

Summary
In higher eukaryotic interphase nuclei, the 100s-1000s fold linear compaction of chromatin is difficult to reconcile with its function as a template for transcription, replication, and repair. It is challenging to imagine how DNA and RNA polymerases with their associated molecular machinery would move along the DNA template without transient decondensation of observed large-scale chromatin “chromonema” fibers [1]. Transcription or replication “factory” models [2] in which polymerases remain fixed while DNA is reeled through are similarly difficult to conceptualize without transient decondensation of these chromonema fibers. Here we show how a dynamic plasticity of chromatin folding within large-scale chromatin fibers allows DNA replication to take place without significant changes in the global large-scale chromatin compaction or shape of these large-scale chromatin fibers. Time-lapse imaging of lac-operator tagged chromosome regions shows no major change in the overall compaction of these chromosome regions during their DNA replication. Improved pulse-chase labeling of endogenous interphase chromosomes yields a model in which global compaction and shape of large Mbp chromatin domains remains largely invariant during DNA replication, with DNA within these domains undergoing significant movements and redistribution as they move into and then out of adjacent replication foci. In contrast to hierarchical folding models, this dynamic plasticity of large-scale chromatin organization explains how localized changes in DNA topology allow DNA replication to take place without an accompanying global unfolding of large-scale chromatin fibers while suggesting a possible mechanism for maintaining epigenetic programming of large-scale chromatin domains throughout DNA replication.
eTOC Blurb
Deng et al. reveal a chromatin “dynamic plasticity” explaining how large-scale chromatin domains preserve their overall compaction and shape during DNA replication. DNA is pulled out of condensed chromatin domains into adjacent replication foci, replicates, and then with increasing chase time returns back into and disperses within these domains.
Results and Discussion
Two very different models have been proposed for DNA replication. Replication factory models propose that polymerases are immobilized in a nuclear body, while DNA is reeled into and out of these bodies as it is replicated. In early replication factory models, replication foci were interpreted as corresponding to special structures well separated from and independent of the pre-existing interphase chromosome structure (Fig. 1A). The more traditional replication-in-place model instead proposes that initiation of DNA replication occurs within the interphase chromosome structure with DNA replication proceeding through movement of polymerases, together with their associated very large, multi-protein assemblies, along the DNA template folded within this interphase chromosome structure. In the simplest replication-in-place model, DNA replication foci and incorporation of labeled nucleotides appear within the pre-existing interphase chromosome structure, which maintains its overall conformation before, during, and after DNA replication (Fig. 1B). In this model, “replication foci” simply correspond to PCNA accumulation or nucleotide incorporation over replicating DNA within pre-existing, condensed interphase chromosome structures. Early pulse labeling experiments revealing compact regions of DNA replication were interpreted as supporting a replication factory model due to the implicit assumption that interphase chromatin was dispersed and relatively uncondensed [3, 4]. Later pulse-chase experiments supported a DNA replication-in-place model (Fig. 1B), due to their visualization of condensed chromosome foci that behaved as stable, structural units through the cell cycle [5–8]. This visualization of stable chromosome foci by pulse-chase replication labeling introduces new constraints with regard to either replication factory or replication-in-place models. If replication factories exist, they should be located adjacent or within condensed interphase chromosome structures (Fig. 1C), rather than apart from decondensed chromatin as envisioned in the original model (Fig. 1A). In the case of a replication-in-place model, any interphase chromosome structural changes coordinated with DNA replication must preserve the overall structural integrity of these units of chromosome replication before and after replication- for instance by a transient uncoiling of a hierarchical folded structure prior to DNA replication, replication within this transiently unfolded structure (Fig. 1D), and then re-coiling of this structure after replication.
Fig. 1. Live-cell microscopy reveals minimal global changes in large-scale chromatin structure of tagged PDC chromosome loci during DNA replication.

(A–D) Four models for DNA replication spatial organization- DNA (grey), replication foci (green): (A) Replication factory model- dispersed DNA is reeled in and out of replication foci during replication. (B) Replication-in-place model- replication foci correspond to pre-existing Mbp-scale units of large-scale chromatin compaction undergoing replication. (C) Modified replication factory model- replication factories lie immediately adjacent to pre-existing Mbp-scale units of large-scale chromatin compaction. (D) Modified replication-in-place model- replication foci correspond to units of large-scale chromatin compaction that transiently decondense during in-place DNA replication. (E) Mitotic spread (DAPI, blue) of PDC βA2 cell clone shows three gene-amplified chromosome regions (mCherry-LacI, red) named in order of decreasing size (locus 1, long arrow; locus2, short arrow; locus 3, arrow head). (F) Tagged (red, mCherry-LacI) chromosome regions (loci 1–3, long arrow, short arrow, arrowhead, respectively) and GFP-PCNA foci (green) visualized as 2D projections as a function of time (hrs) after release from late G1/early S phase block. PCNA foci adjacent or near amplified chromosome loci are only seen in early to middle S-phase. (G) Fraction (LabeledRatio, y-axis) of labeled mitotic spreads (blue) or chromosome loci (locus 1-orange, locus 2-green, locus 3-dotted orange) as a function of chase time after EdU pulse labeling reveals early (loci 1–3) to mid-S phase (loci 1) labeling. (H) Locus 2 DNA replication occurs without global decondensation of large-scale chromatin fiber as shown by doubling of mCherry-LacI signal from 15 mins to 4–5 hrs (H, red) without significant change in projected area (H, black). (I) Visualization of locus 2 (red, mCherry-LacI), as seen in projections at 15 min intervals (left to right, top to bottom) after release from late G1/early-S phase block. GFP-PCNA foci (green) adjacent to locus 2 are observed in optical sections from 15 min (begin) to 4 hrs 45 mins (end). (J) Images and intensity line-scans (along white arrows) from orange-framed panels (I) showing GFP-PCNA (green, image and line-scans) associated with mCherry-LacI (red, image and line-scans) signals. (K) Histogram of measured peak-to-peak distances between GFP-PCNA and mCherry-LacI signals for 28 foci from (I). Scale bars: 5 μm (E,F), 1 μm (I). See also Fig. S1 and Fig. S2.
Distinguishing between replication factory and replication-in-place models is considerably complicated by the high compaction of metazoan interphase chromosomes. In typical mammalian nuclei, most 100–200 kb genomic regions appear as a near diffraction-limited spot by fluorescence in situ hybridization (FISH). This level of compaction is poorly understood, although light and electron microscopy approaches have revealed condensed, linear “chromonema” fiber-like segments with diameters in the range of 100 nm and estimated compaction ratios of ~1–3 Mbp per micron. These fiber-like structures have been observed even for transcriptionally active chromosome regions [1, 9–11].
Although the replication-in-place model was supported by the demonstration of stable, condensed chromosomal foci after pulse-chase labeling, more recent live-cell imaging experiments revealed PCNA foci just several hundred nm in diameter marking sites of active DNA replication smaller than those previously described chromosomal units of replication [12]. These PCNA foci could represent special replication factories into which DNA is reeled from adjacent interphase chromosomes for replication (Fig. 1C). However, they could also simply represent regions of condensed interphase chromosomes actively undergoing DNA replication (Fig. 1D). Clusters of 2–10 replicons (mode 4) are known to replicate synchronously [5]. Each replicon measures 25–325 kb in size (mode 125kb). These replicon clusters packaged into large-scale chromatin fibers with 1–3 Mbp per micron compaction levels therefore would give rise to structures of roughly comparable size to the observed PCNA foci.
Conspicuously missing from these previous studies has been the simultaneous imaging of a clearly demarcated chromosome region together with PCNA replication foci. Also missing has been the use of pulse-chase labeling conditions of sufficient temporal resolution to determine the spatial relationships between sites of DNA replication, recently replicated DNA, and the interphase chromosome region undergoing DNA replication. Here we tackle both needs, visualizing GFP-PCNA replication foci relative to a several Mbp labeled chromosome region and establishing pulse-chase conditions producing effective pulse durations much shorter than those used previously.
We used gene amplification of cDNA DHFR transgenes in Chinese Hamster Ovary (CHO) cells to create large lac operator/repressor tagged chromosome regions [13], choosing the PDC gene-amplified cell clone for further analysis. PDC cells contain large lac operator-tagged chromosome regions forming linear chromonema fibers of ~120–180 nm diameter, as demonstrated by both conventional [13] and in vivo immunogold labeling [11], resembling endogenous chromosome regions [11, 13–15]. We isolated PDC cell clone βA2 stably expressing EGFP-PCNA and mCherry-lac repressor. PDC βA2 mitotic chromosome spreads show three distinct amplified chromosome regions (Fig. 1E) which replicate during the first ~4 hours of S phase, as revealed by live-cell microscopy (Fig. 1F), pulse-chase EdU labeling of mitotic cells (Fig. S1A–C, Fig. 1G), and pulse labeling of interphase nuclei (Fig. S1D–F). The two smaller regions (loci 2&3) begin replication early in S phase, whereas the largest region (locus 1) begins replication ~1–2 hrs into S phase (Fig. 1G). This replication timing is consistent with the localization of PCNA foci near the edge of or within these PDC chromosome regions in cells with replication patterns [16] typical of early (loci 1–3) to mid-S phase (locus 1) (Fig. 1F,I–K, Fig. S1D–F, Fig. S2B,D–H).
From previous estimations of metaphase chromosome compaction in CHO cells [17], loci 1&2 must be tens of Mbp in size (Fig. 1E). The high compaction of DNA and density of lac operator repeats within these amplified chromosome regions allow us to visualize large chromosome regions as near contiguous fiber-segments of lac repressor staining within interphase nuclei as they undergo DNA replication (Fig. 1F).
We observed individual replication foci closely associated with PDC chromosome regions over periods of ~15–60 mins (2–4 time points) (data not shown), consistent with the previously reported 30–180 minute lifetimes of PCNA foci [12]. These replication foci appeared shortly before and during the several hours that the mCherry-LacI signal over these PDC regions doubled in intensity (Fig. 1H, Fig. S2A,C,I). Thus three independent methods for determining replication timing- pulse-chase labeling of mitotic spreads, stable association of GFP-PCNA foci, and doubling of mCherry-LacI signal- all show these amplified chromosome regions as replicating during the first half of S-phase. Locus 1 continues replication into the beginning of replication pattern 3 in middle S-phase, as inferred by the stable association of replication foci with this locus during this time (Fig. S2G–H, 5 hrs) and the 1–2 hour delays in replication timing, relative to the other loci, demonstrated by the mitotic pulse-chase (Fig. 1G) and doubling of the mCherry-LacI signal measurements (Fig. S2I).
Importantly, these PDC chromosome regions typically showed no major, global decondensation in large-scale chromatin structure throughout the periods of S phase in which they replicated (Fig. 1H–I, Fig. S2A–D, H–I). Replication foci frequently appeared at the edge of larger segments of large-scale chromatin foci/fibers (Fig. 1I–J, Fig. S2B,D,E–H), as supported by the skewed distribution of the distance between the PCNA foci centers from the center of the adjacent mCherry-LacI signals (Fig. 1K). Transient localized extension of large-scale chromatin loci was sometimes observed but this occurred after DNA replication, as timed by the disappearance of associated PCNA foci and doubling of the mCherry-LacI signal (Fig. S2H-large arrow, I- red line). No significant decondensation coinciding with DNA replication was observed as would be expected from models in which a hierarchically folded large-scale chromatin fiber uncoiled to a lower level of compaction (Fig. 1D).
Our imaging results instead favor the adjacent replication factory model (Fig. 1C) in which DNA segments, tens of kb in size, move smaller distances from within condensed large-scale chromatin fibers into discrete replication foci immediately adjacent to and/or within these fibers. No new fiber formation or change in overall compaction accompanies DNA replication of these PDC loci, implying that this newly synthesized DNA must exit these replication foci back into the same large-scale chromatin fiber following their replication. This “dynamic chromatin plasticity” replication model would require a relatively disordered, dynamic internal structure for these large-scale chromatin fibers, very different from previously proposed hierarchical folding models [14, 15], but in line with conclusions drawn from analysis of lac operator repeat pairing within BAC transgene tandem arrays [18].
To test this dynamic chromatin plasticity model, we visualized newly replicated DNA of endogenous chromosomes. Preliminary experiments in CHO cells showed that newly replicated DNA exits replication foci after an ~ 9 min pulse. We adjusted EdU labeling conditions until we established significantly improved EdU labeling conditions with an effective pulse duration of 9 min.
We determined the effective pulse duration by measuring for how long the EdU total incorporation per nucleus continued to increase after a pulse and then chase. We measured total EdU fluorescence per nucleus using either deconvolution light microscopy (“image analysis”, Fig. 2A–B) or flow cytometry. A constant EdU concentration produced a linear increase versus time in EdU incorporation (Fig. 2A). In CHO cells, EdU incorporation plateaued 9 mins after a 30 sec pulse of 10 μM EdU (Fig. 2B). This 9 mins effective pulse duration in CHO cells is consistent with the time required for EdU cytosolic concentrations, measured by liquid chromatography and mass spectroscopy (LC-MS/MS), to decrease substantially after the same 30 sec, 10 μM EdU pulse (Fig. 2C).
Fig. 2. Visualizing nascent DNA emerging from replication foci and incorporating into large-scale chromatin structure with improved pulse-chase conditions.

(A,F) EdU incorporation increases linearly with duration of pulse labeling time (mins, x-axis) for both CHO (A) and HT1080 cells (F) as measured by image analysis (solid squares and line, y axis-left) or flow cytometry (hollow squares, dashed line, y axis-right). (B,G) EdU incorporation plateaus in CHO cells (B) at ~9 min after 30 sec EdU pulse and in HT1080 cells (G) at ~6 min after 10 sec EdU pulse. (C,H) EdU cellular pool concentration by LC-MS/MS after 30 sec pulse in CHO cells (C) or 10 sec pulse in HT1080 cells (H). EdU concentrations were undetectable at longer chase times (18 and 36 mins for CHO cells; 9, 18, and 36 mins for HT1080 cells). (D,I) Exit of EdU (red) from GFP-PCNA foci becomes obvious at chase-times of ~9 min in CHO cells (D) and ~6 min in HT1080 cells (I). Scale bars = 0.5 μm. (E,J) EdU signals (red) versus GFP-PCNA foci (green) and DAPI (blue) after chase-times of 9 min, 18 min, 36 min, and 1 hr in CHO cells (E) or 6 min, 12 min, 18 min, and 36min chase in HT1080 cells (J). DNA (blue) colocalizes with GFP-PCNA (green) foci at 9 min (CHO) or 6 min (HT1080), is still compact in shape but has largely separated from GFP-PCNA foci by 18 min (CHO) or 12 min (HT1080), and has spread out within DAPI-dense regions at 36 min (CHO) or 18 min (HT1080) with spreading continuing through to 1 hr (CHO) or 36 min (HT1080). Longer chase times give increased colocalization of EdU signal (red outlines) with DAPI-rich regions, while PCNA foci (green outlines) remain over DAPI-poor regions. EdU area increases in CHO cells (E) from 9 min to 1 hr chase-time and from 6 min to 36 min chase-time in HT1080 cells (J). Panels (below) show enlargements of regions in square white boxes (top). Scale bars = 5 μm (top panels), 1 μm (bottom panels). See also Fig. S3.
Changes in the size or shape of the EdU foci after 9 mins chase therefore represent conformational changes rather than new EdU incorporation. The EdU signal co-localized with PCNA immunstaining with short pulse times of ~8 mins or less, as visualized by 3D SIM (Structured Illumination Microscopy) super-resolution light microscopy (Fig. 2D). Newly labeled DNA, still organized in punctate foci, shows increasing separation from partially overlapping PCNA foci from ~9–18 mins chase times (Fig. 2D&E, Fig. S3A–B). Separation between EdU and PCNA foci is also apparent from the progressive drop in the Manders’ overlap coefficient[19], measuring the proportion of EdU image signal overlapping with GFP-PCNA signal (Fig. S3D). By 36–60 mins of chase, newly labeled DNA has expanded noticeably from small punctate foci to larger, elongated fiber-like segments that are largely non-overlapping with adjacent PCNA foci (Fig. 2E, Fig. S3C). These adjacent PCNA foci likely represent some combination of old and new replication foci from what existed at the time of the EdU pulse, with the proportion of new replication foci increasing with increasing chase time. The appearance of GFP-PCNA foci adjacent to EdU fiber-like signals after a long (1 hr) pulse suggests that new GFP-PCNA foci may form and replicate additional replicon clusters contained within the same large-scale chromatin fiber, consistent with our live-cell imaging showing replication foci appearing and disappearing at different locations on the periphery of the same mCherry-LacI foci (Fig. 1I, Fig. S2B,D,H). Both sets of observations suggest intermingling of neighboring replicon clusters with asynchronous replication timing, each several hundred kb in size, within the same chromonema fiber segment. Our results do not distinguish between random firing of replicon clusters along a chromonema fiber segment versus a domino model[20] in which completion of replication in one replication focus triggers initiation of replication in a neighboring new replication focus. Our results do exclude a model involving a continuous progression in replication timing along the length of a chromonema fiber.
This transition from small, punctate EdU foci overlapping with PCNA foci to larger, elongated, fiber-like segments adjacent to PCNA foci is paralleled by an increase in the co-localization of EdU with DAPI-dense regions (Fig. 2E). After 36–60 mins chase, the EdU signal largely overlaps bright DAPI-stained regions, while PCNA foci lie on the periphery or adjacent to these DAPI/EdU bright regions, like the distribution of GFP-PCNA foci adjacent to the PDC chromosome loci visualized in live cells.
These results strongly support the adjacent replication factory model (Fig. 1C) in which DNA moves into replication foci lying immediately adjacent to or overlapping with large-scale chromatin domains, replicates, and then moves back into and spreads within these large-scale chromatin domains with increasing chase time. We suggest that poorer temporal resolution due to continued nucleotide incorporation after traditional pulse-chase experiments have led instead to the replication-in-place models proposed in previous studies [5–8].
Indeed, earlier experiments of ours in human HT1080 fibroblasts had also suggested a “replication-in-place” model for DNA replication. Using traditional pulse-chase conditions, SIM images showed abundant short, fiber-like segments of EdU incorporation after 10 μM EdU pulses of 20–40 mins (data not shown). We subsequently determined that separation of DNA from replication foci in HT1080 cells occurs within ~6 mins. In HT1080 cells, we used a 10 sec, 40 μM EdU exposure to create an effective pulse time of 6 min (Fig. 2F–H).
Using these modified pulse-chase conditions, we observed a faster but otherwise similar transition as seen in CHO cells. Punctate EdU incorporation extensively overlapped PCNA foci after 3–6 mins chase (Fig. 2I). EdU incorporation began to separate from PCNA foci at later times, changing to larger, fiber-like segments of EdU incorporation adjacent to PCNA foci after 18–36 mins chase (Fig. 2J, Fig. S3E–H). PCNA foci are smaller and more closely spaced in HT-1080 cells (0.27 μm mean separation) than in CHO cells (0.50 μm mean separation). Using traditional pulse durations of 20–40 mins, EdU incorporation from adjacent replication foci instead would merge into labeled fiber segments.
To confirm our light microscopy observations, we used transmission electron microscopy (TEM) to visualize EdU spreading after a pulse-chase in CHO cells. After 9 mins chase, immunogold staining shows EdU incorporation in small foci lying over or at the immediate periphery of large-scale chromatin domains (Fig. 3A–B). After 36 min chase, this EdU incorporation spreads throughout large-scale chromatin fiber segments (Fig. 3C–D).
Fig. 3. Spreading of newly replicated DNA within large-scale chromatin fibers in CHO cells visualized by TEM.

Immunogold staining against EdU-Alexa488. 30 sec EdU pulse followed by 9 min (A–B) or 36 min (C–D) chase. At 9min chase, EdU labeled newly-replicated DNA (black, silver-enhanced gold particles) localizes at the immediate periphery of large-scale chromatin domains (grey) (A,B). After 36min chase (C,D), EdU pulse incorporation has spread within large-scale chromatin domains. Scale bars = 1 μm (A,C), 0.5 μm (B,D). See also Fig. S4.
We were unable to achieve adequate immunogold visualization of EdU incorporation in HT-1080 cells using the 6 min “effective” pulse conditions described above for SIM visualization. However, we were able to visualize EdU incorporation by immunogold labeling using traditional 15 min EdU pulse labeling (Fig. S4). Using these conditions, EdU immunogold staining revealed small foci of EdU incorporation centered over large-scale chromatin structures after 15 mins pulse (Fig. S4F). Correlative light and electron microscopy showed the alignment of these small, closely spaced foci into linear patterns, giving rise to the fiber-like appearance (Fig. S4B,D). After 1–3 hr chase times, this EdU incorporation coalesced into more obvious fiber-like regions (Fig. S4A,C,E,G). Thus our immunogold visualization of EdU incorporation in HT1080 cells also supports our dynamic plasticity model in which DNA replicated in small foci spreads out after replication throughout large-scale chromatin fibers.
In summary, our SIM and TEM pulse-chase labeling combined with our live imaging results lead to a dynamic chromatin plasticity model for DNA replication within the context of large-scale chromatin compaction. Traditional effective pulse times of tens of minutes would have suggested a replication-in-place model (Fig. 4A). In contrast, our improved pulse-chase conditions reveal how DNA from multiple replicons contained within a common large-scale chromatin domain/fiber must move into and concentrate in replication foci that lie adjacent to this domain/fiber (Fig. 4B, upper left). Newly replicated DNA then exits these foci, returning into and spreading throughout these large-scale chromatin structures without any major global large-scale chromatin decondensation accompanying the DNA replication of these regions (Fig. 4B, arrows indicate order of increasing chase time).
Fig. 4. Movement of replicated DNA out of replication foci and into large-scale chromatin fibers/domains and model for how this chromatin dynamic plasticity may explain inheritance of distinct epigenetic states over large chromatin domains.
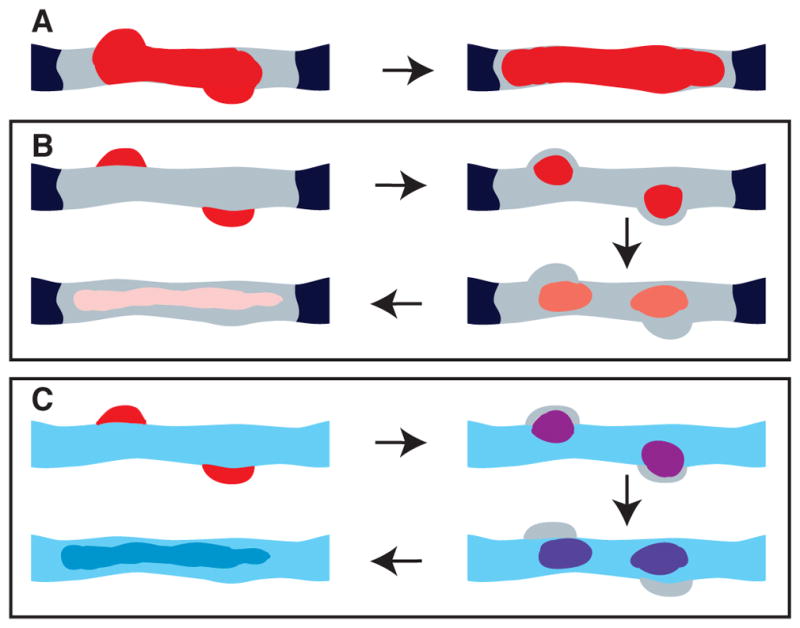
(A) Traditional labeled nucleotide pulse times of tens of minutes will result in labeling (red) of both replication foci (bumps on edge of fiber) and adjacent large-scale chromatin fibers/domains (grey). The predicted invariance of this labeling pattern with additional chase time would be interpreted as consistent with a replication-in-place model. (B) Using shorter effective pulse times will result in labeling (red) first over replication foci (upper left) adjacent to large-scale chromatin fiber/domain (grey), followed by exit of labeled DNA out of the replication foci into the large-scale chromatin fiber. With increasing chase times (arrows), a constant amount of labeled DNA will spread out within large-scale chromatin fiber (pink). (C) Model for epigenetic inheritance of marks over large chromatin domains: Newly replicated DNA (red, upper left corner) in replicated foci will incorporate new histones. With chase out of replication foci into large-scale chromatin fiber, newly replicated DNA as it inserts into large-scale chromatin fiber/domain will now accumulate new marks through contact with surrounding, not yet replicated chromatin with distinctive epigenetic marks and machinery for writing these marks (blue). If this acquisition of new marks (red to purple to blue transition) occurs sufficiently fast, the last replicating DNA in each replicon will reinsert into large-scale chromatin domains after earlier replicated regions of these replicons have assumed these marks and machinery for writing these marks.
These results also confirm the folding of most DNA, including early replicating, gene-rich, euchromatic chromosomal regions, into large-scale chromatin fibers in CHO and HT1080 cells. We had previously inferred this from our studies of transcriptionally active multi-copy BAC transgenes[10], but our current results now demonstrate this for endogenous chromosomes.
Our dynamic chromatin plasticity model has several potential biological implications. First, it explains how DNA replication, executed by large, macromolecular complexes, can occur in the context of highly condensed, large-scale chromatin organization. Second, our model suggests a speculative model for inheritance of distinct epigenetic states over large chromatin domains such as TADs [21–24], LOCKs [25], and LADs [26, 27]. Our results show that newly replicated DNA near newly activated origins would still be within replication foci within ~6–9 mins after replication, depending on the cell line (Fig. 4C, upper left panel). At later times, replicated DNA would begin dispersing throughout condensed large-scale chromatin domains (Fig. 4C, arrows show progression over time), thus becoming surrounded by unreplicated chromatin still containing its full repertoire of epigenetic marks and chromatin modifying machinery. Close contact with this unreplicated DNA would then facilitate duplication of similar marks over the newly replicated DNA (Fig. 4C). The estimated 30–180 mins lifetime of individual replication foci[12] would allow sufficient time first for the transfer of these epigenetic marks from the unreplicated DNA to the earliest replicating DNA and then the repeated transfer of these epigenetic marks from this newly replicated DNA to the last DNA to replicate within the chromatin domain. Moreover, DNA from several neighboring clusters of replicons, each cluster replicating at slightly different times in different PCNA foci, might physically intermingle through this dynamic plasticity behavior within the same large-scale chromatin fiber-segment, extending the time even further for inheritance of chromatin domain epigenetic memory. Finally, our dynamic plasticity model suggests how long-range DNA looping can occur within the context of large-scale chromatin domains/fibers, possibly reconciling predictions of extensive long-range looping from 3C experiments with the observations of extended chromonema fibers visualized by microscopy.
Experimental Procedures
More detailed experimental procedures are provided in the Supplemental Experimental Procedures. Briefly, CHO DG44 derivatives, CHO-K1, and HT-1080 cells were grown using standard protocols for these cells. Live imaging was done using cell clones stably expressing EGFP-PCNA and mCherry-LacI using an Applied Precision OMX V3 microscope with EMCCD cameras and short exposures to minimize photo-toxicity. Click-iT (Invitrogen) labeling of EdU with Alexa Fluor 488 or Alexa Fluor 594 followed the manufacturer’s suggested procedure. Anti-PCNA staining used mouse monoclonal anti-PCNA PC-10 (Abcam) primary antibody at 1:2000 dilution and Alexa Fluor 488 goat anti-mouse IgG secondary antibody (Life Technologies) at 1:1000 dilution. Cell synchronization used a mitotic shake-off followed by a hydroxyurea (HU) late G1/early S phase block followed by release for PDC cells, an isoleucine minus G0 block followed by a HU block and then release for CHO-K1 cells, and a double-thymidine block for HT1080 cells. Immunogold pre-embedding labeling used EdU Click-iT labeling with Alexa 488-azide, an anti-Alexa 488 primary antibody (1:1000), and a secondary goat anti-mouse Nanogold-coupled Fab′ (Nanoprobes). SIM imaging was done using an Applied Precision OMX V3 (Fig. 2, SFig. 3) or Nikon N-SIM microscopes (SFig. 4); deconvolution wide-field microscopy was done using an Applied Precision Personal Deltavision.
Supplementary Material
Highlights.
Overall large-scale chromatin compaction does not change during DNA replication
Replicated DNA moves out of PCNA foci and into adjacent large-scale chromatin fibers
Replicated DNA spreads within large-scale chromatin fibers with increasing time
Chromatin “dynamic plasticity” explains how condensed templates are replicated
Acknowledgments
This work was supported by NIH grant R01 GM58460 to ASB and RFBR grant 13-04-00885 to IIK. We thank Christina Cardoso for the pENeGFPPCNA plasmid. We thank Furong Sun (UIUC, Laboratory for Mass Spectroscopy) for technical support with LC-MS/MS.
Footnotes
Author Contributions: XD created stable PDC cell lines expressing fluorescent proteins, carried out live-cell microscopy, determined effective pulse conditions for pulse-chase experiments and carried out pulse-chase experiments by wide-field light microscopy. OAZ carried out CLEM and TEM of EdU incorporation together with OAZ, VDC, XD and OSS. XD and OAZ carried out SIM light microscopy. Experiments were designed through discussions among XD, OAZ, IIK, and ASB.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Belmont AS. Large-scale chromatin organization: the good, the surprising, and the still perplexing. Curr Opin Cell Biol. 2014;26:69–78. doi: 10.1016/j.ceb.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cook PR. The organization of replication and transcription. Science. 1999;284:790–1795. doi: 10.1126/science.284.5421.1790. [DOI] [PubMed] [Google Scholar]
- 3.Nakamura H, Morita T, Sato C. Structural organizations of replicon domains during DNA synthetic phase in the mammalian nucleus. Exp Cell Res. 1986;165:291–297. doi: 10.1016/0014-4827(86)90583-5. [DOI] [PubMed] [Google Scholar]
- 4.Hozak P, Jackson DA, Cook PR. Replication factories and nuclear bodies: the ultrastructural characterization of replication sites during the cell cycle. J Cell Sci. 1994;107(Pt 8):2191–2202. doi: 10.1242/jcs.107.8.2191. [DOI] [PubMed] [Google Scholar]
- 5.Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol. 1998;140:1285–1295. doi: 10.1083/jcb.140.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sparvoli E, Levi M, Rossi E. Replicon clusters may form structurally stable complexes of chromatin and chromosomes. J Cell Sci. 1994;107(Pt 11):3097–3103. doi: 10.1242/jcs.107.11.3097. [DOI] [PubMed] [Google Scholar]
- 7.Sadoni N, Langer S, Fauth C, Bernardi G, Cremer T, Turner BM, Zink D. Nuclear organization of mammalian genomes. Polar chromosome territories build up functionally distinct higher order compartments. J Cell Biol. 1999;146:1211–1226. doi: 10.1083/jcb.146.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferreira J, Paolella G, Ramos C, Lamond AI. Spatial organization of large-scale chromatin domains in the nucleus: a magnified view of single chromosome territories. J Cell Biol. 1997;139:1597–1610. doi: 10.1083/jcb.139.7.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Belmont AS, Hu Y, Sinclair PB, Wu W, Bian Q, Kireev I. Insights into interphase large-scale chromatin structure from analysis of engineered chromosome regions. Cold Spring Harb Symp Quant Biol. 2010;75:453–460. doi: 10.1101/sqb.2010.75.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu Y, Kireev I, Plutz MJ, Ashourian N, Belmont AS. Large-scale chromatin structure of inducible genes- transcription on a linear template. J Cell Biol. 2009;185:87–100. doi: 10.1083/jcb.200809196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kireev I, Lakonishok M, Liu W, Joshi VN, Powell R, Belmont AS. In vivo immunogold labeling confirms large-scale chromatin folding motifs. Nat Methods. 2008;5:311–313. doi: 10.1038/nmeth.1196. [DOI] [PubMed] [Google Scholar]
- 12.Leonhardt H, Rahn H-P, Weinzierl P, Sporbert A, Cremer T, Zink D, Cardoso MC. Dynamics of DNA Replication Factories in Living Cells. J Cell Biol. 2000;149:271–280. doi: 10.1083/jcb.149.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinett CC, Straight A, Li G, Willhelm C, Sudlow G, Murray A, Belmont AS. In vivo localization of DNA sequences and visualization of large-scale chromatin organization using lac operator/repressor recognition. J Cell Biol. 1996;135:1685–1700. doi: 10.1083/jcb.135.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belmont AS, Bruce K. Visualization of G1 chromosomes: a folded, twisted, supercoiled chromonema model of interphase chromatid structure. J Cell Biol. 1994;127:287–302. doi: 10.1083/jcb.127.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kireeva N, Lakonishok M, Kireev I, Hirano T, Belmont AS. Visualization of early chromosome condensation: a hierarchical folding, axial glue model of chromosome structure. J Cell Biol. 2004;166:775–785. doi: 10.1083/jcb.200406049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Keefe RT, Henderson SC, Spector DL. Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific alpha-satellite DNA sequences. J Cell Biol. 1992;116:1095–1110. doi: 10.1083/jcb.116.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li G, Robinett C, Sudlow G, Willhelm C, Belmont AS. Interphase dynamics of a late replicating, heterochromatic chromosome HSR. Mol Biol Cell. 1995;6(supplement):71a. [Google Scholar]
- 18.Sinclair P, Bian Q, Plutz M, Heard E, Belmont AS. Dynamic plasticity of large-scale chromatin structure revealed by self-assembly of engineered chromosome regions. J Cell Biol. 2010;190:761–776. doi: 10.1083/jcb.200912167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci. 1992;103(Pt 3):857–862. doi: 10.1242/jcs.103.3.857. [DOI] [PubMed] [Google Scholar]
- 20.Sporbert A, Gahl A, Ankerhold R, Leonhardt H, Cardoso MC. DNA polymerase clamp shows little turnover at established replication sites but sequential de novo assembly at adjacent origin clusters. Mol Cell. 2002;10:1355–1365. doi: 10.1016/s1097-2765(02)00729-3. [DOI] [PubMed] [Google Scholar]
- 21.Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148:458–472. doi: 10.1016/j.cell.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 22.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hou C, Li L, Qin ZS, Corces VG. Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol Cell. 2012;48:471–484. doi: 10.1016/j.molcel.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wen B, Wu H, Shinkai Y, Irizarry RA, Feinberg AP. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat Genet. 2009;41:246–250. doi: 10.1038/ng.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peric-Hupkes D, van Steensel B. Role of the nuclear lamina in genome organization and gene expression. Cold Spring Harb Symp Quant Biol. 2010;75:517–524. doi: 10.1101/sqb.2010.75.014. [DOI] [PubMed] [Google Scholar]
- 27.Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.