

Abstract
A highly stereoselective three-component C(sp2)–H bond addition across alkene and polarized π-bonds is reported for which Co(III) catalysis was shown to be much more effective than Rh(III). The reaction proceeds at ambient temperature with both aryl and alkyl enones employed as efficient coupling partners. Moreover, the reaction exhibits extremely broad scope with respect to the aldehyde input with electron rich and poor aromatic, alkenyl, and branched and unbranched alkyl aldehydes all coupling in good yield and with high diastereoselectivity. Multiple directing groups participate in this transformation, including pyrazole, pyridine, and imine functionalities. Both aromatic and alkenyl C(sp2)–H bonds undergo the three-component addition cascade, and the alkenyl addition product can readily be converted to diastereomerically pure 5-membered lactones. Additionally, the first asymmetric reactions with Co(III)-catalyzed C–H functionalization are demonstrated with three-component C–H bond addition cascades employing N-tert-butanesulfinyl imines. These examples represent the first transition metal catalyzed C–H bond additions to N-tert-butanesulfinyl imines, which have proven to be versatile and extensively used intermediates for the asymmetric synthesis of amines.
Keywords: C–H activation, Homogeneous catalysis, Multicomponent reactions, Diastereoselectivity
Graphical abstract

A highly stereoselective three-component C(sp2)–H bond addition across alkene and polarized π-bonds is reported. Co(III) catalysis was much more effective than Rh(III) providing products in good yield and with high diastereoselectivity for an extremely broad range of aldehydes. Additionally, the first asymmetric reactions employing Co(III)-catalyzed C–H functionalization are reported for three-component C–H bond addition cascades with N-tert-butanesulfinyl imines.
C–H functionalization has emerged as a core strategy to access synthetically useful structural motifs, many of which are found in drugs and natural products.1,2 Of the many transition metal catalysts that facilitate this process, Rh(III) catalysts have been demonstrated to be among the most useful due to their unique reactivity and high functional group compatibility.3 Following the seminal reports of Co(III)-catalyzed C–H functionalization by Kanai and Matsunaga in 2013,5ac a number of groups including our own have demonstrated that earth abundant Co(III) catalysts can provide analogous reactivity to their second row Rh(III) congeners.4,5 For some transformations divergent reactivity is displayed between Cp*Co(III) and Cp*Rh(III) catalysts,6 but significant differences in non-annulative redox-neutral couplings have yet to be reported. Recently, we described preliminary results on a three-component C–H functionalization cascade via Rh(III)-catalysis (Figure 1a).7 Though the transformation proceeded in good yield, very poor diastereoselectivity was observed. After thorough investigation, only ethyl glyoxylate proved to be reactive in the three-component transformation (Figure 1a–b).
Figure 1.

C–H bond enabled three-component coupling.
Here we report that Cp*Co(III)-catalysts not only facilitate three-component additions to a wide array of aldehydes, including electron poor and rich aromatic, alkenyl, and branched and unbranched alkyl derivatives, but that the transformation also proceeds with high diastereoselectivities ranging from 87:13 to >98:2 dr (Figure 1c).8.9 Moreover, we disclose the first asymmetric C–H bond functionalization reaction employing Co(III) with three-component C–H bond addition cascades to N-tert-butanesulfinyl imines (Figure 1d).
Experimental Section
Our initial investigation of non-activated aldehydes in the three-component coupling reaction began with the conditions previously developed for Rh(III) catalysis using ethyl glyoxylate (see Supplemental Optimization Table in Supporting Information).7 Unfortunately, attempts at obtaining the desired alcohol products such as 4a were unsuccessful leading only to high yields of the two-component product 5a (Table 1). We next investigated Co(III)-catalysis and first evaluated Kanai and Matsunaga's preformed catalyst [Cp*Co(C6H6)][PF6]2 because this complex had previously been shown to facilitate C–H bond additions to electron deficient alkenes.5ac The desired product 4a was formed in 65% yield and with high diastereoselectivity (95:5 dr) (entry 1, Table 1). Notably, for ethyl vinyl ketone (2a) a single regioisomer of the three-component product was obtained, establishing that equilibration to regioisomeric enolates does not occur after addition to the enone. In an effort to enhance the diastereoselectivity of this transformation we next ran the reaction at ambient temperature (23 °C), but noticed minimal conversion (entry 2). In contrast, employing the preformed catalyst [Cp*Co(C6H6)][B(C6F5)4)]2 previously developed in our laboratory for the direct C–H bond addition to aldehydes,5v we found that the reaction proceeded at ambient temperature in 61% yield and with an improved 96:4 dr (entry 3). Exploring other acetate salts such as cesium or lithium acetate provided similar yields (entries 4–5). Furthermore, upon lowering the equivalents of benzaldehyde (3a) with LiOAc as the additive, the yield was maintained along with a slight improvement in diastereoselectivity (entry 6).
Table 1.

| Entry | M(III)-catalyst | Additive (mol %) | 3a (equiv) | Yield 4ab | Yield 5ab |
|---|---|---|---|---|---|
| 1c | [Cp*Co(C6H6)][PF6]2 | KOAc (20) | 5 | 65% (95:5 dr) | 21% |
| 2 | [Cp*Co(C6H6)][PF6]2 | KOAc (20) | 5 | <5% | <5% |
| 3 | [Cp*Co(C6H6)][B(C6F5)4]2 | KOAc (20) | 5 | 61% (96:4 dr) | 33% |
| 4 | [Cp*Co(C6H6)][B(C6F5)4]2 | CsOAc (20) | 5 | 57% (96:4 dr) | 43% |
| 5 | [Cp*Co(C6H6)][B(C6F5)4]2 | LiOAc (20) | 5 | 63% (96:4 dr) | 37% |
| 6 | Cp*Co(C6H6)][B(C6F5)4]2 | LiOAc (20) | 3 | 70% (97:3 dr) | 27% |
| 7d | [Cp*Co(CO)I2] / AgB(C6F5)4 | LiOAc (20) | 3 | 63% (97:3 dr) | 32% |
| 8d,e | [Cp*RhCl2]2 / AgB(C6F5)4 | LiOAc (20) | 3 | <5% | 77% |
Conditions: 1a (1.0 equiv), 2a (1.2 equiv), 3a (3–5 equiv) for 20 h in 1,4-dioxane (2.0 M).
Determined by 1H NMR analysis relative to 1,3,5-trimethoxybenzene as an external standard.
Reaction run at 50 °C.
20 mol % of AgB(C6F5)4.
5 mol % of [Cp*RhCl2]2 dimer.
Employing the precatalyst [Cp*Co(CO)I2] in conjunction with AgB(C6F5)4 under the optimal conditions provided comparable yield and diastereoselectivity to that achieved with the preformed catalyst (entry 7). However, because the preformed catalyst [Cp*Co(C6H6)][B(C6F5)4)]2 simplifies reaction set up and also is prepared without the use of any precious metals,5v this preformed catalyst was used in all subsequent experiments. Finally, when the precatalyst [Cp*RhCl2]2 with AgB(C6F5)4 was used under the optimal reaction conditions for Co(III)-catalysis, the desired product 4a was obtained in only trace amounts with high yields of the two-component addition product 5a instead obtained (entry 8). This result highlights the vast superiority of Co(III) over Rh(III) catalysis for this three-component coupling reaction. When the Co(III) catalyst is absent no reaction is observed, while the removal of the LiOAc additive results in some product but with reduced yields and conversion for most substrates (see Supplemental Optimization Table).
After determining optimal reaction conditions, we next turned our attention to evaluating the scope of the three-component reaction (Table 2). Initially, a variety of enones were investigated. Phenyl vinyl ketone (2b) proved to be an effective coupling partner leading to high yields of products 4b and 4c with good diastereoselectivity. Methyl vinyl ketone also displayed good reactivity furnishing product 4d in 69% yield and 95:5 dr. Following this, a series of aldehydes were then subjected to the three-component reaction. Electron-rich heteroaryl aldehydes coupled efficiently to give products 4e and 4f in good yields and with 98:2 dr. For aryl aldehydes, strong and weakly donating substituents provided products 4g and 4j, respectively, in good yield and with high diastereoselectivity. Benzaldehydes substituted with weakly and strongly electron deficient groups such as 4-bromo (4h), 4-methyl ester (4i), and 2-fluoro (4k) groups also coupled in good yields though with modestly lower diastereoselectivities. Use of the highly activated ethyl glyoxylate provided product 4l in 51% yield and 75:25 dr, with the lower yield attributed to a competitive direct arene C–H bond addition to the aldehyde (see Supporting Information for details).
Table 2.

|
Conditions: 1 (1.0 equiv), 2 (1.2 equiv), and 3 (3.0 equiv) for 20 h in 1,4-dioxane (2.0 M).
Isolated yield after silica gel chromatography.
In addition to aryl and heteroaryl aldehydes, reactions with more challenging alkenyl and alkyl aldehydes proceeded smoothly. When crotonaldehyde was employed product 4m was obtained in 66% yield and 96:4 dr. Importantly, competitive 1,4-addition into crotonaldehyde was not observed. The activated benzyloxy acetaldehyde displayed good reactivity providing product 4n in 65% and 94:6 dr. Linear and branched alkyl aldehydes also underwent successful three-component coupling to give products 4o–s in high yield and diastereoselectivity. These examples clearly demonstrate that self-condensation side reactions do not compete for enolizable alkyl aldehyde derivatives. Three-component addition product 4q was isolated as a single diastereomer in 81% yield with the relative stereochemistry for this product rigorously established by X-ray crystallography.10
Different directing groups were also explored. In addition to pyrazole, pyridine was an effective directing group for the heteroaromatic C–H functionalization of isoquinolinones (Scheme 1). Notably, both aryl and alkyl aldehydes coupled efficiently to furnish products 4t and 4u in good yield and selectivity. Ketimine 1c also directed the three-component C–H bond addition cascade (Scheme 2). Interestingly, the expected alcohol underwent an additional intramolecular cyclization to provide spirohemiaminal ether 6 in 95:5 dr.
Scheme 1.
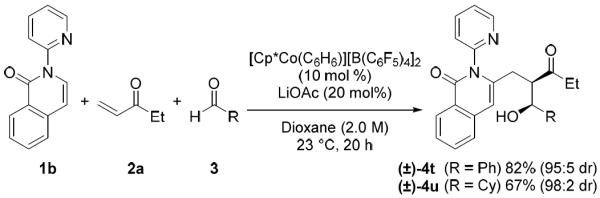
Three-component coupling with pyridylisoquinolinone.
Scheme 2.
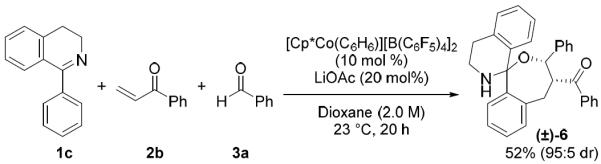
Use of ketimine directing group to access a spirohemiaminal ether.
In addition to the functionalization of aryl C(sp2)–H bonds, participation of alkenyl C(sp2)–H bonds in the three-component coupling was also demonstrated (Scheme 3). Reaction of alkenyl pyrazole 1d with ethyl vinyl ketone (2a) and benzaldehyde (3a) gave 4v in 77% yield and with 87:13 diastereoselectivity. Further elaboration of 4v was readily accomplished by ozonolysis to afford a lactol intermediate, which was conveniently oxidized with Fétizon's reagent to provide lactone 7 in diastereomerically pure form after chromatography (Scheme 4). This three-component coupling cascade consequently provides rapid access to gamma lactones, which are present in numerous drugs and natural products, from readily available α,β-unsaturated ketones and aldehydes.
Scheme 3.
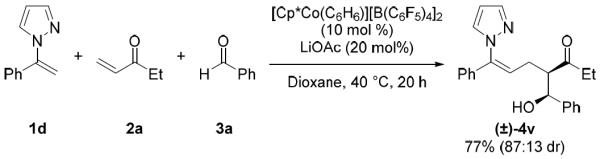
Alkenyl C–H bond three-component coupling.
Scheme 4.
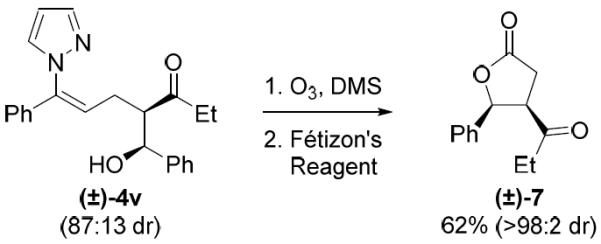
Employing a cleavable directing group for further functionalization.
A number of experiments were also conducted to provide insight into the mechanism of the Co(III)-catalyzed three-component reaction (Scheme 5). In the first mechanistic experiment, 5a was subjected to the optimal reaction conditions in the presence of benzaldehyde (3a) (Scheme 5a). The alcohol product 4a resulting from the three-component transformation was not observed and instead quantitative recovery of 5a was obtained suggesting that upon release from the catalyst, 5a does not participate in the catalytic cycle. Next, the three-component coupling of pyrazole 1e with phenyl vinyl ketone (2b) and benzaldehyde (3a) was evaluated in the presence of 5a (Scheme 5b). Under these conditions the expected alcohol product 4c was formed in only a slightly slower yield than previously observed in the absence of 5a (Table 2), and 5a was recovered in 95% yield. This result conclusively indicates that the released conjugate addition product 5a does not participate in the catalytic cycle, and therefore, the cobalt enolate that reacts with the aldehyde only arises through initial C–H bond addition to enone 2. To evaluate whether or not alcohol 4a exists in equilibrium with the conjugate addition product 5a, alcohol 4a was subjected to the optimal reaction conditions (Scheme 5c). Complete recovery of 4a was obtained, and without any loss in stereochemical purity, thereby establishing irreversible formation of the alcohol.
Scheme 5.
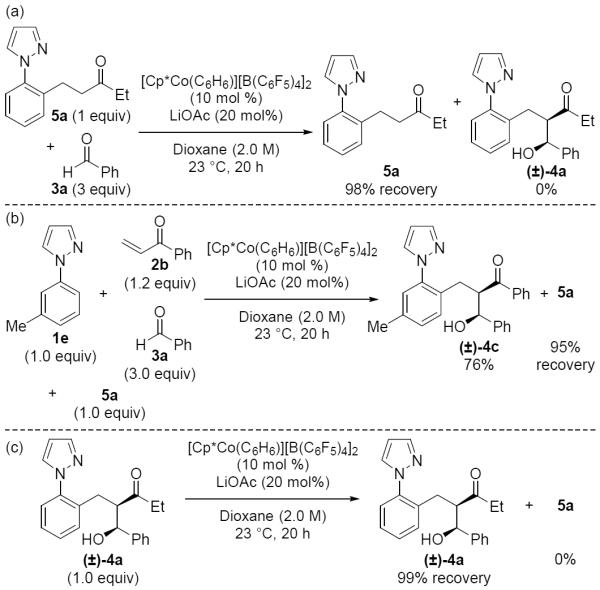
Mechanistic Experiments.
A plausible mechanism for the reaction is depicted in Scheme 6. Coordination of the directing group to the cationic Co(III) catalyst, followed by directed C–H bond metallation provides the active cobaltacycle 8. Conjugate addition of 8 into the enone coupling partner 2 provides the racemic cobalt enolate 9.11 Diastereoselective addition of the cobalt enolate 9 to aldehyde 3 via a chair transition state generates cobalt alkoxide 10. Release of the product upon proto-demetallation yields the desired alcohol 4 while regenerating the cationic Co(III) catalyst. The high observed diastereoselectivity of this transformation could alternatively be explained by generation of a Z-Co(III) enolate that reacts with the aldehyde via a boat transition state with likely coordination of the directing group nitrogen. Mechanistic studies are planned that should provide better resolution of the reaction pathway and origin of stereoselectivity.
Scheme 6.
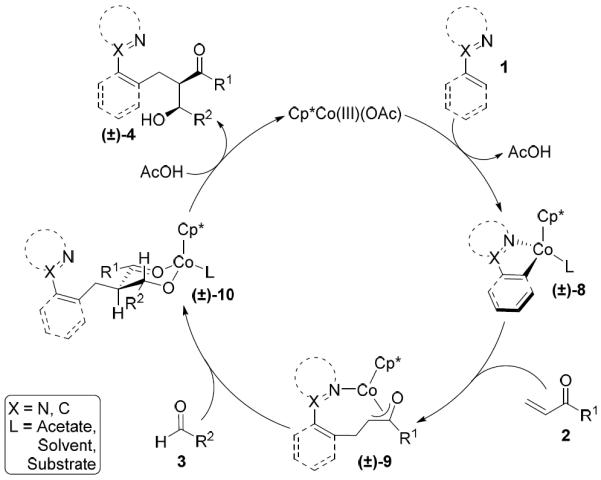
Proposed mechanism for the Co(III)-catalyzed three-component transformation.
Following our investigation of aldehydes in the three-component reaction, we next explored additions to imines to provide access to branched amine products 12 (Scheme 7). Given the extensive use of nucleophilic additions to N-tert-butanesulfinyl imines for the asymmetric synthesis of amines,12 we evaluated the viability of using N-tert-butanesulfinyl imines in this Co(III)-catalyzed three-component coupling process. In very promising preliminary results, asymmetric three-component addition to N-sulfinyl trifluoroacetaldimine 11a gave the amine product 12a with high diastereoslectivity and with the absolute configuration rigorously established by X-Ray crystallography.10 Furthermore, asymmetric three-component addition to sulfinyl imino ester 11b proceeded to give α-amino ester 12b also with high diastereoselectivity. To our knowledge these are the first asymmetric reactions employing Co(III)-catalyzed C–H functionalization and also represent the first examples of transition metal catalyzed C–H bond addition to N-tert-butanesulfinyl imines.
Scheme 7.
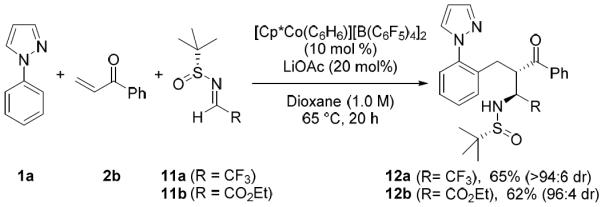
Three-component coupling with N-tert-butanesulfinyl imines.
In summary, we have developed an efficient route for Co(III)-catalyzed three-component coupling of aromatic and alkenyl C(sp2)–H bonds, enones, and aldehydes or N-tert-butanesulfinyl imines. Significantly, this method highlights major reactivity and stereoselectivity differences between Cp*Rh(III) and Cp*Co(III) catalyst systems. Here, we have showcased that Cp*Co(III)-three-component coupling is effective for aryl and alkyl enone coupling partners and displays extremely broad aldehyde scope. The 1-pyrazole, 2-pyridine, and imine directing groups each afforded products in good yield and with high diastereoselectivity. A cleavable alkenyl pyrazole can also be used in the three-component coupling to efficiently provide diastereomerically pure lactones. Additionally, we have provided preliminary data on an asymmetric variant of the reaction through the first C–H bond additions to N-tert-butanesulfinyl imines. Broadening enone scope by including β-substitution is currently underway as a means of introducing an additional stereocenter. Asymmetric additions to a range of N-tert-butanesulfinyl imines as well as chiral aldehydes are under active investigation, as is the development and evaluation of chiral Co(III) catalysts for this three-component cascade process.
Supplementary Material
Acknowledgements
This work was supported by the NIH (R01GM069559). We gratefully acknowledge Dr Brandon Mercado (Yale University) for solving the crystal structures of 4q and 12a.
Footnotes
Dedication: This work is dedicated to K. Barry Sharpless on occasion of his 75th birthday.
References
- [1].For recent reviews on C–H functionalization, see: Gensch T, Hopkinson MN, Glorius F, Wencel-Delord J. Chem. Soc. Rev. 2016 doi: 10.1039/c6cs00075d. Advance Article [DOI: 10.1039/C6CS00075D] Yang L, Huang H. Chem. Rev. 2015;115:3468. doi: 10.1021/cr500610p. Wencel-Delord J, Glorius F. Nat. Chem. 2013;5:369. doi: 10.1038/nchem.1607. Arockiam PB, Bruneau C, Dixneuf PH. Chem. Rev. 2012;112:5879. doi: 10.1021/cr300153j. Li B-J, Shi Z-J. Chem. Soc. Rev. 2012;41:5588. doi: 10.1039/c2cs35096c. Yamaguchi J, Yamaguchi AD, Itami K. Angew. Chem. Int. Ed. 2012;51:8960. doi: 10.1002/anie.201201666. Engle KM, Mei T-S, Wasa M, Yu J-Q. Acc. Chem. Res. 2012;45:788. doi: 10.1021/ar200185g. Colby DA, Tsai AS, Bergman RG, Ellman JA. Acc. Chem. Res. 2012;45:814. doi: 10.1021/ar200190g. Ackermann L. Chem. Rev. 2011;111:1315. doi: 10.1021/cr100412j. Cho SH, Kim JY, Kwak J, Chang S. Chem. Soc. Rev. 2011;40:5068. doi: 10.1039/c1cs15082k.
- [2].For reviews on C–H bond additions to polarized π-bonds, see: Shi X-Y, Han W-J, Li C-J. Chem. Rec. 2016 doi: 10.1002/tcr.201500250. Early View [DOI: 10.1002/tcr.201500250] Zhang X-S, Chen K, Shi Z-J. Chem. Sci. 2014;5:2146. Yan G, Wu X, Yang M. Org. Biomol. Chem. 2013;11:5558. doi: 10.1039/c3ob40652k.
- [3].For recent reviews on Rh(III)-catalyzed C–H bond functionalization, see: Song G, Wang F, Li X. Chem. Soc. Rev. 2012;41:3651. doi: 10.1039/c2cs15281a. Patureau FW, Wencel-Delord J, Glorius F. Aldrichimica Acta. 2012;45:31. Satoh T, Miura M. Chem.–Eur. J. 2010;16:11212. doi: 10.1002/chem.201001363.
- [4].For recent reviews on Co(III)-catalyzed C–H bond functionalization, see: Moselage M, Li J, Ackermann L. ACS Catal. 2016;6:498. Wei D, Zhu X, Niu J-L, Song M-P. ChemCatChem. 2016;8:1242. Yoshikai N. ChemCatChem. 2015;7:732.
- [5].For examples of Co(III)-catalyzed C–H bond additions to π-bonds, see: Lerchen A, Vasquez-Cespedes S, Glorius F. Angew. Chem. Int. Ed. 2016;55:3208. doi: 10.1002/anie.201510705. Zhang S-S, Liu X-G, Chen S-Y, Tan D-H, Jiang C-Y, Wu J-Q, Li Q, Wang H. Adv. Synth. Catal. 2016 Early View [DOI: 10.1002/adsc.201600025] Wang H, Moselage M, Gonzalez MJ, Ackermann L. ACS Catal. 2016;6:2705. Zhang Z-Z, Liu B, Xu J-W, Yan S-Y, Shi B-F. Org. Lett. 2016;18:1776. doi: 10.1021/acs.orglett.6b00494. Sen M, Emayavaramban B, Barsu N, Premkumar JR, Sundararaju B. ACS Catal. 2016;6:2792. Sivakumar G, Vijeta A, Jeganmohan M. Chem.–Eur. J. 2016;22:5899. doi: 10.1002/chem.201600471. Wang F, Wang H, Wang Q, Yu S, Li X. Org. Lett. 2016 ASAP [DOI: 10.1021/acs.orglett.6b00227] Kong L, Yu S, Zhou X, Li X. Org. Lett. 2016;18:588. doi: 10.1021/acs.orglett.5b03629. Nguyen TT, Grigorjeva L, Daugulis O. ACS Catal. 2016;6:551. doi: 10.1021/acscatal.5b02391. Prakash S, Muralirajan K, Cheng C-H. Angew. Chem. Int. Ed. 2016;55:1844. doi: 10.1002/anie.201509316. Barsu N, Sen M, Premkumar JR, Sundararaju B. Chem. Commun. 2016;52:1338. doi: 10.1039/c5cc08736h. Muralirajan K, Kuppusamy R, Prakash S, Cheng C-H. Adv. Synth. Catal. 2016;358:774. Liang Y, Jiao N. Angew. Chem. Int. Ed. 2016;55:4035. doi: 10.1002/anie.201511002. Liu X-G, Zhang S-S, Jiang C-Y, Wu J-Q, Li Q, Wang H. Org. Lett. 2015;17:5404. doi: 10.1021/acs.orglett.5b02728. Hummel JR, Ellman JA. Org. Lett. 2015;17:2400. doi: 10.1021/acs.orglett.5b00910. Sen M, Kalsi D, Sundararaju B. Chem.–Eur. J. 2015;21:15529. doi: 10.1002/chem.201503643. Gensch T, Vasquez-Cespedes S, Yu D-G, Glorius F. Org. Lett. 2015;17:3714. doi: 10.1021/acs.orglett.5b01701. Li J, Ackermann L. Angew. Chem. Int. Ed. 2015;54:8551. doi: 10.1002/anie.201501926. Wang H, Koeller J, Liu W, Ackermann L. Chem.–Eur. J. 2015;21:15525. doi: 10.1002/chem.201503624. Ma W, Ackermann L. ACS Catal. 2015;5:2822. Suzuki Y, Sun B, Yoshino T, Kanai M, Matsunaga S. Tetrahedron. 2015;71:4552. Hummel JR, Ellman JA. J. Am. Chem. Soc. 2015;137:490. doi: 10.1021/ja5116452. Li J, Ackermann L. Angew. Chem., Int. Ed. 2015;54:3635. doi: 10.1002/anie.201409247. Yu D-G, Gensch T, de Azambuja F, Vasquez-Cespedes S, Glorius F. J. Am. Chem. Soc. 2014;136:17722. doi: 10.1021/ja511011m. Grigorjeva L, Daugulis O. Angew. Chem., Int. Ed. 2014;53:10209. doi: 10.1002/anie.201404579. Grigorjeva L, Daugulis O. Org. Lett. 2014;16:4684. doi: 10.1021/ol502005g. Grigorjeva L, Daugulis O. Org. Lett. 2014;16:4688. doi: 10.1021/ol502007t. Yoshino T, Ikemoto H, Matsunaga S, Kanai M. Chem.–Eur. J. 2013;19:9142. doi: 10.1002/chem.201301505. Yoshino T, Ikemoto H, Matsunaga S, Kanai M. Angew. Chem., Int. Ed. 2013;52:2207. doi: 10.1002/anie.201209226.
- [7].a Lu Q, Vasquez-Cespedes S, Gensch T, Glorius F. ACS Catal. 2016;6:2352. [Google Scholar]; b Park J, Chang S. Angew. Chem. Int. Ed. 2015;54:14103. doi: 10.1002/anie.201505820. [DOI] [PubMed] [Google Scholar]; c Sun B, Yoshino T, Kanai M, Matsunaga S. Angew. Chem. Int. Ed. 2015;54:12968. doi: 10.1002/anie.201507744. [DOI] [PubMed] [Google Scholar]; d Suzuki Y, Sun B, Sakata K, Yoshino T, Matsunaga S, Kanai M. Angew. Chem. Int. Ed. 2015;54:9944. doi: 10.1002/anie.201503704. [DOI] [PubMed] [Google Scholar]; e Zhao D, Kim JH, Stegemann L, Strassert CA, Glorius F. Angew. Chem. Int. Ed. 2015;54:4508. doi: 10.1002/anie.201411994. [DOI] [PubMed] [Google Scholar]; f Ikemoto H, Yoshino T, Sakata K, Matsunaga S, Kanai M. J. Am. Chem. Soc. 2014;136:5424. doi: 10.1021/ja5008432. [DOI] [PubMed] [Google Scholar]
- [7].Boerth JA, Ellman JA. Chem. Sci. 2016;7:1474. doi: 10.1039/c5sc04138d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].For a review on cascade Michael/aldol reactions, see: Gorobets EV, Miftakhov MS, Valeev FA. Russ. Chem. Rev. 2000;69:1001.
- [9].For relevant metal-catalyzed cascade Michael/aldol reactions, see the following references. For Rh(I)-catalyzed examples, see: Cauble DF, Gipson JD, Krische MJ. J. Am. Chem. Soc. 2003;125:1110. doi: 10.1021/ja0211095. Huddleston RR, Krische MJ. Org. Lett. 2003;5:1143. doi: 10.1021/ol0300219. For Cu(II)-catalyzed examples, see: Alexakis A, Trevitt GP, Bernardinelli G. J. Am. Chem. Soc. 2001;123:4358. doi: 10.1021/ja005911n. Arnold LA, Naasz R, Minnaard AJ, Feringa BL. J. Am. Chem. Soc. 2001;123:5841. doi: 10.1021/ja015900+. Feringa BL, Pineschi M, Arnold LA, Imbos R, de Vries AHM. Angew. Chem. Int. Ed. 1997;36:2620. For a Co(II)-catalyzed example, see: Baik TG, Luis AL, Wang JC, Krische MJ. J. Am. Chem. Soc. 2001;123:5112. doi: 10.1021/ja0040971. For a Ni(0)-catalyzed example, see: Montgomery J, Oblinger E, Savchenko AV. J. Am. Chem. Soc. 1997;119:4911.
- [10].Synthetic details, characterization, and molecular structures obtained by X-ray structural analysis are described in the Supporting Information. CCDC 1471274 (4q) and 1471275 (12a) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
- [11].For the Cp*Rh(III)-catalyzed three-component coupling reported in reference 7, a related active rhodium enolate has been characterized by X-Ray crystallography.
- [12].a Robak MT, Herbage MA, Ellman JA. Chem. Rev. 2010;110:3600. doi: 10.1021/cr900382t. [DOI] [PubMed] [Google Scholar]; b Ferreira F, Botuha C, Chemla F, Pérez-Luna A. Chem. Soc. Rev. 2009;38:1162. doi: 10.1039/b809772k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.