [Google Scholar]
- 3.Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol. 2005;64:479–489. doi: 10.1093/jnen/64.6.479. [DOI] [PubMed] [Google Scholar]
- 4.Yoshimoto K, Dang J, Zhu S, Nathanson D, Huang T, Dumont R, Seligson DB, Yong WH, Xiong Z, Rao N, et al. Development of a real-time RT-PCR assay for detecting EGFRvIII in glioblastoma samples. Clin Cancer Res. 2008;14:488–493. doi: 10.1158/1078-0432.CCR-07-1966. [DOI] [PubMed] [Google Scholar]
- 5.Mitchell DA, Xie W, Schmittling R, Learn C, Friedman A, McLendon RE, Sampson JH. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro Oncol. 2008;10:10–18. doi: 10.1215/15228517-2007-035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masui K, Cloughesy TF, Mischel PS. Review: Molecular pathology in adult high-grade gliomas: From molecular diagnostics to target therapies. Neuropathol Appl Neurobiol. 2012;38:271–291. doi: 10.1111/j.1365-2990.2011.01238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, Agarwalla PK, Chheda MG, Campos B, Wang A, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012;26:756–784. doi: 10.1101/gad.187922.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 9.Park JK, Hodges T, Arko L, Shen M, Dello Iacono D, McNabb A, Olsen Bailey N, Kreisl TN, Iwamoto FM, Sul J, et al. Scale to predict survival after surgery for recurrent glioblastoma multiforme. J Clin Oncol. 2010;28:3838–3843. doi: 10.1200/JCO.2010.30.0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Bigner DD. Tumor-specific immunotherapy targeting the EGFRvIII mutation in patients with malignant glioma. Semin Immunol. 2008;20:267–275. doi: 10.1016/j.smim.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Herndon JE, II, Lally-Goss D, McGehee-Norman S, Paolino A, Reardon DA, Friedman AH, et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther. 2009;8:2773–2779. doi: 10.1158/1535-7163.MCT-09-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dix AR, Brooks WH, Roszman TL, Morford LA. Immune defects observed in patients with primary malignant brain tumors. J Neuroimmunol. 1999;100:216–232. doi: 10.1016/S0165-5728(99)00203-9. [DOI] [PubMed] [Google Scholar]
- 13.Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest. 2010;120:1368–1379. doi: 10.1172/JCI41911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuppner MC, Hamou MF, Sawamura Y, Bodmer S, de Tribolet N. Inhibition of lymphocyte function by glioblastoma-derived transforming growth factor beta 2. J Neurosurg. 1989;71:211–217. doi: 10.3171/jns.1989.71.2.0211. [DOI] [PubMed] [Google Scholar]
- 15.Bodmer S, Strommer K, Frei K, Siepl C, de Tribolet N, Heid I, Fontana A. Immunosuppression and transforming growth factor-beta in glioblastoma. Preferential production of transforming growth factor-beta 2. J Immunol. 1989;143:3222–3229. [PubMed] [Google Scholar]
- 16.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 17.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm0902-1039c. [DOI] [PubMed] [Google Scholar]
- 18.Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, Herndon JE, II, Bigner DD, Dranoff G, Sampson JH. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66:3294–3302. doi: 10.1158/0008-5472.CAN-05-3773. [DOI] [PubMed] [Google Scholar]
- 19.Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006;8:261–279. doi: 10.1215/15228517-2006-008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou M, Wiemels JL, Bracci PM, Wrensch MR, McCoy LS, Rice T, Sison JD, Patoka JS, Wiencke JK. Circulating levels of the innate and humoral immune regulators CD14 and CD23 are associated with adult glioma. Cancer Res. 2010;70:7534–7542. doi: 10.1158/0008-5472.CAN-10-0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schlehofer B, Blettner M, PrestonMartin S, Niehoff D, Wahrendorf J, Arslan A, Ahlbom A, Choi WN, Giles GG, Howe GR, et al. Role of medical history in brain tumour development. Results from the international adult brain tumour study. Int J Cancer. 1999;82:155–160. doi: 10.1002/(SICI)1097-0215(19990719)82:2<155::AID-IJC1>3.3.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 22.Lachance DH, Yang P, Johnson DR, Decker PA, Kollmeyer TM, McCoy LS, Rice T, Xiao Y, AliOsman F, Wang F, et al. Associations of high-grade glioma with glioma risk alleles and histories of allergy and smoking. Am J Epidemiol. 2011;174:574–581. doi: 10.1093/aje/kwr124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, Grau S, Hiraoka N, Eckstein V, Ecker RC, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-β. Clin Cancer Res. 2011;17:4296–4308. doi: 10.1158/1078-0432.CCR-10-2557. [DOI] [PubMed] [Google Scholar]
- 24.Kmiecik J, Poli A, Brons NH, Waha A, Eide GE, Enger PØ, Zimmer J, Chekenya M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol. 2013;264:71–83. doi: 10.1016/j.jneuroim.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 25.Vauléon E, Tony A, Hamlat A, Etcheverry A, Chiforeanu DC, Menei P, Mosser J, Quillien V, Aubry M. Immune genes are associated with human glioblastoma pathology and patient survival. BMC Med Genomics. 2012;5:41. doi: 10.1186/1755-8794-5-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pino PA, Cardona AE. Isolation of brain and spinal cord mononuclear cells using percoll gradients. J Vis Exp. 2011;pii:2348. doi: 10.3791/2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Randolph GJ, Jakubzick C, Qu C. Antigen presentation by monocytes and monocyte-derived cells. Curr Opin Immunol. 2008;20:52–60. doi: 10.1016/j.coi.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26:453–479. doi: 10.1146/annurev.immunol.26.021607.090357. [DOI] [PubMed] [Google Scholar]
- 29.Huettner C, Paulus W, Roggendorf W. Messenger RNA expression of the immunosuppressive cytokine IL-10 in human gliomas. Am J Pathol. 1995;146:317–322. [PMC free article] [PubMed] [Google Scholar]
- 30.Samaras V, Piperi C, Korkolopoulou P, Zisakis A, Levidou G, Themistocleous MS, Boviatsis EI, Sakas DE, Lea RW, Kalofoutis A, Patsouris E. Application of the ELISPOT method for comparative analysis of interleukin (IL)-6 and IL-10 secretion in peripheral blood of patients with astroglial tumors. Mol Cell Biochem. 2007;304:343–351. doi: 10.1007/s11010-007-9517-3. [DOI] [PubMed] [Google Scholar]
- 31.Rossol M, Heine H, Meusch U, Quandt D, Klein C, Sweet MJ, Hauschildt S. LPS-induced cytokine production in human monocytes and macrophages. Crit Rev Immunol. 2011;31:379–446. doi: 10.1615/CritRevImmunol.v31.i5.20. [DOI] [PubMed] [Google Scholar]