

Abstract
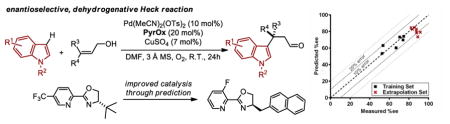
An enantioselective, intermolecular dehydrogenative Heck arylation of trisubstituted alkenes to construct remote quaternary stereocenters has been developed. Using a new chiral pyridine oxazoline ligand, good to high enantioselectivity is achieved for various combinations of indole derivatives and trisubstituted alkenes. However, some combinations of substrates led to lower enantioselectivity, which provided the impetus to use structure enantioselectivity correlations to design a better performing ligand.
Graphical Abstract
Since the early years in the development of the Heck reaction, the formal addition of an arene to an alkene has been sought rather than using a preformed aryl organometallic under oxidative conditions.1 This transformation has been termed an oxidative dehydrogenative Heck reaction (Figure 1a).2 While many impressive and important reports have detailed this process, several key limitations have arisen including the general need of an electron deficient alkene and/or a terminal alkene, and often the use of forcing conditions.2,3 Indeed, the use of electron rich multi-substituted alkenes has been restricted with issues associated with addition to a specific carbon on the alkene and overall reactivity.4 Therefore, there is a significant need to develop systems to facilitate the direct addition of an arene to multi-substituted, electron rich alkenes.5
Figure 1.

a) General dehydrogenative Heck reactions. b) Proposed direct indole addition and resultant relay Heck reaction.
In this regard, we desired to integrate our recently developed enantioselective redox relay Heck reactions of alkenols with the direct addition of a nucleophilic arene thus avoiding the need of a preformed organometallic reagent (Figure 1b).6,7 In particular, our previous reports demonstrate that heteroaromatic boronic acid derivatives are relatively poor coupling partners in these reactions especially when using trisubstituted alkenes.7 Therefore, our initial goal was to evaluate indole derivatives as coupling partners in the reaction of various trisubstituted alkenol alcohols. This would allow us to both determine if dehydrogenative Heck reactions of these relatively unreactive alkenes are possible and to develop a method to enantioselectively form quaternary centers containing a heterocycle remote from a carbonyl.8–11 Herein, we describe the successful development of an enantioselective oxidative dehydrogenative Heck arylation of trisubstituted alkenols. Catalysis is performed at ambient temperature, a range of indole derivatives, an important pharmacophore under study in our lab,12 can be incorporated, and new chiral pyridine oxazolines are discovered and designed for improved catalysis by using the relationship of enantioselectivity to a simple ligand structural parameter.
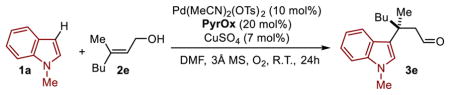
On the basis of our previous reports,6,7 we initiated the study by evaluating PyrOx ligand L1, which is optimal in a wide range of enantioselective redox relay Heck reactions, for the Pd(II) catalyzed addition of 1-methyl-indole (1a) to a simple trisubstituted homoallylic alkenol (2a) under aerobic conditions (Table 1). Successful conversion to product is observed albeit in low yield and encouraging enantioselectivity. To initially improve the catalysis, smaller substituents were incorporated into the oxazoline as it was reasoned that the formation of a requisite Pd-aryl species (or direct nucleopalladation of the alkene) may be thwarted by the larger t-butyl substituent. As anticipated, an improvement in conversion and yield is observed but at the cost of reduced enantioselectivity (L1–L3, Table 1). Therefore, it was rationalized that we may need alternative non-covalent interactions to improve enantioselectivity. This hypothesis was explored by incorporating various aryl groups on the oxazoline that may engage in putative π-interactions (L4–L9, Table 1). Interestingly, the ligand containing a naphthyl ring (L9) provided by far the best result, wherein a 96:4 er was observed in 77% yield.
Table 1.
Ligand optimization.

|
Reaction conditions: 1a (1.25 mmol, 5.0 eq), 2 (0.25 mmol, 1.0 eq), Pd(MeCN)2(OTs)2 (0.025 mmol, 10 mol%), CuSO4 (0.0175 mmol, 7 mol%), PyrOx (0.05 mmol, 20 mol%), DMF (3 mL), 3Å Ms (50 mg), O2(1 atm), room temperature, 24 h. Reported yields are of isolated material and enantioselectivity determined by SFC.
The scope of different trisubstituted alkenyl alcohols was investigated with 2-methylindole as the heteroaromatic nucleophile and using this ligand 2-NapPyrOx (L9) (Table 2a). In general, a broad range of trisubstituted alkenes are compatible with the reaction in terms of yield although the conversion to product with more hindered alkenes is reduced. An interesting observation is that alkenes incorporating a methyl group (3e–3j) rather than an ethyl group (3a–3d) undergo the reaction in reduced enantioselectivity. It should be noted that the reaction was initially optimized for a substrate containing the latter. Of additional importance, the reaction of homoallylic alcohols is possible in reduced yields (3k–3m). Excellent enantioselectivity is observed in this case when using the cis-alkene (3l, 96:4 er) in lieu of the trans-alkene.
Table 2.
Scope and limitations of the enantioselective dehydrogenative Heck reaction.

|
Standard reaction conditions: 1a (1.25 mmol, 5.0 eq), 2 (0.25 mmol, 1.0 eq), Pd(MeCN)2(OTs)2 (0.025 mmol, 10 mol%), CuSO4 (0.0175 mmol, 7 mol%), 2-NapPyrOx (0.05 mmol, 20 mol%), DMF (3 mL), 3Å Ms (50 mg), O2 (1 atm), room temperature, 24 h. Reported yields are of isolated material and enantioselectivity determined by SFC.
Various indole derivatives were evaluated in the reaction (Table 2b). The use of electron-rich and modestly electron-deficient substituted indoles is tolerated. Similar to above, tri-substituted alkenyl alcohols containing an ethyl group leads to improved enantioselectivity with the exception of an N-substituted with an isopropyl group (3n, 3v), which leads to the high enantioselectivity of 96:4 er for 3n. Clearly, the enantioselectivity is sensitive to both the nature of the indole and the alkene substituents. More complex indoles containing fused rings (3u, 3x) are compatible with the reaction and the use of a 2-substituted indole is modestly effective with low yield of the desired product (3y). Unfortunately, other electron rich aromatics such as pyrroles, furans, and anisoles are poor substrates under the current reaction conditions.
Two reasonable pathways are possible for the formal addition of indole to the alkene: 1) direct palladation through an electrophilic aromatic substitution type process or 2) a Wacker type addition. The results of the scope investigation suggest that an electron rich arene is required, which could be consistent with either path. Therefore, we performed a simple experiment by evaluating both an indole (4) and a boronic ester containing a similar indole (6), which likely forms an intermediate similar to A (Figure 2). We rationalized that the enantioselectivity should be quite different as a function of a Wacker type mechanism as compared to a Heck type insertion. In the event, nearly the same magnitude of asymmetric induction is achieved to yield 5 consistent with a Heck type mechanism.
Figure 2.

Exploring the nature of palladation.
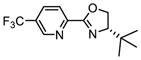
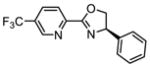
As noted above, the enantioselectivity is especially sensitive to the nature of both reagents. In particular, methyl substituted alkenes have reduced enantioselectivity as compared to the simple change to an ethyl substituted variant (compare 3a and 3e, Table 2a). As the ligand was optimized for the formation of 3a, we evaluated the same initial ligand set for the construction of 3e (Table 3). Unfortunately, the optimal ligand for 2a, 2-NapPyrOx, is also the best evaluated for the reaction of 2e (entry 6, Table 3) and no obvious trends to improve the performance of this substrate were qualitatively observed. Therefore, we sought to improve the catalysis through the relationship of enantioselectivity to parameters describing the ligand substituents. The goal was to use this correlative approach to predict new, possibly non-intuitive, ligand structures. To accomplish this, each ligand was geometrically optimized using M06-2x/def2tzvp13 level of theory to obtain steric and electronic parameters including Natural Bond Orbital (NBO) charges,14 IR frequencies and intensities,15 and Sterimol values.16
Table 3.
Empirical results of training set and exploration set to optimize ligand performance.
| |||||
|---|---|---|---|---|---|
| entry | PyrOx | er | yield (%) | Measured ΔΔC≠ | Predicted ΔΔG≠ |
| Training Set | |||||
| 1 |
|
87.0:13.0 | 41 | 1.13 | 1.02 |
| 2 |

|
76.8:23.2 | 72 | 0.71 | 0.88 |
| 3 |
|
76.2:23.8 | 85 | 0.69 | 0.71 |
| 4 |
|
83.4:16.6 | 55 | 0.96 | 0.83 |
| 5 |
|
85.2:14.8 | 83 | 1.04 | 1.10 |
| 6 |
|
87.4:12.6 | 79 | 1.14 | 1.13 |
|
| |||||
| Extrapolation Set | |||||
| 7 |
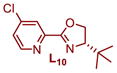
|
94.6:5.4 | 9 | 1.69 | 1.11 |
| 8 |
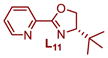
|
93.9:6.1 | 19 | 1.62 | 1.25 |
| 9 |
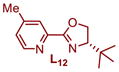
|
95.8:4.2 | 6 | 1.85 | 1.27 |
| 10 |
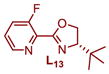
|
93.8:6.2 | 33 | 1.61 | 1.40 |
| 11 |

|
93.8:6.2 | 20 | 1.60 | 1.31 |
| 12 |
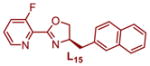
|
93.0:7.0 | 69 | 1.53 | 1.48 |
| 13 |
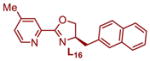
|
91.8:8.2 | 14 | 1.43 | 1.40 |
| 14 |
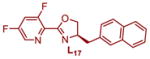
|
91.1:8.9 | 59 | 1.38 | 1.47 |
Reaction conditions: 1a (1.25 mmol, 5.0 eq), 2 (0.25 mmol, 1.0 eq), Pd(MeCN)2(OTs)2 (0.025 mmol, 10 mol%), CuSO4 (0.0175 mmol, 7 mol%), PyrOx (0.05 mmol, 20 mol%), DMF (3 mL), 3Å Ms (50 mg), O2 (1 atm), room temperature, 24 h. Reported yields are of isolated material and enantioselectivity determined by SFC. Note that some ligands are used in their enantiomeric form.
We then utilized linear regression analysis to relate the effect of these ligand parameters to the enantioselectivity. A simple correlation was achieved between the enantioselectivity and NBO charge on the oxazoline nitrogen of the training set of 5CF3 PyrOx ligands (black squares, Figure 3a). This relationship shows that a more electronegative NBO charge on the oxazoline nitrogen leads to higher enantioselectivity, which may be attributed to the modulation of the cationic nature of Pd. NBO charge calculations were then performed on a variety of proposed PyrOx ligands with emphasis on including unique and accessible substituents on the pyridine. Simply, the goal was to determine PyrOx ligands with a more negative NBO charge on the oxazoline nitrogen. Indeed, several possible ligands were virtually identified that meet this criterion. Eight of these predicted ligands were synthesized and evaluated in the dehydrogenative Heck reaction of 1a and 2e. The results are presented in Table 3 under the heading of extrapolation set and high-lighted as red crosses in Figure 3a. To our delight, most of the PyrOx ligands evaluated resulted in improved enantioselectivities and were generally well-predicted albeit the yields were highly dependent on the nature of the ligand with electron poor ligands containing a smaller oxazoline substituent leading to the best results.
Figure 3.

Correlation of NBO charge with enantioselectivity and resultant predictions.
Plotting the predicted %ee versus measured %ee (Figure 3b) demonstrated the effectiveness of this linear regression model to accurately predict the enantioselectivity of the extrapolation set within absolute error of 10% ee except for two ligands, L10 and L12 (see Table S4 in the Supporting Information). In sum, this structure function correlation has guided us to L15 (entry 12), with a 3-fluoro substituent on the pyridine ring, as the best combination of enantioselectivity and yield, wherein a 93:7 er in 69% yield is achieved. This is a ligand that has not been previously prepared and would not have necessarily been an obvious one to include in a screening set making the tactic of relating enantioselectivity to a parameter more compelling for ligand optimization protocols. It should be noted that this strategy improved the enantioselectivity by ~0.5 kcal/mol, which is significant in terms of synthetic utility. Finally, the enantioselectivity is nearly unaffected with steric variations in oxazoline substitutions, but remote electronic effects from the pyridine ring appears to have the most impact.
To assess the effectiveness of this new ligand (L15), various tri-substituted alkenyl alcohols and indoles were evaluated (Table 4). Improved enantioselectivity is achieved for substrates previously evaluated, which was highlighted by the formation of 3i in 90% ee as compared to 72% ee using ligand 2-NapPyrOx. Furthermore, the N-benzyl group of 3z can be removed (see SI) to afford the free N-indole product. Finally, using L15 as the ligand, Et-substituted alkenyl alcohols (3b and 3c, Table 4) give modestly improved enantioselectivity for poorer performing substrates.
Table 4.
Selected scope using new ligand, L15.

|
Standard reaction conditions: 1a (1.25 mmol, 5.0 eq), 2 (0.25 mmol, 1.0 eq), Pd(MeCN)2(OTs)2 (0.025 mmol, 10 mol%), CuSO4 (0.0175 mmol, 7 mol%), L15 (0.05 mmol, 20 mol%), DMF (3 mL), 3Å Ms (50 mg), O2 (1 atm), room temperature, 24 h. Reported yields are of isolated material and enantioselectivity determined by SFC.
er when using 2-NapPyrOx as a comparison.
In conclusion, we have developed a rare variant of a dehydrogenative Heck reaction of trisubstituted, electron rich alkenes to construct quaternary stereocenters containing a heterocycle remote to a carbonyl. This reaction was rendered enantioselective using a new PyrOx ligand containing a naphthyl group but initially the enantioselectivity was moderate for a particular substrate class. Therefore, structure enantioselectivity relationships were developed and used to virtually evaluate computationally suggested improved ligands. These ligands were validated providing enhanced asymmetric catalysis. An interesting feature of this reaction beyond the useful bond construction is that all components have a subtle impact on yield and enantioselectivity providing the basis to probe the non-covalent interactions responsible for enantioselection.17 Further development of mild, enantioselective dehydrogenative Heck reactions are ongoing.
Supplementary Material
Acknowledgments
The synthetic aspects were supported by National Institute of Health (R01GM063540) and the modeling was supported by NSF (CHE-1361296). We gratefully acknowledge the Center for High Performance Computing (CHPC) at the University of Utah. We would like to dedicate this paper to the memory of Richard Heck who has inspired so much of this field.
Footnotes
Supporting Information. Experimental procedures, analytical data, NMR spectra, and computational methods. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a) Heck RF. Acc Chem Res. 1979;12:146. [Google Scholar]; b) Crisp GT. Chem Soc Rev. 1998;27:427. [Google Scholar]; c) Irina P, Beletskaya IP, Cheprakov AV. Chem Rev. 2000;100:3009. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]
- 2.Bras JL, Muzart J. Chem Rev. 2011;111:1170. doi: 10.1021/cr100209d. [DOI] [PubMed] [Google Scholar]
- 3.For selected recent reviews, see: Lyons TW, Sanford MS. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e.Yeung CS, Dong VM. Chem Rev. 2011;111:1215. doi: 10.1021/cr100280d.. For selected recent examples, see: Lu Y, Wang DH, Engle KM, Yu JQ. J Am Chem Soc. 2010;132:5916. doi: 10.1021/ja101909t.Feng C, Loh TP. J Am Chem Soc. 2010;132:17710. doi: 10.1021/ja108998d.Wasa M, Engle KM, Yu JQ. J Am Chem Soc. 2010;132:3680. doi: 10.1021/ja1010866.Wang DH, Engle KM, Shi BF, Yu JQ. Science. 2010;327:315. doi: 10.1126/science.1182512.Hu P, Huang S, Xu J, Shi ZJ, Su W. Angew Chem Int Ed. 2011;50:9926. doi: 10.1002/anie.201103380.Ye M, Gao GL, Yu JQ. J Am Chem Soc. 2011;133:6964. doi: 10.1021/ja2021075.Huang L, Qi J, Wu X, Wu W, Jiang H. Chem Eur J. 2013;19:15462. doi: 10.1002/chem.201302962.Vora HU, Silvestri AP, Engelin CJ, Yu JQ. Angew Chem Int Ed. 2014;53:2683. doi: 10.1002/anie.201310539.Tang RY, Li G, Yu J-Q. Nature. 2014;507:215. doi: 10.1038/nature12963.
- 4.Czernecki S, Dechavanne V. Can J Chem. 1983;61:533. [Google Scholar]
- 5.For selected reviews on asymmetric C-H alkylation reaction, see: Giri R, Shi BF, Engle KM, Maugel N, Yu JQ. Chem Soc Rev. 2009;38:3242. doi: 10.1039/b816707a.Ye B, Cramer N. Acc Chem Res. 2015;48:1308. doi: 10.1021/acs.accounts.5b00092.. For selected examples about asymmetric C-H alkylation reaction, see: Thalji RK, Ellman JA, Bergman RG. J Am Chem Soc. 2004;126:7192. doi: 10.1021/ja0394986.Wilson RM, Thalji RK, Bergman RG, Ellman JA. Org Lett. 2006;8:1745. doi: 10.1021/ol060485h.Han X, Widenhoefer RA. Org Lett. 2006;8:3801. doi: 10.1021/ol061441b.Sevov CS, Hartwig JF. J Am Chem Soc. 2013;135:2116. doi: 10.1021/ja312360c.Song G, Wylie WNO, Hou Z. J Am Chem Soc. 2014;136:12209. doi: 10.1021/ja504995f.Shibata T, Ryu N, Takano H. Adv Synth Catal. 2015;357:1131.Filloux CM, Tomislav Rovis T. J Am Chem Soc. 2015;137:508. doi: 10.1021/ja511445x.Shirai T, Yamamoto Y. Angew Chem Int Ed. 2015;54:9894. doi: 10.1002/anie.201504563.
- 6.(a) Werner EW, Mei TS, Burckle AJ, Sigman MS. Science. 2012;338:1455. doi: 10.1126/science.1229208. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mei TS, Werner EW, Burckle AJ, Sigman MS. J Am Chem Soc. 2013;135:6830. doi: 10.1021/ja402916z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang C, Santiago CB, Kou L, Sigman MS. J Am Chem Soc. 2015;137:7290. doi: 10.1021/jacs.5b04289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mei TS, Patel HH, Sigman MS. Nature. 2014;508:340. doi: 10.1038/nature13231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For review about asymmetric Heck reactions see: Cartney DM, Guiry PJ. Chem Soc Rev. 2011;40:5122. doi: 10.1039/c1cs15101k.
- 9.For selected examples of using a redox relay strategy, see: McDonald RI, Stahl SS. Angew Chem, Int Ed. 2010;49:5529. doi: 10.1002/anie.200906342.McDonald RI, White PB, Weinstein AB, Tam CP, Stahl SS. Org Lett. 2011;13:2830. doi: 10.1021/ol200784y.Aspin S, Goutierre AS, Larini P, Jazzar R, Baudoin O. Angew Chem, Int Ed. 2012;51:10808. doi: 10.1002/anie.201206237.Stokes BJ, Opra SM, Sigman MS. J Am Chem Soc. 2012;134:11408. doi: 10.1021/ja305403s.Weinstein AB, Stahl SS. Angew Chem, Int Ed. 2012;51:11505. doi: 10.1002/anie.201206702.Renata H, Zhou Q, Baran PS. Science. 2013;339:59. doi: 10.1126/science.1230631.
- 10.For recent examples of isomerization/migration in Heck-type reactions, see: Crawley ML, Phipps KM, Goljer I, Mehlmann JF, Lundquist JT, Ullrich JW, Yang C, Mahaney PE. Org Lett. 2009;11:1183. doi: 10.1021/ol900036y.Kochi T, Hamasaki T, Aoyama Y, Kawasaki J, Kakiuchi F. J Am Chem Soc. 2012;134:16544. doi: 10.1021/ja308377u.
- 11.For the review about synthesis all-carbon quaternary stereogenic centers, see: Das JP, Marek I. Chem Commun. 2011;47:4593. doi: 10.1039/c0cc05222a.. For the selected methods for the synthesis quaternary stereogenic centers, see: Taylor MS, Jacobsen EN. J Am Chem Soc. 2003;125:11204. doi: 10.1021/ja037177o.Mermerian AH, Fu GC. J Am Chem Soc. 2003;125:4050. doi: 10.1021/ja028554k.Zhang P, Le H, Kyne RE, Morken JP. J Am Chem Soc. 2011;133:9716. doi: 10.1021/ja2039248.Jung B, Hoveyda A. J Am Chem Soc. 2012;134:1490. doi: 10.1021/ja211269w.Liu WB, Reeves CM, Stoltz BM. J Am Chem Soc. 2013;135:17298. doi: 10.1021/ja4097829.Krautwald S, Sarlah D, Schafroth MA, Carreira EM. Science. 2013;340:1065. doi: 10.1126/science.1237068.Dabrowski JA, Villaume MT, Hoveyda AH. Angew Chem Int Ed. 2013;52:8156. doi: 10.1002/anie.201304035.Masarwa A, Didier D, Zabrodski T, Schinkel M, Ackermann L, Marek I. Nature. 2014;505:199. doi: 10.1038/nature12761.
- 12.Pathak TP, Gligorich KM, Welm BE, Sigman MS. J Am Chem Soc. 2010;132:7870. doi: 10.1021/ja103472a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Zhao Y, Truhlar DG. Theor Chem Acc. 2008;120:215. [Google Scholar]; b) Zhao Y, Truhlar DG. Acc Chem Res. 2008;41:157. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 14.a) Reed AE, Weinhold F. J Chem Phys. 1983;78:4066. [Google Scholar]; b) Reed AE, Weinstock RB, Weinhold F. J Chem Phys. 1985;83:735. [Google Scholar]; c) Gross KC, Seybold PG, Hadad CM. Int J Quant Chem. 2002;90:445. [Google Scholar]
- 15.Milo A, Bess EN, Sigman MS. Nature. 2014;507:210. doi: 10.1038/nature13019. [DOI] [PubMed] [Google Scholar]
- 16.a) Verloop A. In Drug Design. Academic Press; New York: 1976. [Google Scholar]; b) Harper KC, Bess EN, Sigman MS. Nat Chem. 2012;4:366. doi: 10.1038/nchem.1297. [DOI] [PubMed] [Google Scholar]
- 17.Milo A, Neel AJ, Toste FD, Sigman MS. Science. 2015;347:737. doi: 10.1126/science.1261043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.