

Abstract
Orexins (hypocretins) are neuropeptides that regulate multiple homeostatic processes, including reward and arousal, in part by exciting serotonergic dorsal raphe neurons, the major source of forebrain serotonin. Here, using mouse brain slices, we found that, instead of simply depolarizing these neurons, orexin-A altered the spike encoding process by increasing the postspike afterhyperpolarization (AHP) via two distinct mechanisms. This orexin-enhanced AHP (oeAHP) was mediated by both OX1 and OX2 receptors, required Ca2+ influx, reversed near EK, and decayed with two components, the faster of which resulted from enhanced SK channel activation, whereas the slower component decayed like a slow AHP (sAHP), but was not blocked by UCL2077, an antagonist of sAHPs in some neurons. Intracellular phospholipase C inhibition (U73122) blocked the entire oeAHP, but neither component was sensitive to PKC inhibition or altered PKA signaling, unlike classical sAHPs. The enhanced SK current did not depend on IP3-mediated Ca2+ release but resulted from A-current inhibition and the resultant spike broadening, which increased Ca2+ influx and Ca2+-induced-Ca2+ release, whereas the slower component was insensitive to these factors. Functionally, the oeAHP slowed and stabilized orexin-induced firing compared with firing produced by a virtual orexin conductance lacking the oeAHP. The oeAHP also reduced steady-state firing rate and firing fidelity in response to stimulation, without affecting the initial rate or fidelity. Collectively, these findings reveal a new orexin action in serotonergic raphe neurons and suggest that, when orexin is released during arousal and reward, it enhances the spike encoding of phasic over tonic inputs, such as those related to sensory, motor, and reward events.
SIGNIFICANCE STATEMENT Orexin peptides are known to excite neurons via slow postsynaptic depolarizations. Here we elucidate a significant new orexin action that increases and prolongs the postspike afterhyperpolarization (AHP) in 5-HT dorsal raphe neurons and other arousal-system neurons. Our mechanistic studies establish involvement of two distinct Ca2+-dependent AHP currents dependent on phospholipase C signaling but independent of IP3 or PKC. Our functional studies establish that this action preserves responsiveness to phasic inputs while attenuating responsiveness to tonic inputs. Thus, our findings bring new insight into the actions of an important neuropeptide and indicate that, in addition to producing excitation, orexins can tune postsynaptic excitability to better encode the phasic sensory, motor, and reward signals expected during aroused states.
Keywords: arousal, narcolepsy, reward, SK channels, slow AHP, spike frequency adaptation
Introduction
Orexin neuropeptides (orexin-A and -B), also known as hypocretin-1 and -2 (de Lecea et al., 1998; Sakurai et al., 1998), act via two G-protein-coupled receptors (Sakurai et al., 1998) to influence multiple homeostatic systems, including those controlling reward and arousal (Carter et al., 2009). Indeed, orexins are critical for normal waking and sleep because the loss of orexin signaling results in the sleep disorder narcolepsy with cataplexy in animals and humans (Chemelli et al., 1999; Lin et al., 1999; Peyron et al., 2000; Thannickal et al., 2000).
The dorsal raphe (DR) and other monoaminergic nuclei receive strong orexinergic innervation (Peyron et al., 1998), express orexin receptors (OxRs) (Trivedi et al., 1998; Marcus et al., 2001) and are targets through which orexin promotes arousal, suppresses sleep (Piper et al., 2000), and prevents cataplexy in narcolepsy (Hasegawa et al., 2014). Serotonergic (5-HT) DR neurons have extensive ascending projections, where 5-HT acts to regulate mood, reward, feeding, arousal and motor behavior, and is implicated in the etiology and treatment of psychiatric disorders, including depression (for review, see Jacobs and Fornal, 1999; Lowry et al., 2005). Thus, factors influencing output from these neurons will widely impact CNS function.
5-HT DR neurons fire slowly at tonic rates that vary with behavioral state, having their highest rates in active waking and near-zero rates during REM sleep (for review, see Monti, 2011). Some 5-HT neurons also show a “cluster” or “burst” firing, which augments 5-HT release (Hajós et al., 2007) and occurs mostly during waking (Sakai, 2011), when orexin neurons are most active (Lee et al., 2005b; Mileykovskiy et al., 2005). 5-HT neurons also phasically encode sensory/motor events and reward magnitude (Ranade and Mainen, 2009; Liu et al., 2014), and the majority of these neurons burst during sucrose reward acquisition (Li et al., 2016). While orexin neurons fire during reward acquisition (Hassani et al., 2016), how orexins might influence these firing patterns of 5-HT DR neurons is unknown. Orexins produce slow depolarizations throughout the CNS (for review, see Leonard and Kukkonen, 2014), which result mainly from a noisy cation current in DR 5-HT neurons (Brown et al., 2002; Liu et al., 2002; Kohlmeier et al., 2008). Orexins can also regulate transmitter release, which is associated with increased GABAergic input (Liu et al., 2002) and decreased excitatory input (Haj-Dahmane and Shen, 2005) to 5-HT neurons, suggesting a multifaceted role in regulating DR output. The firing of DR 5-HT neurons is strongly regulated by a postspike afterhyperpolarization (AHP) (Aghajanian and Vandermaelen, 1982), mediated by apamin-sensitive, SK-type, Ca2+-dependent K+ channels (Freedman and Aghajanian, 1987; Pan et al., 1994; Scuvée-Moreau et al., 2004; Crawford et al., 2010; Alix et al., 2014) that promotes slow firing (Rouchet et al., 2008).
Here we show that orexin-A enhances this AHP and that this orexin-enhanced AHP (oeAHP) results from two distinct mechanisms, including both an increased SK current and the induction of a novel slow-AHP-like current. Moreover, our functional studies establish that the oeAHP promotes slower and more regular firing than expected for depolarizing orexin currents, and that it preserves responsiveness to phasic inputs while attenuating responsiveness to tonic inputs. Collectively, our findings reveal a new modulatory action of orexin that plays a previously unexpected role in shaping excitability of 5-HT DR neurons to better encode phasic sensory, motor, and reward signals expected during aroused states.
Materials and Methods
All procedures complied with National Institutes of Health guidelines and were approved by New York Medical College Institutional Animal Care and Use Committee.
Mice and genotyping.
Brain slices were prepared from young male and female C57BL/6N mice (Charles River) and OX1 receptor null, OX2 receptor null, or double OxR knock-out (DKO) mice (P14-P26). Receptor knock-out mice were offspring from homozygous null breeders having a mixed C57BL6 and 129/Sv genetic background (kindly provided by Drs. M. Yanagisawa and R. Chemelli). Use of these mice has been described previously (Willie et al., 2003; Mieda et al., 2011; Kohlmeier et al., 2013). To confirm the genotype of each mouse, tail biopsies were obtained during slice preparation and analyzed by PCR to determine whether each receptor was wild-type or knock-out. The three primers for OX1 consisted of a common primer (5′ CTCTTTCTCCACAGAGCCCAGGACTC 3′), a knock-out primer (5′ TGAGCGAGTAACAACCCGTCGGATTC 3′), and a wild-type primer (5′ gCAAGAATGGGTATGAAGGGAAGGGC 3′). The expected product sizes were ∼320 bp for the wild-type allele and ∼500 bp for the knock-out allele. The three primers for OX2 consisted of a common primer (5′ CTGGTGCAAATCCCCTGCAAA 3′), a knock-out primer (5′ GGTTTTCCCAGTCACGACGTTGTA 3′), and a wild-type primer (5′ AATCCTTCTAGAGATCCCTCCTAG 3′). The expected product sizes were ∼620 bp for wild-type allele and ∼300 bp for the knock-out allele. These two sets of primers for different OxRs were processed separately, and PCR was performed using 35 cycles of 30 s at 94°C, 30 s at 62°C and 1 min at 72°C, followed by one cycle at 72°C for 10 min. The result of each PCR was then fractionated on a 2% agarose gel, and the PCR product was visualized by ethidium bromide staining.
Brain slice preparation.
Mice were decapitated following induction of deep anesthesia with isofluorane. A block of brain with the target structure was rapidly removed and incubated and then cut in ice-cold ACSF, which contained the following (in mm): 124 NaCl, 5 KCl, 1.2 NaH2PO4, 2.7 CaCl2, 1.2 MgSO4, 26 NaHCO3, and 10 dextrose (295–305 mOsm) and was oxygenated with carbogen (95% O2/5% CO2). Brain slices (250 μm) were cut with a Leica vibratome (VT1000S) and were then incubated at 35°C for 15 min in oxygenated ACSF. They were then stored in continuously oxygenated ACSF at room temperature until use. For recordings, slices were submerged in a chamber that was perfused at 1–2 ml/min with continuously oxygenated ACSF, which was at room temperature (23 ± 2°) for all experiments, except those examining the firing properties with current clamp and dynamic clamp. For those experiments, slices were superfused with ACSF maintained at 32 ± 1° (see Figs. 9; 10).
Figure 9.

The oeAHP slows the firing rate resulting from the orexin-mediated depolarization. A, Repetitive firing evoked by current clamp (CC) delivery of a previously recorded “noisy” orexin current (A1) was compared with firing achieved by bath application of orexin-A (A2) in the same neuron (Orx A, 300 nm). Top traces, Instantaneous firing rate (FR, red before; blue during Orx A) in spikes/s (sps). Middle traces, Vm. Bottom traces, Injected current. Vertical scales are the same in A1 and A2, while the time base differs, as indicated. A2, Filled round symbols represent when the AHP was tested before (red) and after (blue) orexin (5 pulse protocol). The resulting AHPs are superimposed on the right (boxed inset, red before; blue during Orx A). A2, Arrow indicates application of hyperpolarizing current to return Vm to baseline. A1, A2, Dashed horizontal line indicates mean firing rate resulting from injected current in A1. A3, Comparison of the mean ± SD of steady-state firing rate (ssFR, mean ± SD) for this neuron produced by orexin-A (Orx A, filled triangle) and by a range of scaled recorded orexin currents (CC; open horizontal diamonds). B, Repetitive firing evoked by dynamic clamp (DC) addition of a virtual “noisy” orexin conductance (B1) was compared with the firing rate achieved by bath application of orexin-A (B2). Traces, scaling, and markings as in A. B3, Comparison of steady-state firing rate for this neuron produced by orexin-A (filled triangle) with a range of virtual orexin conductances (DC; open vertical diamonds). C1, Summary of mean ssFR produced by orexin-A (actual) and the corresponding CC (open horizontal diamonds; n = 5) and DC (open vertical diamonds; n = 6) simulated orexin currents (virtual). *p < 0.05, the ssFR firing produced by orexin-A (Actual) was significantly slower than firing produced by corresponding CC and DC virtual current. C2, Summary of firing rate variability, measured by the SD of steady-state firing rate produced by orexin-A (Actual) and by the corresponding virtual orexin conductances (Virtual). *p < 0.05, the SD produced by orexin-A was significantly lower than that produced by the corresponding DC virtual current.
Figure 10.

Orexin-A increased spike-frequency adaptation and produced a delayed decrease in firing fidelity. A, Repetitive firing (100 pA, 5 s) before (top trace, Control, red) and during Orx A action (300 nm; bottom trace, Orx A, blue). Arrow indicates −65 mV in both traces. Calibration bars apply to both traces. B, Summary of initial firing rate (computed from the first interval; mean ± SEM) versus injected current (I; Control, open circles, red; orexin, filled triangles, blue; n = 5). C, Summary of steady-state firing (mean ± SEM; control, red open circles; orexin, blue filled triangles; n = 5). *p < 0.05. D, Firing evoked by 10 s duration current pulse-trains (5 ms; 0.5–20 Hz) before (top; Control, red) and after orexin (bottom; Orx A, blue). E, Summary (mean ± SEM) of steady-state firing fidelity (ss Fidelity; percentage pulses that fired a spike in the second half of the train; n = 5) versus input pulse frequency (Hz) before (red open circles) and during orexin action (blue filled triangles). *p < 0.05, significant difference in orexin. F, Summary (mean ± SEM) of steady-state output firing-frequency (n = 5) versus input pulse-frequency before (Control SS, red open circles) and during orexin action (Orx A SS, blue filled triangles). *p < 0.05, significant difference between control and orexin. Dashed line indicates the initial input–output relation, which was 1:1.
Drugs and experimental solutions.
Ionotropic receptor antagonists DNQX (15 μm, Sigma), AP5 (50 μm, Sigma, Tocris Bioscience), and SR-95531 (gabazine, 20 μm, Sigma) with strychnine (2.5 μm, Sigma) were added to ACSF in all experiments to block synaptic potentials. In voltage-clamp experiments, TTX (500 nm, Alomone Labs) was added to block voltage-gated sodium channels. Orexin-A (Peptides International, 3–300 nm) was diluted into ACSF to final concentration from frozen aliquots just before use. Apamin (100 or 300 nm, Tocris Bioscience), cyclopiazonic acid (CPA; 10 μm, Sigma), phenylephrine (PE; 300 nm to 30 μm, Sigma), and prazosin (100 nm, Tocris Bioscience) were dissolved in ultrapure water at stock concentration and then dissolved in ACSF the day of the experiment. UCL2077 (10 μm, Tocris Bioscience) was dissolved in DMSO at 5000 times final concentration. Adenylyl cyclase activator forskolin (10 μm, Tocris Bioscience) was also dissolved in DMSO at 2000 times final concentration. Protein kinase C inhibitor peptide 19–36 (PKCIP; 2 μm, Calbiochem) was dissolved in pipette solution at 200 μm and then further dissolved to final concentration in pipette solution on the day of the experiment. PKC inhibitor bisindolylmaleimide I (Bis I; 1 μm, Calbiochem) was dissolved in water at a stock concentration. PKC activator phorbol 12,13-dibutyrate (PDBu; 1 μm, Tocris Bioscience) was dissolved in DMSO at 104 times final concentration. Phospholipase C (PLC) inhibitor U73122 (1 μm, Tocris Bioscience) and U73343 (1 μm, Tocris Bioscience), the inactive analog of U73122, were dissolved in DMSO at 1000 times final concentration just before use, then diluted in pipette solution at final concentration. 5-HT (30 μm, Tocris Bioscience) was dissolved in ultrapure water at stock concentration and then dissolved in ACSF at final concentration the day of the experiment. Xestospongin C (XeC; 1 μm, Tocris Bioscience) and 2-aminoethoxydiphenylborane (2-APB; 50 μm, Tocris Bioscience) were dissolved in DMSO at 1000 and 2000 times final concentration, then diluted in pipette solution at final concentration, respectively. Ruthenium Red (RuR, 100 μm, Sigma) was dissolved in ultrapure water at 10 mm and then dissolved in pipette solution at final concentration the day of the experiment. Voltage-dependent potassium channel blocker 4-aminopyridine (4-AP; 5 mm, 0.5 mm, Sigma) was directly dissolved in ACSF at final concentration.
Whole-cell electrophysiological recording, imaging, IP3, and Ca2+ uncaging.
Whole-cell recordings (seals > 3 GΩ) with simultaneous Ca2+ imaging were obtained from neurons in the DR and other structures (tuberomammilary nucleus [TMN] or laterodorsal tegmental nucleus [LDT]) as previously described (Kohlmeier et al., 2008). Briefly, borosilicate micropipettes (2–4 MΩ; catalog #8050, AM Systems) were used and neurons were visualized for whole-cell recordings at 160 × magnification using visible or near IR light, differential interference contrast optics, and a Nuvicon tube camera (Dage VE-1000) mounted on a fixed-stage microscope (Olympus, BX50WI).
Our normal pipette solution contained the following (in mm): 144 K-gluconate, 3 MgCl2, 10 HEPES, 0.3 NaGTP, and 4 Na2ATP (310 mOsm), and Bis-fura-2 (50 μm; Invitrogen) was added for calcium-imaging experiments. For strong and fast calcium buffering, the pipette solution contained 104 K-gluconate, 3 MgCl2, 10 HEPES, 0.3 NaGTP, 4 Na2ATP, and 10 K4-BAPTA. Biotinylated Alexa-594 (25 μm; Invitrogen) was included in all experiments for cell identification.
To isolate and measure whole-cell Ca2+ currents, the pipette solution contained the following (in mm): 144 Cs-methanesulfonate, 3 MgCl2, 10 HEPES, 0.3 NaGTP, and 4 Na2ATP, pH adjusted to 7.3 with CsOH, and the ACSF contained the following (in mm): 124 NMDG, 5 KCl, 1.25 NaH2PO4, 0.25 CaCl2, 3.65 MgSO4, 26 NaHCO3, and 10 dextrose (295–305 mOsm).
To isolate and measure whole-cell K+ currents, the normal pipette solution was used and the ACSF contained the following (in mm): 125.25 NaCl, 5 KCl, 2.7 CoCl2, 1.2 MgCl2, 26 NaHCO3, and 10 dextrose. Cobalt was added to the ACSF just before each experiment.
Gigaseals were obtained using an Axopatch 200B amplifier (Molecular Devices) operated in voltage-clamp mode, filtered at 2 or 5 kHz, and sampled at 20 kHz. Imaging experiments were conducted using a 40× water-immersion objective (0.8 NA) with a MicroMax camera (Roper Scientific) equipped with an EEV 57 frame-transfer chip (field size = 160 μm/side). Bis-fura-2 was excited at 380 nm using a shuttered 75 W xenon lamp.
Whole-cell recordings were either conducted in voltage-clamp or “I-clamp fast” mode, following pipette capacitance compensation; the quality of the recording was assayed by monitoring holding current, access resistance and input resistance as determined by the voltage or current response to a brief, negative going step. These parameters were monitored throughout the recording, and data were discarded from analysis if access resistance became unstable or changed by ∼>20% between measurements. Recordings were also terminated if cell parameters became unstable. In most experiments, current and voltage traces were digitized and command pulses were generated with custom software (TIWB) (Inoue et al., 1998) run on a Mac OS computer controlling an ITC-18 interface (Instrutech-HEKA) that ensured precise synchronization between electrophysiological and optical signals.
Images were binned on the chip at 4 × 4 and read out through a 1 MHz, 14 bit A/D converter. Images were acquired every 50 ms, a rate fast enough to monitor changes in [Ca2+]i accompanying rapid alterations of the membrane potential. Changes in fluorescence (dF/F) were quantified from the average pixel values of regions of interest (ROIs) placed on the fluorescence images. dF was calculated as the difference between the background-subtracted ROI value for each frame minus the background-subtracted baseline fluorescence. The background was taken from the value of an ROI positioned remotely from the filled cell. Baseline fluorescence was the average ROI values measured over the first few frames of each sequence before stimulation. dF was then divided by the background-subtracted baseline fluorescence to compute dF/F. Because Bis-Fura-2 fluorescence resulting from excitation at 380 nm decreases with calcium binding, dF/F responses have been inverted for clarity.
Postspike AHPs were evoked by firing single action potentials with brief (2–3 ms) current pulses adjusted to be suprathreshold, or by a 5 spike protocol consisting of a train of 5 suprathreshold current pulses (2–3 ms each at 20 Hz). To compare these AHPs, baseline membrane potential was adjusted to identical values of −65 or −70 mV by direct current injection.
AHP currents were evoked with a 5 pulse protocol consisting of a train of 5 depolarizing voltage steps to −5 mV (10 ms duration, 20 Hz) from a holding potential of −65 mV and delivered every ∼30 s. To compute the AHP current altered by orexin, leak-corrected membrane current evoked by the 5 pulse protocol was subtracted from the leak-corrected current evoked by the 5 pulse protocol during orexin action. Because AHP currents were measured at the holding potential, leak correction was accomplished by subtracting the average holding current just before delivering each 5 pulse protocol.
Isolated Ca2+ and K+ currents were evoked and recorded using PCLAMP 8 software (Molecular Devices, RRID: SCR_011323) running on a PC using a Digidata 1322 (Molecular Devices). Voltage and current traces were low pass filtered at 2 kHz and sampled at 20 kHz. Leakage currents and uncompensated capacitive components were subtracted using a −P/4 method, implemented in Clampex 8, and both the subtracted and raw current traces were acquired.
Isolated Ca2+ currents were monitored before and after orexin application by 30 ms step depolarizations to −25 mV from a holding potential of −65 mV, delivered every 30 s, and confirmed by blockade with extracellular Co2+ (2.7 mm). To measure isolated K+ currents, three sets of voltage-clamp experiments were performed before and after orexin application. From a holding potential of −65 mV, delayed rectifier current was elicited by a series of activation pulses from −95 mV to −5 mV in 10 mV increments following a 300 ms inactivation pulse to −45 mV. To elicit a transient outward current, the same protocol was repeated with a 150 ms deinactivating prepulse to −105 mV preceding the activation steps. The transient currents were then computed by subtracting the first series of currents from the second series of current. A third series of pulses to measure the voltage dependence of inactivation was delivered from a holding potential of −65 mV. An initial 150 ms inactivating pulse to −45 mV was followed by a 150 ms prepulse that varied from −105 mV to −35 mV in 10 mV increments and was followed by a 150 ms test pulse to −15 mV. The transient current was computed by subtracting the delayed rectifier current elicited by a test pulse preceded by a prepulse to −35 mV. For both Ca2+- and K+-current recordings, uncompensated series resistance was between 4 and 11 MΩ and was electronically corrected by 40%–75%.
Repetitive firing was elicited with constant current steps of 5 s duration and amplitudes from 50 to 300 pA, delivered once every 20 s. The ability to fire spikes in response to different frequency trains of stimuli was assessed with 10 s duration trains of brief (5 ms) current pulses delivered at 0.5–20 Hz and adjusted in strength (∼250 pA) to produce a spike by 100% of the pulses delivered at 5 Hz in control conditions.
To simulate the effect of the noisy orexin current on firing, two approaches were used. In the first approach, a previously recorded inward orexin current (holding potential of −65 mV) was scaled and used as the command current in current-clamp recording experiments. In the second approach, a noisy orexin conductance was added by dynamic clamp, which was implemented using QuB software as modified by Dr. Lorin S. Milescu (Milescu et al., 2008), running on a PC (ASL, Marquis M517-T) controlling a National Instruments multifunction PCIe card (NI PCIe-6251). The virtual orexin conductance was derived from a typical orexin current recorded at −65 mV from a DR neuron having an average conductance of 0.5 nS and an estimated reversal potential of −15 mV. The normalized conductance fluctuations were computed from the current fluctuations divided by the driving force and were well fit with a Gaussian function having a mean of 1.0 and a SD of 0.183. The virtual orexin current injected by the dynamic clamp was determined by the equation Iorx = Gave × Gfluct × (Vm − Vrev), where Gave ranged between 0.5 and 2 nS, Gfluct is the normalized conductance wave, Vm is the membrane potential, and Vrev is the orexin reversal potential (−15 mV).
To uncage IP3 inside recorded neurons, D-myo-inositol 1,4,5-trisphosphate, P4(5)-(1-(2-nitrophenyl)ethyl) ester, tris(triethylammonium) salt (NPE-Caged Ins 1,4,5-P3; Invitrogen) was dissolved in normal patch pipette solution to a final concentration of 100 μm. To uncage Ca2+ inside recorded neurons, 2 mm DMNP-EDTA (1-(4,5-dimethoxy-2-nitrophenyl)-1,2-diaminoethane-N,N,N′,N′-tetraacetic acid, ThermoFisher, D6814; DMNP) with 0.75 mm CaCl2 was dissolved in normal patch solution containing 100 μm Oregon Green BAPTA 2. Using Patcher's Power Tools (version 2.19) XOP (http://www3.mpibpc.mpg.de/groups/neher/index.php?page=aboutppt) for Igor Pro 6 (Wavemetrics, RRID:SCR_000325), we estimated a resting free Ca2+ concentration of ∼70 nm. Both solutions were prepared in low light, and patching was done using IR illumination. Neurons were filled for >5 min before experiments began. For caged IP3, photolysis was accomplished using flashes of 380 nm light provided by our xenon arc lamp with exposure time controlled by the shutter system. For caged Ca2+, photolysis was accomplished using a Prizmatix Mic-LED emitting at 385 nm (15 nm FWHM) with exposures determined by computer-generated TTL pulses gating the controller. To minimize uncaging within the pipette, the neuron was positioned so that the pipette was mostly occluded by the edge of the aperture in the light path. DMNP uncaging pulses were delivered at intervals of >2 min and were interleaved with three 5 pulse protocols.
Data analyses.
Data were analyzed and figures prepared using Igor Pro 6. All reported values of Vm have been corrected by −15 mV to compensate for liquid junction potentials, which were measured for each patch solution and ACSF combination (range: −14.7 to −16.4 mV). Action potential and AHP amplitudes were measured from the baseline membrane potential. Rinput was measured in current clamp with −10 or −20 pA pulses or in voltage clamp with −10 mV pulses. AHP duration was measured as time to 50% recovery (T50). Spike-frequency adaptation (SFA) was determined from the initial (first interspike interval) and steady-state firing frequency (last 3–5 intervals) produced by 5 s current pulses. Curve fitting was done using Igor Pro. Single and double exponentials were fit to average currents to estimate time constants (tau). A Boltzmann function (1/(1 + exp (V1/2 − Vm/k))) was fit to the average G/Gmax and I/Imax curves measured for the transient outward current to determine V1/2 and the slope factor, k. All fit coefficients are reported as the coefficient ± the 95% confidence interval. Other results are reported as mean ± SEM. Groups were compared using two-tailed t tests, ANOVAs, or repeated-measures ANOVAs (Data Desk 6 and 7, Data Description). Results from these tests are reported as the value of the t statistic or the F ratio with corresponding degrees of freedom in parentheses. Following a significant ANOVA, group differences were compared using Fisher's least significant difference post hoc tests. Significance was set at p < 0.05. To minimize variability from possible drifts in experimental conditions (e.g., different lots of orexin), treatment and control responses were obtained from interleaved recordings.
Immunofluorescence.
To identify serotonergic neurons, slices were immersion fixed (4% PFA) overnight and cryoprotected (30% sucrose in PBS). Slices were then resectioned (40 μm) on a freezing microtome, and free-floating sections were immunolabeled with antibodies against tryptophan hydroxylase (TpH; Abcam, 3907, RRID:AB_304147; and Covance PSH-327P, RRID:AB_10063449, sheep polyclonals, 1:400). Serotonergic neurons were identified by immunofluorescence using an Alexa-488-conjugated secondary antibody (Invitrogen, A11015, donkey anti-sheep).
Results
Large neurons (long axis > ∼25 μm) in the dorsal and ventromedial portions of the DR nucleus were chosen for recording (n = 292) and had an average input resistance of 670.2 ± 38.9 MΩ (n = 44), which is comparable with previous measurements (e.g., Rood et al., 2014).
Orexin enhances the late AHP
Orexin-A (300 nm) application produced membrane depolarization, a clearly discernible increase in membrane potential noise and firing. To examine the AHP, we repolarized the neurons (Fig. 1A, −Iinj) to baseline with DC current and then compared the AHP produced by the 5 spike protocol (Fig. 1A, right blue box) to that evoked before orexin-A (Fig. 1A, left, red box). Superimposing the traces (Fig. 1A, right) revealed that, following orexin application, the peak of the AHP was larger (control: 6.6 ± 0.5 mV; orexin: 12.7 ± 1.4 mV, n = 21; paired t test: t(20) = 4.76, p < 0.05), later, and slower to recover (control T50: 0.62 ± 0.06 s; orexin T50: 2.2 ± 0.24 s, n = 21; paired t test: t(20) = −7.16, p < 0.05). Of the 22 DR neurons studied in this way, one did not show this AHP effect. Thirteen neurons that showed this oeAHP were recovered after recording, and 12 of 13 were TpH-immunopositive (Fig. 1B). Moreover, an oeAHP was observed in all cells for which orexin produced a depolarization. It was activated over the same orexin dose range (Fig. 1C) and developed over the same time course (Fig. 1D) as the depolarization.
Figure 1.

Orexin-A enhances a late AHP in serotonergic DR neurons. A, Orexin-A (Orx A) depolarized DR neurons and greatly enhanced the AHP resulting from 5 spikes delivered at 20 Hz (Control: red dashed box left; Orexin, Orx A: blue dashed box right, following return of Vm to baseline with direct current, −Iinj). AHPs are expanded and superimposed on the right. B, Cell recorded in A, visualized with Alexa-594 (Alexa), was immunopositive for TpH. Scale bar, 30 μm. C, Orexin-enhanced AHP (mean ± SEM time to 50% recovery, 50% Rec, filled triangles) and orexin-mediated depolarization (mean ± SEM current required to keep Vm at baseline, Iinj, filled circles) versus Orexin-A (Orx A) concentration. D, Average time course of Orx A actions on DR neurons (n = 7). The mean ± SEM AHP parameters time to AHP peak (Peak), AHP amplitude (Amp), and duration of the AHP to 50% recovery (50% Rec) are shown with changes in input resistance (Rin) and the current injected to keep membrane potential at baseline (Iinj). E, An average of three 5 spike-evoked AHPs before (Control, red) and after (Orx A, blue) orexin-A (300 nm) in ACSF containing prazosin (100 nm). F, The 5 spike-evoked AHP before (Control, red), during PE (3 μm, green) and during PE + orexin-A (300 nm, PE + Orx A, blue). Scale bars in E apply to F.
Previous studies showed that adrenergic α-1 receptor activation increases the AHP in DR neurons (Freedman and Aghajanian, 1987; Pan et al., 1994); and because orexins can release norepinephrine (Hirota et al., 2001), we tested whether the oeAHP might result from indirect activation of α-1 receptors. We confirmed that the α-1 agonist PE (300 nm to 30 μm) depolarized and enhanced the AHP in mouse DR neurons (3 μm PE, AHP amplitude: 139.0 ± 17.0% of control; AHP duration: 315.4 ± 63.9% of control, n = 5) but found that prasozin (100 nm), which completely blocked the PE effects, failed to block the oeAHP (n = 11; Fig. 1E). Nevertheless, PE (3 μm) enhancement of the AHP was sufficient to occlude any further enhancement by orexin-A (300 nm; AHP amplitude in orexin: 102.1 ± 3.8% of AHP in PE, paired t test: t(4) = 0.54, p > 0.05; AHP duration in orexin: 94.4 ± 9.3% of AHP in PE; paired t test: t(4) = −1.57, p > 0.05, n = 5; Figure 1F).
Because there are sex differences in 5-HT DR neurons and baseline firing rates are higher in male rats (Klink et al., 2002), we examined whether we could detect a sex difference in the late AHP or in the oeAHP. The control AHPs evoked with the 5 spike protocol were not different in either amplitude (male: 7.22 ± 0.79 mV, n = 11; female: 5.66 ± 0.75 mV, n = 7; unpaired t test: t(16) = −1.35, p = 0.19) or in T50 (male: 0.56 ± 0.06 s, n = 11; female: 0.68 ± 0.15 s, n = 7; unpaired t test: t(16) = −0.83, p = 0.42). Moreover, orexin-A (300 nm) strongly increased the amplitude and duration of the late AHP in slices from both sexes, with a trend toward larger increases in slices from females. Nevertheless, no statistically significant difference between males and females was found in either the amplitude increase (male: 155.5 ± 19.4%, n = 11; female: 222.8 ± 39.2%, n = 7; unpaired t test: t(16) = 0.17, p > 0.05) or duration increase (male: 385.0 ± 69.1%, n = 11; female: 459.0 ± 90.9%, n = 7; unpaired t test: t(16) = −0.66, p > 0.05).
Either OxR alone can mediate the oeAHP in DR neurons
Because mRNA for both OX1 and OX2 is expressed in the DR (Trivedi et al., 1998; Marcus et al., 2001), and either receptor is sufficient to produce the orexin-mediated inward current (Kohlmeier et al., 2013), we tested whether each receptor was competent to produce the oeAHP using slices from mice lacking one receptor. Application of 300 nm orexin-A evoked a depolarization and an unambiguous oeAHP in DR neurons recorded in slices from OX1−/− (Fig. 2A) and OX2−/− (Fig. 2B) mice. Moreover, neither the increase in AHP amplitude (OX1−/−: 165.6 ± 23.4%, n = 5; OX2−/−: 152.1 ± 24.2%, n = 5) nor increase in AHP duration (OX1−/−: 345.1 ± 98.6%, n = 5; OX2−/−: 322.8 ± 63.2%, n = 5) was different from those recorded in control DR neurons (amplitude: 194.2 ± 18.4%, n = 21; duration: 416.1 ± 49.9%, n = 21) in slices from C57BL6 mice (Fig. 2C; Amplitude, ANOVA: F(2,28) = 0.76, p > 0.05; Duration, ANOVA: F(2,28) = 0.50, p > 0.05). To confirm that these OxRs were, indeed, necessary, we also recorded DR neurons in slices from DKO mice (n = 5 from 3 DKO mice). Orexin-A (300 nm) produced neither a depolarization nor a change in the AHP (Fig. 2D, top traces; amplitude: 97.9 ± 5.6% of control, n = 5; duration: 99.66 ± 2.6% of control, n = 5), although application of PE still produced an increase in amplitude and duration of the AHP in these neurons (Fig. 2D, bottom traces). Thus, both OX1 and OX2 are competent to mediate the oeAHP in DR neurons.
Figure 2.
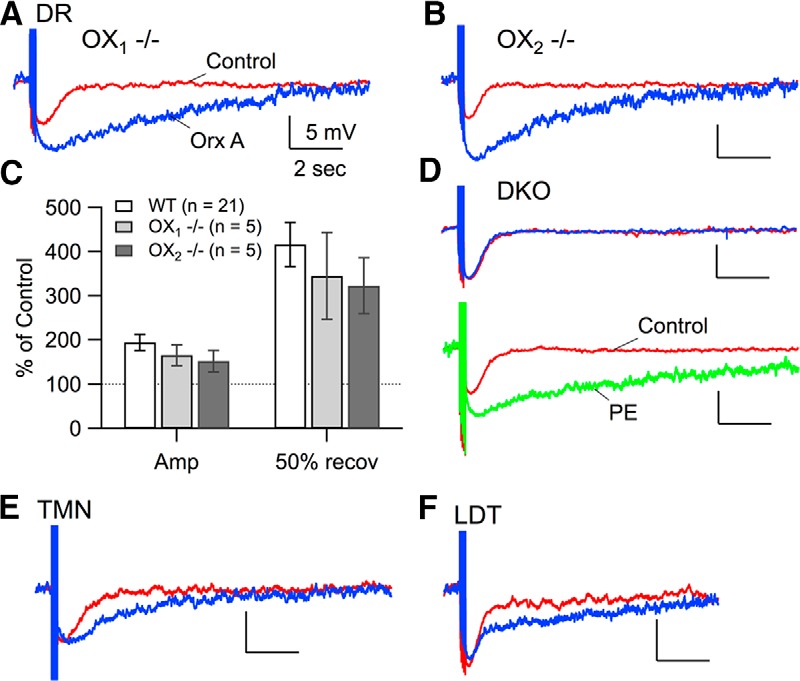
Activation of either OxR alone produces the oeAHP. A, B, An average of three 5 spike-evoked AHPs before (Control, red) and after orexin-A (300 nm; Orx A, blue) in DR neurons recorded in slices from OX1 null mice (A) and OX2 null mice (B). C, The effects of orexin-A (300 nm; mean percentage of Control ± SEM) on the amplitude (Amp) and duration (50% recov) of the 5 spike-evoked AHP. D, An average of three 5 spike-evoked AHPs before (red) and after orexin-A (300 nm; blue) in a DR neuron from a DKO mouse (top traces). An average of three 5 spike-evoked AHPs before (Control, red) and after PE (10 μm; lower traces, PE, green) in a DR neuron from a DKO mouse. E, F, An average of three 5 spike-evoked AHPs before (red) and after orexin-A (300 nm; blue) in a TMN neuron (E) and a LDT neuron (F). All scale bars correspond to those in A.
This suggests that orexin could produce an oeAHP in neurons normally expressing predominantly OX1 or OX2 receptors. To test this idea, and to determine whether neurons other than DR neurons expressed this type of orexin action, we examined the effect of orexin-A on the late AHP of two other arousal-linked targets: the TMN, where the orexin-mediated depolarization is primarily mediated by OX2 (Eriksson et al., 2001; Willie et al., 2003); and the LDT, where the orexin-mediated depolarization is primarily mediated by OX1 (Kohlmeier et al., 2013). We found orexin-A (300 nm) enhanced the AHP duration evoked with the 5 spike protocol in TMN neurons (control: 0.6 ± 0.18 s; orexin: 1.05 ± 0.11 s, n = 3 of 3; paired t test: t(2) = −3.03, p < 0.05; Fig. 2E) and in LDT neurons (control: 0.56 ± 0.12 s; orexin: 2.06 ± 0.83 s, n = 7/11; paired t test: t(6) = 1.14, p < 0.05; Fig. 2F). However, unlike in the DR, the average amplitude of the late AHP was not increased in TMN neurons (control: 5.0 ± 0.8 mV; orexin: 5.0 ± 1.0 mV, n = 3 of 3; paired t test: t(2) = −0.04, p > 0.05) or LDT neurons (control: 6.8 ± 1.4 mV; orexin: 9.0 ± 1.8 mV, n = 7 of 11; paired t test: t(6) = −1.99, p > 0.05), suggesting that different mechanisms may be involved. Nevertheless, this indicates that the AHP enhancing action of orexin is not restricted to DR neurons and can be observed in neuronal populations naturally expressing a predominance of either receptor.
The AHP enhanced by orexin requires elevation of [Ca2+]i
Because the postspike AHP in DR neurons is Ca2+-dependent, and orexin enhances somatic Ca2+ transients produced by long depolarizing pulses in DR neurons (Kohlmeier et al., 2008), we examined whether the oeAHP resulted from enhanced spike-evoked Ca2+ transients. Somatic Ca2+ transients resulting from single spikes (Fig. 3A, top traces) or the 5 spike protocol (Fig. 3A, bottom traces) were readily detected and orexin enhanced the AHP following both stimuli. However, orexin did not increase the average somatic Ca2+ transient produced by single spikes (orexin: 92.6 ± 3.1% of control, n = 7) or by 5 spikes (orexin: 96.7 ± 10.0% of control, n = 20; Fig. 3B).
Figure 3.
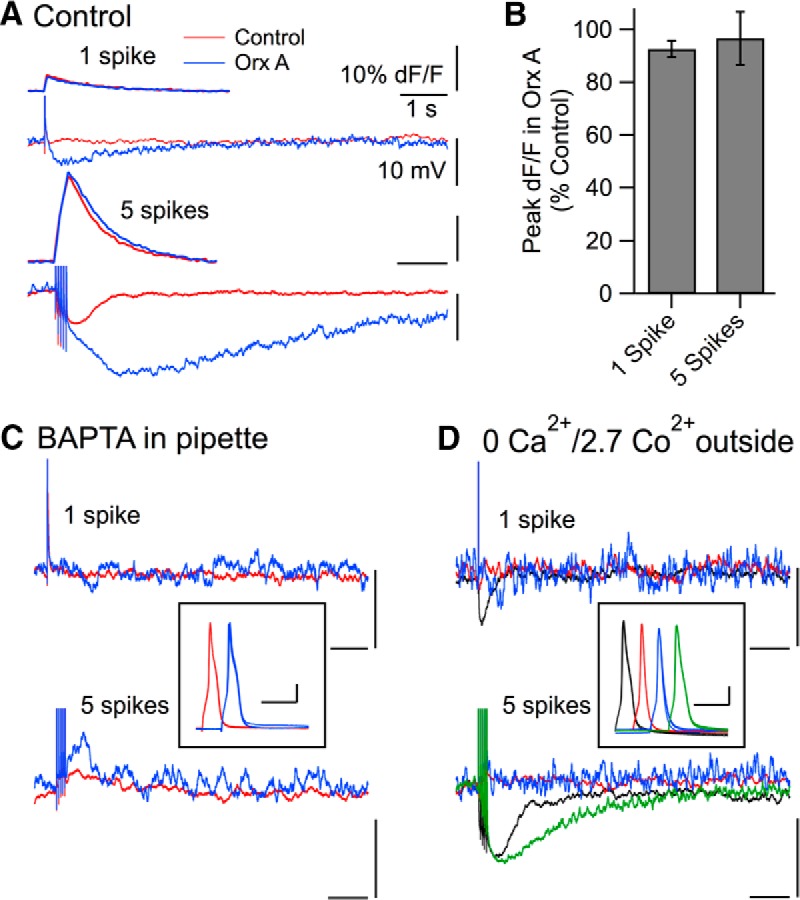
The oeAHP requires Ca2+ influx and the elevation of [Ca2+]i but is not associated with increased somatic Bis-fura-2 transients. A, Ca2+-dependent fluorescence changes from an ROI encompassing the soma (top traces, dF/F) and membrane potential (bottom traces, mV) were measured concurrently while 1 or 5 spikes were evoked before (red, Control) and after orexin-A (blue, Orx A, 300 nm) application. B, Summary (mean ± SEM) of peak somatic Ca2+ transients (dF/F) produced by 1 and 5 spikes as a percentage of control following orexin-A treatment. C, Membrane potential following 1 and 5 spikes before (red) and after (blue) orexin-A (300 nm) recorded with normal ACSF and 10 mm BAPTA in the pipette (average of 3 trials). Boxed inset, Three superimposed spikes in each condition staggered in time for comparison. Calibration: Inset, 10 ms, 20 mV. D, Membrane potential following 1 and 5 spikes recorded in control ACSF (black), in ACSF containing no added Ca2+, and 2.7 mm Co2+ (0 Ca2+/2.7 Co2+; red), following orexin-A (300 nm) in this ACSF (blue) and after reintroduction of control ACSF (green; 5 spikes only). Inset, Three superimposed spikes in each condition staggered in time for comparison. Calibration: Inset, 10 ms, 20 mV. All calibration, except for insets, as in A.
To determine whether elevation of intracellular [Ca2+] was necessary for the oeAHP, we recorded with a patch solution containing the fast calcium buffer BAPTA (10 mm). Under these conditions, the late AHP following 1 and 5 spikes was undetectable (Fig. 3C), even though recording conditions remained excellent, as indicated by normal input resistance and action potential amplitude (Fig. 3C, inset). Moreover, despite an intact orexin-evoked depolarization and large increase in membrane noise (data not shown), no enhanced AHP following 1 or 5 spikes emerged following the application of orexin. This indicates that the AHP enhanced by orexin requires elevation of intracellular Ca2+ and did not result from a Ca2+-independent current that was switched on by OxR activation.
Using a normal internal solution, we next applied ACSF having Co2+ substituted for Ca2+ to block voltage-gated Ca2+ channels and thus determine whether Ca2+ influx was necessary for the oeAHP. Switching into this ACSF completely blocked the late AHP following either 1 or 5 spikes (Fig. 3D). Again, despite an intact orexin-evoked depolarization and a large increase in membrane noise, neither 1 nor 5 spikes elicited an oeAHP following orexin application (n = 5), even though recording conditions remained excellent and action potentials retained their normal amplitude (Fig. 3D, inset). Moreover, enabling Ca2+ influx by reintroducing normal Ca2+ ACSF greatly prolonged the late AHP compared with control, even though the orexin action was subsiding, as indicated by a recovering inward current. Thus, the oeAHP requires Ca2+ influx and elevation of intracellular Ca2+.
Orexin enhances an apamin-sensitive K+ current and a slower apamin-insensitive current that is insensitive to UCL2077
We next conducted voltage-clamp recordings to measure the oeAHP current (IoeAHP). Following spike blockade with TTX, AHP currents were evoked using the 5 pulse protocol delivered every 30 s before and after orexin-A application (300 nm; Fig. 4A). As the noisy inward orexin current developed, a prolonged AHP current emerged (Fig. 4B; compare top traces). Subtracting these currents, following leak-correction, yielded the IoeAHP (Fig. 4B, bottom trace), which decays more slowly than the AHP current in control condition and underlies the oeAHP observed in current-clamp experiments.
Figure 4.
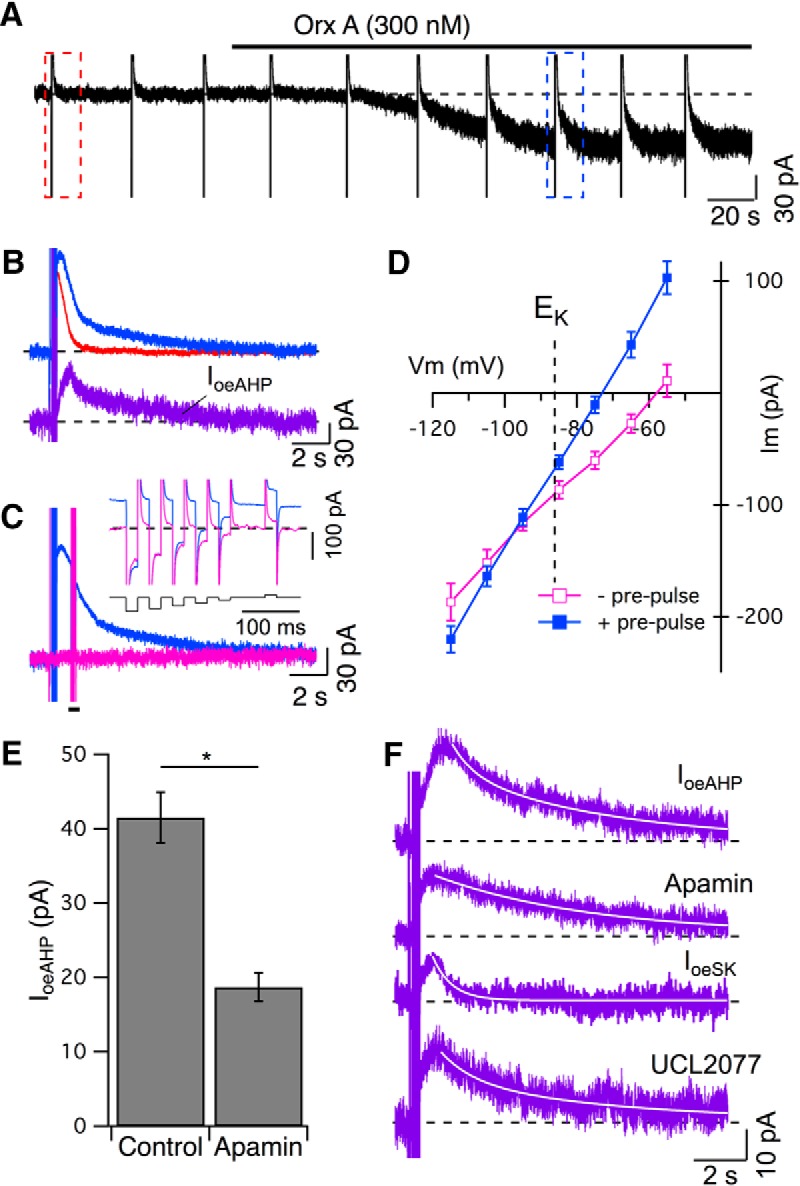
The oeAHP current (IoeAHP) reverses near the potassium equilibrium potential (EK) and has a biphasic decay mediated by a medium duration SK current and a slower apamin-insensitive current. A, Continuous recording of membrane current in voltage-clamp mode (holding potential −65 mV) before and during application of orexin-A (Orx A, 300 nm). Intermittent 5 pulse protocols (delivered ∼30 s intervals) evoked AHP currents before (left, red dashed box) and after Orx A (right, blue dashed box). B, Top traces, Leak-corrected currents from A before (red) and after (blue) Orx A (3 traces averaged). Bottom trace (purple), IoeAHP, computed as the difference between the top traces. C, Membrane current in orexin (magenta) is superimposed on the membrane current evoked by the 5 pulse protocol (blue) in orexin. Brief (20 ms) voltage pulses (−115 mV to −55 mV in 10 mV increments, bottom black trace, inset) were delivered during both traces at a time corresponding to the peak of the oeAHP (bar). The current at the end of each brief step (top traces, inset) was used to estimate the instantaneous I–V relation. D, Resulting instantaneous current (mean ± SEM, Im)–voltage (Vm) relations for traces without the 5 pulse protocol (− prepulses, open squares, magenta) and traces with the 5 pulse protocol (+ prepulses, filled squares, blue) cross near the calculated potassium reversal potential (EK; n = 7). E, Apamin (100, 300 nm) reduced the amplitude (mean ± SEM) of IoeAHP (n = 23). *p < 0.05. F, The average IoeAHP (purple) under control conditions (n = 7; top) had a biphasic decay that was well fit with the sum of a medium and slow-decaying exponential (superimposed white trace). The average IoeAHP in apamin (n = 7; second trace) decayed with a single slow exponential (superimposed white trace). The average orexin-enhanced SK current (IoeSK, n = 5; third trace) was well fit with a medium-decaying single exponential (superimposed white trace). The average IoeAHP in UCL2077 (n = 4; fourth trace) also had a biphasic decay that was well fit with the sum of a medium and slow-decaying exponential (superimposed white trace).
To estimate the reversal potential of this current, we measured the instantaneous current-voltage curve resulting from brief voltage steps timed to occur at the peak of IoeAHP, and delivered both with and without a preceding 5 pulse protocol (Fig. 4C, inset). These curves crossed between −95 and −100 mV (Fig. 4D; n = 7) near the computed EK (−86 mV, dashed line). Shifting EK by ∼18 mV with ACSF containing either 2.5 or 10 mm K+ shifted this estimated reversal potential by −24 mV (n = 4) and 19 mV (n = 5), respectively (data not shown), indicating K+ channel involvement in IoeAHP.
The Ca2+ dependence and shift in reversal with EK suggest involvement of Ca2+-activated K+ channels. Two slow Ca2+-dependent K+ currents with different kinetics commonly mediate a medium (ImAHP) and slow AHP (sAHP, IsAHP) in neurons. Channels mediating ImAHP are mostly blocked by apamin, a selective blocker of channels containing SK1–3 subunits (for review, see Stocker et al., 2004). In contrast, the IsAHP is strongly inhibited by some neurotransmitters, but the underlying channels are generally unidentified, often insensitive to channel blockers and may be different in different neurons (Vogalis et al., 2003). To test for SK channel involvement, we applied apamin, which blocks most of the postspike AHP in control condition and the AHP enhancement produced by PE (Freedman and Aghajanian, 1987; Pan et al., 1994). Following pretreatment with apamin (100 or 300 nm), our control AHP was blocked by ∼90% (100 nm: 88% blocked, n = 11, paired t test: t(10) = 11.98, p < 0.05; 300 nm: 91% blocked, n = 12, paired t test: t(11) = 6.34, p < 0.05), yet after orexin, IoeAHP amplitude (18.7 ± 1.9 pA, n = 23) was only reduced by approximately half (45.0% of control amplitude: 41.5 ± 3.4 pA; n = 11; unpaired t test: t(32) = −6.34, p < 0.05; Fig. 4E). This indicates that both an apamin-sensitive SK current (ISK) and an apamin-insensitive current are involved. Involvement of two currents was also suggested by the often-observed biphasic decay kinetics of IoeAHP. To estimate the time course of each component, we averaged IoeAHP from control (n = 7) and apamin pretreated (n = 7) recordings. In the control average, the decay was well fit by a medium (tau = 0.8 ± 0.08 s) and slow exponential (tau = 6.0 ± 0.02 s; Fig. 4F, top). In the apamin average, the decay was well fit by a single slow exponential (tau = 5.9 ± 0.01 s; Fig. 4F, second trace). This suggests that the oeAHP results from both an increase in the medium-duration ISK present before orexin application and the emergence of a longer-duration apamin-insensitive current that was absent before orexin application. We confirmed the presence of this enhanced ISK by applying apamin (300 nm) just after emergence of the orexin effect and subtracting the apamin-insensitive component from the initial orexin-enhanced current, to yield the ISK in orexin. From this, we then subtracted the SK current before orexin, to yield the orexin-enhanced SK current (IoeSK). The averaged IoeSK (n = 6) was well fit with a single exponential having a medium decay (tau = 0.71 ± 0.01 s; Fig. 4F, third trace).
The apamin-insensitive part might be mediated by an IsAHP, which is found in many types of neurons. The antiepileptic compound UCL2077, which blocks KCNQ1/2 channels (Soh and Tzingounis, 2010), has recently been found to inhibit the classical IsAHP in hippocampal pyramidal neurons (Shah et al., 2006) and in paraventricular thalamic (PVT) neurons, where orexin inhibits IsAHP (Zhang et al., 2010). We therefore tested whether UCL2077 blocks the oeAHP current in DR neurons. Following preapplication of UCL2077 (10 μm, >7 min), the oeAHP current was still biphasic (medium tau = 1.2 ± 0.03 s; slow tau = 6.5 ± 0.08 s; Fig. 4F, fourth trace) and had an average amplitude of 29.4 ± 6.4 pA (n = 4), which was not significantly different from that in control ACSF (p > 0.05), indicating that the slow part of the oeAHP is resistant to UCL2077 and is mediated by different channels from those mediating the sAHP in hippocampus and PVT neurons.
PLC antagonism blocks the entire IoeAHP and the orexin-mediated inward current, but these currents do not require PKC activation and are insensitive to altered cAMP/PKA signaling
Both OX1 and OX2 can activate PLC, although it has become apparent that these receptors can also couple to other second-messenger systems (Kukkonen and Leonard, 2014). We therefore examined whether intracellular application of U73122, an inhibitor of PLC, could inhibit the oeAHP. As control, we first evaluated the inactive analog U73343 (1 μm) on both the orexin currents and the outward current activated by 5-HT because the latter current does not depend on PLC activation (Innis et al., 1988; Williams et al., 1988; Penington et al., 1993). Under these conditions, orexin and 5-HT produced their expected actions (n = 5; Fig. 5A), whereas the active analog U73122 (n = 6) blocked both the orexin-mediated inward current and the entire IoeAHP,without reducing the 5-HT current (Fig. 5B). Results from ANOVAs (inward current: F(2,19) = 6.61, p < 0.05; IoeAHP: F(2,19) = 5.70, p < 0.05) followed by post hoc testing confirmed that the orexin action was not different from control in recordings with the inactive analog but was blocked in recordings with the active analog (post hoc tests, p < 0.05; Fig. 5C). In contrast, the active analog did not attenuate the outward current evoked by 5-HT (unpaired t test, t(9) = 0.90, p > 0.05, compared with inactive analog; Fig. 5D). Thus, the PLC inhibitor U73122 selectively blocked both the orexin-mediated inward current and the IoeAHP.
Figure 5.

PLC inhibitor U73122 blocked both the orexin-induced inward current and IoeAHP, although both currents were resistant to PKC inhibitors and forskolin despite being abolished by PDBu. A1, Continuous membrane current with intermittent 5 pulse protocols (holding potential = −65 mV) recorded with U73343 (1 μm, inactive analog of U73122) in the pipette before and after application of Orx A (300 nm; left) or serotonin (5-HT; 30 μm; right). A2, IoeAHP (purple) computed from recordings in A1. B1, Membrane current with intermittent 5 pulse protocols (holding potential = −65 mV) recorded with U73122 (1 μm, selective PLC inhibitor) in the pipette before and after application of Orx A (300 nm; left) or serotonin (5-HT; 30 μm; right). B2, IoeAHP (purple) computed from recordings in B1 was completely abolished. C, Summary (mean ± SEM) of currents produced by orexin recorded with either U73343 (n = 5) or U73122 (n = 6) in the pipette. U73122 strongly attenuated the inward current and IoeAHP. *p < 0.05. D, Summary (mean ± SEM) of outward current evoked by 5-HT in recordings with U73343 (n = 5) and U73122 (n = 6). ns, Not significant. E–I, Examples of IoeAHP computed for orexin application (300 nm) recorded under different conditions, including control (E), with the PKCIP in the pipette solution (F), following slice preincubation with the PKC inhibitor Bis I (G), following application of the PKC activator PDBu (H), and following preincubation with the adenylyl cyclase activator forskolin (Forsk) (I). J, Summary of drug actions on the amplitude of IoeAHP (mean ± SEM). **p < 0.05, significant comparisons from post hoc Fischer tests following a significant ANOVA, revealing differences between control and PKC drug groups.
Neurotransmitter inhibition of the classical IsAHP is mediated by both cAMP/PKA signaling (Pedarzani and Storm, 1993) and activation of PLC (Villalobos et al., 2011), with involvement of PKC in some cases (Malenka et al., 1986; Agopyan and Agopyan, 1991). Indeed, whereas the IsAHP in PVT neurons is inhibited by both PKA and PKC activation, orexin-mediated inhibition of the IsAHP was only blocked by PKC inhibitors (Zhang et al., 2010). We therefore tested whether the oeAHP might also involve these signaling pathways. In the first series of experiments, we tested whether PKCIP (2 μm in the patch solution, dialyzed for >10 min) (Hall et al., 1995; Schubert et al., 1999) or the PKC inhibitor Bis I (1 μm, brain slices preincubated for >45 min) altered IoeAHP. Before orexin application, the AHP current produced by the 5 pulse protocol was 67.4 ± 14.8 pA with the PKCIP (n = 4) and 60.4 ± 9.1 pA with Bis I (n = 7). Neither of these was different from the control AHP current (87.4 ± 7.5 pA, n = 11; ANOVA: F(2,19) = 2.64, p > 0.05). Application of orexin-A (300 nm) resulted in an IoeAHP of 36.1 ± 2.5 pA with the PKCIP (Fig. 5F; n = 4) and an IoeAHP of 29.6 ± 5.6 pA with Bis I (Fig. 5G; n = 7). Neither was different from the IoeAHP measured in corresponding control experiments (41.5 ± 3.4 pA, n = 11; Fig. 5E), suggesting that PKC activation is not required for triggering the oeAHP. In contrast, the PKC activator PDBu (1 μm, n = 6) strongly reduced the IoeAHP (4.1 ± 2.0 pA; Fig. 5H) compared with control or PKC inhibitor conditions (Fig. 5J; ANOVA: F(3,24) = 9.70, p < 0.05), suggesting that PKC, when activated, can inhibit the expression of IoeAHP.
In the second series of experiments, we examined involvement of the cAMP/PKA pathway in regulating the late AHP and IoeAHP by applying forskolin (10 μm; >10 min) before orexin application. The average amplitude of the late AHP current produced by the 5 pulse protocol before forskolin application was 125.5 ± 17.1 pA, and it was unchanged by forskolin application (122.1 ± 18.5 pA, paired t test: t(4) = 0.90, p > 0.05, n = 5). Following orexin application with forskolin (300 nm, n = 5), the IoeAHP was still observed (38.6 ± 8.5 pA; Fig. 5I), and it was not different in amplitude from IoeAHP produced without forskolin (unpaired t test: t(14) = 0.39, p > 0.05; Fig. 5E). Thus, unlike neurotransmitter inhibition of the classical IsAHP, IoeAHP in DR neurons does not appear to depend on PKC activation or the cAMP/PKA system.
Ca2+ stores and Ca2+-induced Ca2+ release (CICR) regulate the SK part of the oeAHP
Because the oeAHP requires Ca2+ influx and elevation of [Ca2+]i, and an increase of the apamin-sensitive SK-current contributes to the early part of IoeAHP, we next considered Ca2+-dependent factors that might be regulated by orexin. First, native SK2 channels exist in a complex with casein kinase 2 and protein phosphatase 2A (PP2A) (Bildl et al., 2004), which reciprocally regulate the Ca2+ sensitivity and kinetics of SK2 currents (Bildl et al., 2004; Allen et al., 2007). Because SK2 deactivation kinetics are slowed by PP2A-mediated dephosphorylation, we intracellularly applied an inhibitor of PP2A (okadaic acid; 25 nm; >12 min) to determine whether this would shorten the ISK in DR neurons and perhaps prevent the early, apamin-sensitive part of the oeAHP current. Recordings with okadaic acid in the pipette revealed a baseline AHP current with a magnitude (76.4 ± 13.5 pA, n = 7), similar to that observed with normal pipette solution (87.4 ± 7.5 pA, n = 11, unpaired t test: t(16) = 0.77, p > 0.05), but with a duration (T50: 0.42 ± 0.03 s) that was shorter than that with normal pipette solution (T50: 0.66 ± 0.08 s; unpaired t test: t(16) = 2.33, p < 0.05), consistent with PP2A inhibition and greater SK phosphorylation. However, IoeAHP recorded under these conditions still showed a biphasic decay with an amplitude (48.5 ± 5.1 pA, n = 7) that was not different from control (unpaired t test: t(16) = −1.17, p > 0.05; Fig. 6A). This indicates that IoeAHP was resistant to PP2A inhibition and that prolongation of the SK current by orexin occurred independently of PP2A-mediated dephosphorylation.
Figure 6.

The SK portion of the oeAHP requires SERCA-dependent Ca2+ stores and CICR but not IP3. A, An example of IoeAHP recorded with okadaic acid (25 nm) in the pipette solution. AHP currents were evoked with a 5 pulse protocol before and after orexin-A (300 nm). B, Current-clamp recordings of the AHP evoked by the 5 spike protocol before (black), following SERCA-dependent Ca2+ store depletion with CPA (red) and during Orx A (300 nm) application in CPA (blue). C, Summary of the effect of CPA treatment on orexin-evoked changes in amplitude (Amp) and duration (50% recov) of the AHP. Data (mean ± SEM) are reported as percentage of the AHP measured in CPA before orexin application (control; dotted line = 100%). D, Summary of voltage-clamp experiments following CPA treatment. IoeAHP amplitude (mean ± SEM) was measured in CPA and CPA following blockage of SK currents with apamin (CPA w Apa). Dotted lines indicate mean ± SEM of the IoeAHP recorded under control conditions. *p < 0.05, significant difference from control. E, Examples of IoeAHP recorded with IP3R inhibitors XeC (1 μm) and 2-APB (50 μm) and the ryanodine receptor antagonist RuR (100 μm) included in the pipette solution. Calibration bars for left trace apply to the other two traces. F, Summary of IoeAHP amplitude (mean ± SEM) for recordings made with XeC, 2-APB, RuR, and RuR with apamin pretreatment. *p < 0.05, significant difference compared with Control (mean ± SEM, dotted lines) by post hoc tests following an ANOVA. G, Example membrane currents from a DR neuron (holding potential = −65 mV). The neuron was stimulated by IP3 uncaging with full-field UV flashes (500 ms; left traces) or by the 5 pulse protocol (right traces) both before (red) and after (blue) orexin-A (300 nm). Arrowhead indicates 0 pA. Calibration applies to both pairs of traces. H, Example whole-cell recording from a hippocampal CA1 neuron. Identical IP3 uncaging (UV) evoked an outward current. Calibration in G applies to H. I, Summary of evoked currents by UV uncaging of IP3 in DR neurons before (Control) and after orexin-A application (Orx A) and in hippocampal CA1 neurons (Hipp). *p < 0.05.
Because IoeAHP depends on PLC, but not on PKC activation, we next examined whether it might depend on Ca2+ release from intracellular stores, which could result from PLC-mediated production of IP3. We first applied the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) inhibitor CPA (10 μm, 10 min) to deplete intracellular Ca2+ stores in these neurons (Kohlmeier et al., 2004). In a series of current-clamp experiments, we found that CPA did not significantly change the amplitude of the SK-mediated AHP evoked by the 5 spike protocol (Pre-CPA: 7.8 ± 0.8 mV, CPA: 6.6 ± 0.4 mV, n = 5, paired t test: t(4) = −2.43, p > 0.05), but it did significantly slow its recovery (Pre-CPA T50: 0.7 ± 0.1 s, CPA T50: 1.2 ± 0.1 s, n = 5, paired t test: t(4) = −6.15, p < 0.05). This suggests that SERCA-dependent Ca2+ sequestration normally contributes to recovery of the SK-mediated AHP. Following Ca2+-store depletion with CPA, application of orexin-A (300 nm; Fig. 6B) produced a membrane depolarization that appeared normal and an oeAHP that achieved the same duration as the oeAHP without CPA (oeAHP with CPA T50: 2.2 ± 0.3 s, n = 5; oeAHP no CPA T50: 2.2 ± 0.24 s, n = 21, unpaired t test: t(24) = 0.14, p > 0.05). However in CPA, orexin-A application did not significantly increase the AHP amplitude (AHP in CPA: 6.6 ± 0.4 mV; oeAHP in CPA: 8.1 ± 1.2 mV, n = 5, paired t test: t(4) = 0.11, p > 0.05). We further tested this in series of voltage-clamp experiments where we found that the IoeAHP recorded in CPA (27.9 ± 3.4 pA, n = 7; Fig. 6D) was smaller than that in control conditions but was similar in amplitude to that in apamin alone and in CPA following preblocking SK channels with apamin (19.7 ± 2.9 pA, n = 5). A significant ANOVA (F(3,42) = 11.10, p < 0.05) followed by post hoc testing confirmed that the IoeAHP in CPA was significantly smaller than in Control (p < 0.05) but was not different from the IoeAHP recorded in apamin (p > 0.05) or in CPA with apamin (p > 0.05). These data indicate that store release is necessary for the orexin-mediated enhancement of the apamin-sensitive component, but not for the longer-lasting apamin-insensitive component of IoeAHP.
We next examined whether store involvement was mediated by IP3 receptors (IP3Rs) and/or ryanodine receptors (RyRs). Intracellular application of the IP3R antagonists XeC (1 μm) or 2-APB (50 μm) did not reduce IoeAHP (XeC: 47.0 ± 9.2 pA, n = 6; 2-APB: 52.6 ± 13.7 pA, n = 6; Fig. 6E,F). In contrast, IoeAHP was smaller in recordings obtained with the RyR antagonist RuR (100 μm) in the patch solution (RuR: 23.3 ± 2.9 pA, n = 7; Fig. 6E,F), and this was not further reduced by preblocking SK channels with apamin (21.9 ± 4.9 pA, n = 5; Fig. 6F). An ANOVA (F(4,34) = 4.45, p < 0.05) with post hoc testing of these data, following a log-transform to correct for heterogeneity of variance across recording groups, confirmed that IoeAHP amplitude was not different from control (p > 0.05) in recordings with XeC or 2-APB but was smaller than control in recordings with RuR and RuR with apamin pretreatment (p < 0.05). Moreover, when recorded with RuR, IoeAHP was not different in amplitude from that recorded in apamin alone (unpaired t test: t(28) = 1.23, p > 0.05), and the average decay was well fit by a single exponential (tau = 2.6 ± 0.01 s, n = 7). In addition, apamin pretreatment did not further reduce IoeAHP recorded with RuR. These data suggest that the Ca2+ release underlying ISK enhancement requires RyRs but does not require IP3R activation. This conclusion is tempered by potential off-target actions of these drugs (e.g., see Taylor and Broad, 1998); so to directly test IP3 involvement, we recorded from DR neurons with a patch solution containing caged-IP3 and used full-field UV flashes (500 ms) to release IP3 into the cytoplasm. Uncaging IP3 failed to induce a membrane current in DR neurons either before or after application of orexin (Fig. 6G, left traces), even though the 5 pulse protocol produced an AHP current that was greatly enhanced by orexin in the same neurons (Fig. 6G, right traces). Similarly, when uncaging was paired with the 5 pulse protocol (500 ms UV pulse started simultaneously with the first depolarization), IoeAHP was not altered (paired with UV amplitude 101.2 ± 6.4% of that without UV, n = 5, paired t test: t(4) = −0.70, p > 0.05). To verify that we were successfully uncaging IP3, we conducted identical recordings from hippocampal CA1 pyramidal neurons, where UV flashes produced an outward current (Fig. 6H), as expected from previous studies showing perisomatic IP3 uncaging and ensuing Ca2+ release activates SK channels (Hong et al., 2007; El-Hassar et al., 2011). These results are summarized in Figure 6I. Collectively, these data indicate that enhancement of the SK component of the oeAHP involves activation of ryanodine receptors and Ca2+ release from intracellular stores, presumably via CICR, but that activation of IP3Rs are not necessary or sufficient.
Orexin prolongs the SK component of IoeAHP via inhibition of A-current and a resulting increase in local Ca2+ influx
SK channel gating is determined by the subplasma membrane elevation in Ca2+, which is not effectively detected by our bulk cytoplasmic somatic Ca2+ indicator signals. We therefore reexamined the possibility that orexin increased spike-evoked Ca2+ influx. We first measured the action potential width and found that orexin-A (300 nm) significantly broadened the spike (measured at −25 mV) to 109.0 ± 2.0% of control (paired t test: t(17) = 1.74, p < 0.05; Fig. 7A). We also recorded action potentials with high intracellular BAPTA (10 mm) to block possible Ca2+-dependent K+ repolarization currents and found that orexin-A broadened the spike by 111.5 ± 4.2% of control (paired t test: t(7) = 2.34, p < 0.05). Similarly, in voltage-clamp mode, we found that the poorly clamped Ca2+ action currents (CACs), evoked by our voltage-steps, were also augmented by orexin (Fig. 7B). These orexin effects could result from increasing voltage-gated Ca2+ currents or reducing spike-repolarization currents. We therefore first examined the effect of orexin-A on isolated voltage-gated Ca2+ currents. We monitored the leak-subtracted Ca2+ currents evoked with 30 ms voltage steps from −65 mV to −25 mV. This protocol was delivered every 30 s before and after orexin-A (300 nm) application. This current was relatively stable, although both the peak inward current and tail current showed a slight run-down over the course of the recordings. Nevertheless, the current was not detectably altered by orexin application (Fig. 7C,D).
Figure 7.

Orexin-A broadened action potentials and CACs in DR neurons but did not increase isolated Ca2+ currents. A, Action potentials were evoked by brief suprathrehold current pulses before (red, Con) and after orexin-A (blue, Orx, 300 nm). Three superimposed spikes recorded with normal patch solution and ACSF are shown for each condition. B, CACs recorded (voltage steps from −65 mV to −5 mV, 10 ms) with normal internal solution and ACSF containing TTX and synaptic blockers before (red, Con) and after application of orexin-A (blue, Orx, 300 nm). C, Pharmacologically isolated Ca2+ currents were monitored by stepping Vm from a holding potential of −65 mV to −25 mV for 30 ms every 30 s (Inset) before (red, Con) and orexin-A (blue, Orx, 300 nm). Superimposed Ca2+ currents are from just before orexin-A application and following 2 min of orexin-A application. D, Summary of peak Ca2+ current (mean ± SEM) evoked as in C and measured before (Con) and after orexin application (Orx). The difference was not significant (paired t test, p > 0.05, n = 5).
We next examined whether orexin suppressed voltage-dependent K+ currents. We isolated K+ currents and compared the effect of orexin-A (300 nm) on both the noninactivating and inactivating components in the same neurons. Orexin application did not change the delayed rectifier currents, but it reduced the transient outward current by ∼20% (Fig. 8A–E). Fitting the normalized activation and inactivation curves with Boltzmann functions revealed that the corresponding V1/2 and slope factors (k) were similar before and after orexin (activation before orexin V1/2: −30.7 ± 1.9 mV, k: 8.5 ± 1.7; activation after orexin V1/2: −32.2 ± 1.7 mV, k: 8.0 ± 1.5; inactivation before orexin V1/2: −63.7 ± 1.0 mV, k: −5.6 ± 0.9; inactivation after orexin V1/2: −64.2 ± 0.7 mV, k: −6.5 ± 0.6), indicating that the transient current was reduced without changing voltage dependence (Fig. 8C–E). Because Co2+-ACSF was used to block voltage-gated Ca2+ channels in these recordings, these parameters will be somewhat shifted in the depolarizing direction (Mayer and Sugiyama, 1988). From additional recordings in normal ACSF (data not shown), we estimate that the activation curve was positively shifted by ∼10 mV, which fits well with previous estimates (Mayer and Sugiyama, 1988) and indicates this transient outward current activated below spike threshold. This, and the finding that the current was largely blocked by 5 mm 4-AP (Fig. 8F), which produced a characteristic late slowing of inactivation (Jackson and Bean, 2007), indicates the current is a subthreshold A-current (Jerng et al., 2004).
Figure 8.

The enhanced SK component of IoeAHP results from orexin inhibition of A-current but not delayed rectifier current in DR neurons. A, Leak-subtracted (−p/4) delayed rectifier currents before (left, red) and after (right, blue) application of orexin-A (300 nm; calibrations on right apply to both sets of current). Middle, Voltage protocol and holding potential. Calibration: Middle, 100 ms, 20 mV. B, Summary (mean ± SEM) of the delayed rectifier current steady-state I–V relations before (open squares, red) and after (filled squares, blue) orexin (n = 6). C, Leak-subtracted (−p/4) transient outward currents evoked by the A-current activation protocol (middle traces; holding potential and calibration as in A) before (left, red) and after (right, blue) application of orexin-A (300 nm). D, Leak-subtracted (−p/4) transient outward currents evoked by the A-current deinactivation protocol (middle traces; holding potential and calibration as in A) before (left, red) and after (right, blue) application of orexin-A (300 nm). E, Summary (mean ± SEM) of the voltage dependence of activation and inactivation of the transient outward current before (open symbols, red) and after (filled symbols, blue) orexin application (n = 6), normalized to the control Imax and Gmax before orexin application. F, The transient outward current was monitored by depolarizing test pulses to −15 mV following a 75 ms hyperpolarizing conditioning pulse to −105 mV, showing characteristic blocking by 5 mm 4-AP. Three traces before 4-AP application (black) and three traces following the maximum blockade (red) are superimposed. G, CACs evoked by voltage steps to −5 mV (10 ms duration) from a holding potential of −65 mV from before (Con, black), during (4-AP, red) application of 5 mm 4-AP, and after application of orexin-A (300 nm; Orx, blue). H, SK currents evoked by the 5 pulse protocol (holding potential = −65 mV) from before (Con, black) and after A-current blockade with 5 mm 4-AP (red). I, The average IoeAHP (n = 8) produced by subsequent application of orexin-A (300 nm) in 5 mm 4-AP. The decay was well fit with a single slow exponential (white line). J, Outward currents produced by single full-field UV pulses (100 ms) to uncage Ca2+ from DMNP before (red) and after (blue) application of orexin-A (300 nm). The holding current preceding each trace was subtracted. K, The average IoeAHP (n = 5) computed from currents evoked by UV pulses (top, uvIoeAHP) and from interleaved 5 pulse protocols (bottom, IoeAHP, 5 pulses). uvIoeAHP was well fit with a slow single exponential (white line on top trace) while the IoeAHP (5 pulses) was well fit with a double exponential having medium and slow time constants (white line on bottom trace). Calibration on bottom trace applies to both traces. L, Examples of SK current evoked by a UV pulse (uvISK, UV, 100 ms, left traces) and a 5 pulse protocol (ISK, 5 pulses, right traces) from before (red) and after (blue) application of orexin-A (300 nm). Holding current preceding each trace was subtracted from each trace. Calibration on right applies to both sets of traces.
In addition to its well-known role in lengthening the interval between action potentials (Connor and Stevens, 1971), subthreshold A-currents also function in spike repolarization (Kim et al., 2005; Yuan et al., 2005), and this inhibition by orexin could therefore contribute to spike broadening, increased CACs, and an increase in the SK component of the oeAHP. Consistent with this idea, orexin (300 nm) produced significant broadening of action potentials measured with Ca2+ channels blocked by Co2+-ACSF (spike width at −25 mV in orexin: 108.8 ± 2.9% of control, paired t test: t(5) = 2.57, p < 0.05; data not shown). To more directly test this idea, we applied 4-AP while measuring CACs and the AHP current. 4-AP (5 mm) enhanced the CACs (Fig. 8G) and prolonged the SK-mediated AHP in the same cells (Fig. 8H; Control T50 = 0.48 ± 0.07 s; 4-AP T50 = 0.58 ± 0.09 s, n = 8, paired t test: t(7) = −2.13, p < 0.05). Treatment with 4-AP also occluded further enhancement of CACs by orexin-A (Fig. 8G) and resulted in an IoeAHP whose amplitude was reduced by approximately half (18.2 ± 2.7 pA, n = 8; unpaired t test: t(17) = −4.99, p < 0.05; Fig. 8I) but was not different from that in apamin (unpaired t test: t(29) = 0.14, p > 0.05). Moreover, the average IoeAHP in 4-AP decayed with a single component that was well fit by a single exponential (Fig. 8I; tau = 4.7 ± 0.03 s) like that in apamin. Collectively, these findings suggest that the augmented SK component of the oeAHP results from suppression of an A-current, broader action potentials and enhanced Ca2+ influx, leading to an increase in subplasma membrane Ca2+, which is further enhanced by CICR. In contrast, the slower-decaying, apamin-insensitive component of the oeAHP was insensitive to A-current inhibition, indicating that a different Ca2+-dependent mechanism must be involved.
Because emerging evidence suggests that store filling can be modulated by intracellular signaling pathways (Yamasaki-Mann et al., 2010), additional factors contributing to the augmented ISK might be that orexin enhances Ca2+ sequestration, leading to greater store release in response to Ca2+ influx, or that the Ca2+ sensitivity of RyRs or SK channels is increased by orexin signaling. We reasoned that, if these factors are important, identical Ca2+ transients should trigger enhanced ISK following orexin application. To bypass the orexin-enhanced CACs, we used full-field DMNP Ca2+ uncaging (100–200 ms pulses) before and after orexin-A (300 nm) application and compared the evoked outward currents. We found that identical uncaging pulses produced a reproducible, medium-duration outward current before orexin that was blocked by apamin (300 nm; data not shown), and a more slowly decaying current following orexin application that was reminiscent of IoeAHP (Fig. 8J). The average outward current produced by Ca2+ uncaging before orexin was 16.9 ± 2.8 pA, whereas after orexin, the current was 25.0 ± 3.6 pA (n = 5). We computed IoeAHP resulting from uncaging pulses and found that this average current (uvIoeAHP) was reasonably well fit by a single slow exponential (tau = 4.70 ± 0.012 s, n = 5; Fig. 8K, top), which was similar to the decay of IoeAHP in 4-AP (Fig. 8I). In contrast, the decay of IoeAHP produced by 5 pulses in these same neurons was well fit by a double exponential having medium (tau = 0.36 ± 0.004 s) and slow (tau = 5.02 ± 0.012 s) time constants (Fig. 8K, bottom). Moreover, when we computed the total ISK evoked by UV after orexin, by applying apamin (300 nm) after orexin and then subtracting the apamin-insensitive component, we found that it was not any longer than ISK before orexin (T50 before orexin = 0.34 ± 0.02 s; T50 after orexin = 0.30 ± 0.03 s; t(4) = 1.26, p > 0.05; Fig. 8L, left). This differed from the ISK evoked by the 5 pulse protocol, tested in these same cells, which increased significantly in duration (T50 before orexin = 0.33 ± 0.04 s; T50 after orexin = 0.44 ± 0.05 s; t(4) = −2.79, p < 0.05; Fig. 8L, right). It is noteworthy that the prolongation of ISK by orexin obtained here, using patch solution with DMNP, agrees well with findings obtained using normal patch solution (ISK T50 before orexin = 0.35 ± 0.05 s, T50 after orexin = 0.43 ± 0.07 s; t(5) = 3.28, p < 0.05; data not shown). Collectively, these findings suggest that in order for orexin to prolong the ISK and thereby produce the SK component of the IoeAHP, an enhanced Ca2+ influx is required. Nevertheless, because DMNP alters intracellular Ca2+ buffering, this conclusion should be considered tentative. Future experiments using a wider range of Ca2+ release and buffering conditions would be necessary to fully test this conclusion.
The firing produced by orexin-A was slower and less variable than predicted for the depolarizing orexin current
In the final series of experiments, we examined the impact of orexin and the oeAHP on the firing pattern of DR neurons. Because these experiments were conducted with the ACSF heated to 32°C, we first confirmed that orexin-A (300 nm) produced an oeAHP under these conditions. We found that, although the input resistance of DR neurons was lower (393.6 ± 31.4 MΩ; n = 22; normal ACSF with ionotropic receptor blockers), orexin-A still significantly increased both the amplitude (control: 7.84 ± 0.58 mV; orexin: 9.17 ± 0.71 mV, n = 22; paired t test: t(21) = 4.10, p < 0.05, paired t test) and duration (control: 0.30 ± 0.03 s; orexin: 0.55 ± 0.04 s, n = 22; paired t test: t(21) = −5.37, p < 0.05, paired t test) of the late AHP, although, not surprisingly, these changes were smaller than observed at room temperature. Twelve cells were recovered and 10 of 12 were TpH+.
Increasing the late AHP during the orexin-mediated depolarization might be expected to produce slower firing than the depolarization alone. Testing this prediction is complicated by the lack of blockers specific for just the oeAHP, and by the fact that orexin inhibits the A-current and produces a large increase in membrane noise, both of which could increase firing (White et al., 2000). For the oeAHP to decrease orexin-induced firing, the orexin-mediated depolarization would have to produce interspike intervals short enough to be influenced by the oeAHP, and the oeAHP would have to play a more dominant role than A-current and membrane noise in setting the spike interval. In the first test, we injected a previously recorded noisy orexin current (virtual current) scaled to different average values and compared the resulting firing rate (without an oeAHP; Fig. 9A1) with the firing rate evoked by an actual orexin current (with the oeAHP; Fig. 9A2) produced by application of orexin-A (300 nm). The magnitude of the actual current was estimated by the injected current necessary to return membrane potential to baseline during the peak effect of orexin-A (Fig. 9A2, arrow). The firing rate, measured as spikes per second (sps), achieved during orexin-A application was lower than that produced by the comparable virtual current (Fig. 9A1), and this firing rate (±SD) fell below the rate-current curve produced by a bracketing range of virtual currents (Fig. 9A3). In a second test, we used a dynamic clamp to introduce a virtual orexin conductance (0.5–2 nS) to generate a noisy, depolarizing orexin current (without the oeAHP). This orexin current was comparable with that produced by 300 nm orexin in these and previous DR recordings (Kohlmeier et al., 2008). We compared the firing evoked by this conductance to the firing produced by subsequent application of orexin-A (300 nm). This also showed that the firing rate evoked by the actual orexin-A current (Fig. 9B2) was lower than that produced by the comparable virtual conductance (Fig. 9B1), and that this firing rate fell below the firing rate-average current curve produced by a range of virtual conductances (Fig. 9B3). These results are summarized in Figure 9C1 and indicate that the average steady-state firing rate expected from the virtual inward orexin currents without the oeAHP was significantly higher than the firing rate evoked by orexin-A with the oeAHP. This was true for cells tested with both current clamp (virtual: 2.8 ± 0.5 sps vs actual: 1.9 ± 0.3 sps; paired t test: t(4) = 3.81, p < 0.05, n = 5) and dynamic clamp (virtual: 1.9 ± 0.5 sps vs actual: 1.2 ± 0.5 sps; paired t test: t(5) = 3.63, p < 0.05, n = 6).
We next tested the idea that the oeAHP would promote more regular firing of DR neurons. We compared the SD of the steady-state firing rate produced by orexin-A with that produced by a corresponding virtual orexin conductance delivered by dynamic clamp. Consistent with a role for the oeAHP in promoting more regular firing, the firing rate variability produced by orexin-A was lower than that produced by dynamic clamp (Fig. 9C2, vertical diamonds; virtual SD: 0.73 ± 0.12 vs actual SD: 0.32 ± 0.06, paired t test: t(5) = 3.80, p < 0.05, n = 6). The same decrease in variability was not observed when comparing the firing rate variability produced by orexin-A with that estimated by current clamp (virtual SD: 0.36 ± 0.07 vs actual SD 0.29 ± 0.06, paired t test: t(4) = 1.07, p > 0.05, n = 5) because the current-clamp stimulus produced significantly less firing rate variability than did the virtual conductance (unpaired t test: t(9) = 2.49, p < 0.05). This lower variability possibly reflects more effective low pass filtering of the noisy current command by the membrane because there was not a corresponding fluctuation in conductance as introduced by the dynamic clamp.
Orexin-A slows steady-state firing and decreases steady-state firing fidelity
To examine how orexin alters responsiveness of DR neurons to other excitatory inputs, we examined the repetitive firing evoked by suprathreshold current steps (5 s, 50–150 pA) delivered from the same baseline membrane potential before and after orexin application. DR neurons show rapid SFA, with the initial firing rate decaying to steady-state firing in only a few interspike intervals. If the oeAHP plays an important role in regulating spiking, we would expect orexin application to increase SFA by reducing steady-state firing. Indeed, application of orexin-A (300 nm) increased SFA by increasing the interval between spikes after the first two spikes and by lowering the steady-state rate (Fig. 10A). While orexin-A had no significant effect on the initial rate (Fig. 10B), it significantly lowered steady-state firing for each current tested (Fig. 10C; repeated-measures ANOVA: F(1,29) = 14.81, p < 0.05; post hoc tests p < 0.05; n = 5).
Finally, we examined the effect of orexin on the fidelity with which DR neurons could fire action potentials in response to trains of brief suprathreshold current pulses. If the oeAHP plays an important role in regulating excitability dynamics, we expect that orexin would produce a decrease in firing fidelity following spiking onset and the development of the oeAHP. To test this, the pulse amplitude was adjusted to produce a spike for 100% of pulses delivered at 5 Hz (100% fidelity; Fig. 10D), which is near the average rate during active-waking for mouse 5-HT neurons (Sakai, 2011). As predicted, after the first few spikes, orexin produced a decrease in firing fidelity (i.e., increased spike failures; repeated-measures ANOVA: F(1,49) = 27.32, p < 0.05, n = 5) that was evident at 5 and 10 Hz (Fig. 10E; post hoc tests, p < 0.05, n = 5). This had the effect of compressing the steady-state input–output frequency relationship (Fig. 10F) without changing the initial input–output relation of 1:1 (Fig. 10F, dotted line).
Of these 10 neurons tested at 32°C, and 10 more tested at room temperature, which all showed a significant decrease in steady-state firing, 10 filled neurons were recovered and 8 of 10 were TpH-immunopositive.
Discussion
Here we elucidated a new action of orexin that enhanced the AHP of 5-HT DR neurons via two distinct mechanisms. We then demonstrated how this action impacts the integrative properties of these neurons.
Distinct currents underlie the oeAHP
Complete dependence of the oeAHP on Ca2+ influx and elevation of [Ca2+]i, along with a reversal potential that shifts with EK in a Nernstian manner, indicate involvement of Ca2+-activated K+ channels rather than the electrogenic Na+/K+ ATPase, as found for some Ca2+-independent sAHPs (e.g., Pulver and Griffith, 2010; Gulledge et al., 2013). Application of apamin revealed that approximately half of IoeAHP resulted from augmented ISK (tau ∼0.5 s), whereas the remainder resulted from induction of a slow-AHP-like current (tau ∼5.0 s).
SK augmentation depends on increased spike-evoked Ca2+ influx resulting from A-current inhibition and CICR
SK current likely derives from SK2 and/or SK3 subunits because mRNA and protein for both localize to the DR (Stocker and Pedarzani, 2000; Sailer et al., 2004). Because the PP2A antagonist okadaic acid did not attenuate the SK component of IoeAHP (Fig. 6A), we examined involvement of SK-regulating Ca2+ sources. Like the α-1 adrenoceptor enhancement of SK-AHPs in DR neurons (Pan et al., 1994), the SK component of the oeAHP was inhibited by Ca2+-store depletion. This α-1 action reportedly involves IP3Rs because IP3 injection transiently (<1 min) lengthened the AHP (Freedman and Aghajanian, 1987). This, however, did not increase the AHP amplitude, and this effect was not prolonged by inhibiting IP3 breakdown. In contrast, the orexin-mediated SK enhancement was inhibited by antagonists of RyRs, rather than IP3Rs, and it was not mimicked by IP3 uncaging, which fits with robust RyR2 and minimal IP3R expression within DR (Allen Brain Atlas; www.brain-map.org).
Because these findings suggested CICR involvement, we examined whether orexin enhanced spike-evoked Ca2+ influx and found that orexin broadened both individual action potentials and CACs (Fig. 7). Instead of directly augmenting Ca2+ channel function (Kohlmeier et al., 2008), we found that orexin inhibited a subthreshold A-current (Fig. 8), which is likely critical because 4-AP (Fig. 7) both prolonged baseline ISK and occluded orexin-mediated SK enhancement, whereas ISK produced by Ca2+ uncaging pulses was not enhanced by orexin (Fig. 8). Because Kv4.3 mRNA is differentially expressed in the DR (Serôdio et al., 1998), it is likely that spike broadening and increased Ca2+ influx result from orexin-mediated inhibition of A-current, mediated by Kv4.3 channels. This is a novel orexin action, although orexin can inhibit (Yang and Ferguson, 2003; Murai and Akaike, 2005) or increase (Belle et al., 2014) unidentified delayed-rectifier currents in some other neurons.
Strikingly, α-1 receptors also inhibit A-current in DR neurons (Aghajanian, 1985). This was suggested to increase firing by shortening the interspike interval, although spike width and Ca2+ influx were not examined. We confirmed that PE (3 μm) also increased CACs in our slices (M.I. and C.S.L., unpublished observations), suggesting that A-current inhibition, spike broadening, and increased Ca2+ influx may also contribute to α-1 enhancement of SK-AHPs in these neurons. Indeed, PE occluded the entire oeAHP (Fig. 1E), suggesting that α-1 and OxRs share effectors or signaling components downstream of the receptors because α-1 actions remained intact in the absence of OxRs (Fig. 2D). These findings fit well with the previous demonstration that orexin and α-1 receptor signaling converges in activating inward current in DR neurons (Brown et al., 2002).
The apamin-insensitive component of IoeAHP differs from the classical IsAHP
In contrast to SK enhancement, the apamin-insensitive component of IoeAHP decayed ∼10 times slower (tau ∼5 s; Fig. 4F) and was absent before orexin application. Moreover, it was insensitive to store depletion and was not induced or occluded by blocking A-current, but it was induced by intracellular uncaging of Ca2+, indicating different Ca2+ requirements. Its slow time course is reminiscent of the IsAHP (Vogalis et al., 2003) and, like the sAHP (Lee et al., 2005a), its duration was reduced at elevated temperatures. Channels underlying the sAHP have been difficult to identify, and evidence suggests that different K+ channels are involved (for review, see Vogalis et al., 2003; Andrade et al., 2012). Recently, UCL2077 was shown to block the IsAHP in hippocampal pyramidal neurons (Shah et al., 2006) and the orexin-inhibited IsAHP in PVT neurons (Zhang et al., 2010). UCL2077 blocks KCNQ1/2 channels, and knock-out evidence suggests involvement of KCNQ2/3 in sAHPs of CA3 and dentate gyrus neurons (Tzingounis and Nicoll, 2008; Tzingounis et al., 2010). Nevertheless, KCNQ antagonists are not universal blockers of classical sAHPs (Abel et al., 2004).
Another feature of the classical IsAHP is their suppression by some neurotransmitters. Monoamines suppress it via cAMP/PKA signaling (Madison and Nicoll, 1986; Pedarzani and Storm, 1993), and receptors that reduce cAMP, such as adenosine A1 and GABAB receptors, can modestly increase IsAHP in hippocampal neurons (Haas and Greene, 1984; Gerber and Gähwiler, 1994). However, unlike IsAHP in the hippocampus, forskolin did not alter the oeAHP (Fig. 5I). In the hippocampus, Gq activation by acetylcholine and glutamate also inhibits IsAHP (Krause et al., 2002), and IsAHP is suppressed by orexin in PVT neurons via PKC activation (Zhang et al., 2010). Unlike those sAHPs, PKC inhibitors did not alter IoeAHP or the α-1 augmentation of the SK-AHP (Pan et al., 1994) in DR neurons. Nevertheless, PDBu powerfully suppressed the entire IoeAHP, as reported for α-1 actions (Pan et al., 1994). This suggests that PKC activation is not required for the oeAHP, but that global activation suppresses orexin and α-1-receptor signaling, perhaps via receptor internalization or desensitization (Chuang et al., 1996; Kelly et al., 2008). Thus, while the apamin-insensitive component of IoeAHP shares features with classical sAHPs, insensitivity to forskolin, PKC inhibitors and a KCNQ blocker indicates it is novel.
Signaling cascades mediating IoeAHP
OxRs are generally thought to activate PLC via Gq/11 (Sakurai et al., 1998), although their signaling is less well explored in CNS than expression systems, where OxRs couple to multiple G-proteins and second messenger systems (for review, see Kukkonen and Leonard, 2014). Intracellular application of the PLC inhibitor U73122, but not its inactive analog U73343, completely blocked orexin-mediated inward current and the oeAHP, but not a PLC-independent 5-HT-mediated outward current (Fig. 5A–D). This fits with expression system evidence for prominent PLC activation (Holmqvist et al., 2002) and findings from some other neurons, where PLC inhibitors attenuate orexin actions (Muroya et al., 2004; Borgland et al., 2006). However, because neither the inward current nor IoeAHP depended on IP3 or PKC activation, noncanonical PLC signaling may be involved. While possibilities abound, one consideration is that U73122 has off-target or indirect effects that influence orexin stimulation of PLA2 (Turunen et al., 2012) and that arachidonic acid or its metabolites regulate the inward current and/or the oeAHP. Alternatively, PLC-generated DAG may be converted to the endocannabinoid 2-AG by DAG-lipase, and 2-AG or its metabolites are involved. Expressed OxRs powerfully stimulate 2-AG and arachidonic acid production (Turunen et al., 2012), and inhibition of DAG-lipase blocks OxR-mediated endocannabinoid inhibition of excitatory input to DR neurons (Haj-Dahmane and Shen, 2005). It is therefore noteworthy that both arachidonic acid and 2-AG can directly inhibit Kv4 channels (Amorós et al., 2010) and broaden action potentials (Keros and McBain, 1997).
Functional consequences of the oeAHP
The oeAHP enhanced SFA by reducing steady state, but not initial firing, in response to current steps (Figs. 9; 10), consistent with functions for slow, Ca2+-dependent AHPs (Faber and Sah, 2003). Dynamic clamp studies revealed that the oeAHP decreases firing-rate variability, an effect particularly relevant in vivo, where the coefficient of variation of firing is ∼10-fold greater (coefficient of variation 0.5–1.0; waking) (Sakai, 2011) than in our slices (coefficient of variation 0.06–0.1). Increased SFA implies reduced excitability for tonic input, as demonstrated by late spike-failures for pulse-train inputs (5, 10 Hz; Fig. 10D,E) and a compressed steady-state input–output relation (Fig. 10F). This reflects the high-pass filter function of SFA (Benda and Herz, 2003), which favors spike encoding of rapidly varying inputs. We hypothesize that, during active waking, when orexin neuron firing is highest (Lee et al., 2005b; Mileykovskiy et al., 2005), released orexin will both depolarize and sharpen the high-pass filter characteristics of 5-HT neurons. The oeAHP would help limit and regularize firing in response to tonic input without attenuating the spike encoding of phasic synaptic inputs related to behavioral events (Hajós et al., 2007; Ranade and Mainen, 2009; Sakai, 2011; Liu et al., 2014). This may be especially important for processing reward signals because orexin neurons increase firing during anticipation and acquisition of reward (Hassani et al., 2016) and the firing of most 5-HT DR neurons transition from a tonic increase to a burst during reward acquisition (Li et al., 2016). Thus, orexin inputs could provide an arousal/attention signal that promotes phasic over tonic outflow from raphe neurons, which could affect the relative engagement of postsynaptic and autoreceptors by released 5-HT (Gartside et al., 2000). Accordingly, the loss of orexin signaling in narcolepsy would be expected to impair this signal processing, potentially contributing to the expression of narcolepsy/cataplexy symptoms (Hasegawa et al., 2014) and comorbid symptoms, such as depression, anxiety, and eating disorders (Fortuyn et al., 2011).
Footnotes
This work was supported by Public Health Service Grants NS027881 and HL064150 to C.S.L. We thank Dr. Carl Thompson for statistics advice; Dr. Lorin S. Milescu for assistance in implementing his dynamic clamp software; Aaron Asch for expert programming; Elizabeth Lynn and Dr. Catherine Steger for expert technical assistance; and Dr. Masashi Yanagisawa and Dr. Richard Chemelli for providing orexin receptor null breeder mice.
The authors declare no competing financial interests.
References
- Abel HJ, Lee JC, Callaway JC, Foehring RC. Relationships between intracellular calcium and afterhyperpolarizations in neocortical pyramidal neurons. J Neurophysiol. 2004;91:324–335. doi: 10.1152/jn.00583.2003. [DOI] [PubMed] [Google Scholar]
- Aghajanian GK. Modulation of a transient outward current in serotonergic neurons by a1 adrenoreceptors. Nature. 1985;315:501–503. doi: 10.1038/315501a0. [DOI] [PubMed] [Google Scholar]
- Aghajanian GK, Vandermaelen CP. Intracellular recordings from serotonergic dorsal raphe neurons: pacemaker potentials and the effect of LSD. Brain Res. 1982;238:463–469. doi: 10.1016/0006-8993(82)90124-X. [DOI] [PubMed] [Google Scholar]
- Agopyan N, Agopyan I. Effects of protein kinase C activators and inhibitors on membrane properties, synaptic responses, and cholinergic actions in CA1 subfield of rat hippocampus in situ and in vitro. Synapse. 1991;7:193–206. doi: 10.1002/syn.890070304. [DOI] [PubMed] [Google Scholar]
- Alix P, Venkatesan K, Scuvée-Moreau J, Massotte L, Nguyen Trung ML, Cornil CA, Seutin V. Mechanism of the medium-duration afterhyperpolarization in rat serotonergic neurons. Eur J Neurosci. 2014;39:186–196. doi: 10.1111/ejn.12408. [DOI] [PubMed] [Google Scholar]
- Allen D, Fakler B, Maylie J, Adelman JP. Organization and regulation of small conductance Ca2+-activated K+ channel multiprotein complexes. J Neurosci. 2007;27:2369–2376. doi: 10.1523/JNEUROSCI.3565-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amorós I, Barana A, Caballero R, Gómez R, Osuna L, Lillo MP, Tamargo J, Delpón E. Endocannabinoids and cannabinoid analogues block human cardiac Kv4.3 channels in a receptor-independent manner. J Mol Cell Cardiol. 2010;48:201–210. doi: 10.1016/j.yjmcc.2009.07.011. [DOI] [PubMed] [Google Scholar]
- Andrade R, Foehring RC, Tzingounis AV. The calcium-activated slow AHP: cutting through the Gordian knot. Front Cell Neurosci. 2012;6:47. doi: 10.3389/fncel.2012.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belle MD, Hughes AT, Bechtold DA, Cunningham P, Pierucci M, Burdakov D, Piggins HD. Acute suppressive and long-term phase modulation actions of orexin on the mammalian circadian clock. J Neurosci. 2014;34:3607–3621. doi: 10.1523/JNEUROSCI.3388-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benda J, Herz AV. A universal model for spike-frequency adaptation. Neural Comput. 2003;15:2523–2564. doi: 10.1162/089976603322385063. [DOI] [PubMed] [Google Scholar]
- Bildl W, Strassmaier T, Thurm H, Andersen J, Eble S, Oliver D, Knipper M, Mann M, Schulte U, Adelman JP, Fakler B. Protein kinase CK2 is coassembled with small conductance Ca(2+)-activated K+ channels and regulates channel gating. Neuron. 2004;43:847–858. doi: 10.1016/j.neuron.2004.08.033. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron. 2006;49:589–601. doi: 10.1016/j.neuron.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Brown RE, Sergeeva OA, Eriksson KS, Haas HL. Convergent excitation of dorsal raphe serotonin neurons by multiple arousal systems (orexin/hypocretin, histamine and noradrenaline) J Neurosci. 2002;22:8850–8859. doi: 10.1523/JNEUROSCI.22-20-08850.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter ME, Schaich Borg J, de Lecea L. The brain hypocretins and their receptors: mediators of allostatic arousal. Curr Opin Pharmacol. 2009;9:39–45. doi: 10.1016/j.coph.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/S0092-8674(00)81973-X. [DOI] [PubMed] [Google Scholar]
- Chuang TT, Iacovelli L, Sallese M, De Blasi A. G protein-coupled receptors: heterologous regulation of homologous desensitization and its implications. Trends Pharmacol Sci. 1996;17:416–421. doi: 10.1016/S0165-6147(96)10048-1. [DOI] [PubMed] [Google Scholar]
- Connor JA, Stevens CF. Voltage clamp studies of a transient outward membrane current in gastropod neural somata. J Physiol. 1971;213:21–30. doi: 10.1113/jphysiol.1971.sp009365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford LK, Craige CP, Beck SG. Increased intrinsic excitability of lateral wing serotonin neurons of the dorsal raphe: a mechanism for selective activation in stress circuits. J Neurophysiol. 2010;103:2652–2663. doi: 10.1152/jn.01132.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, 2nd, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hassar L, Hagenston AM, D'Angelo LB, Yeckel MF. Metabotropic glutamate receptors regulate hippocampal CA1 pyramidal neuron excitability via Ca2+ wave-dependent activation of SK and TRPC channels. J Physiol. 2011;589:3211–3229. doi: 10.1113/jphysiol.2011.209783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson KS, Sergeeva O, Brown RE, Haas HL. Orexin/hypocretin excites the histaminergic neurons of the tuberomammillary nucleus. J Neurosci. 2001;21:9273–9279. doi: 10.1523/JNEUROSCI.21-23-09273.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES, Sah P. Calcium-activated potassium channels: multiple contributions to neuronal function. Neuroscientist. 2003;9:181–194. doi: 10.1177/1073858403009003011. [DOI] [PubMed] [Google Scholar]
- Fortuyn HA, Mulders PC, Renier WO, Buitelaar JK, Overeem S. Narcolepsy and psychiatry: an evolving association of increasing interest. Sleep Med. 2011;12:714–719. doi: 10.1016/j.sleep.2011.01.013. [DOI] [PubMed] [Google Scholar]
- Freedman JE, Aghajanian GK. Role of phosphoinositide metabolites in the prolongation of afterhyperpolarizations by alpha 1-adrenoceptors in rat dorsal raphe neurons. J Neurosci. 1987;7:3897–3906. doi: 10.1523/JNEUROSCI.07-12-03897.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartside SE, Hajós-Korcsok E, Bagdy E, Hársing LG, Jr, Sharp T, Hajós M. Neurochemical and electrophysiological studies on the functional significance of burst firing in serotonergic neurons. Neuroscience. 2000;98:295–300. doi: 10.1016/S0306-4522(00)00060-9. [DOI] [PubMed] [Google Scholar]
- Gerber U, Gähwiler BH. GABAB and adenosine receptors mediate enhancement of the K+ current, IAHP, by reducing adenylyl cyclase activity in rat CA3 hippocampal neurons. J Neurophysiol. 1994;72:2360–2367. doi: 10.1152/jn.1994.72.5.2360. [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Dasari S, Onoue K, Stephens EK, Hasse JM, Avesar D. A sodium-pump-mediated afterhyperpolarization in pyramidal neurons. J Neurosci. 2013;33:13025–13041. doi: 10.1523/JNEUROSCI.0220-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas HL, Greene RW. Adenosine enhances afterhyperpolarization and accommodation in hippocampal pyramidal cells. Pflugers Arch. 1984;402:244–247. doi: 10.1007/BF00585506. [DOI] [PubMed] [Google Scholar]
- Haj-Dahmane S, Shen RY. The wake-promoting peptide orexin-B inhibits glutamatergic transmission to dorsal raphe nucleus serotonin neurons through retrograde endocannabinoid signaling. J Neurosci. 2005;25:896–905. doi: 10.1523/JNEUROSCI.3258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajós M, Allers KA, Jennings K, Sharp T, Charette G, Sík A, Kocsis B. Neurochemical identification of stereotypic burst-firing neurons in the rat dorsal raphe nucleus using juxtacellular labelling methods. Eur J Neurosci. 2007;25:119–126. doi: 10.1111/j.1460-9568.2006.05276.x. [DOI] [PubMed] [Google Scholar]
- Hall KE, Browning MD, Dudek EM, Macdonald RL. Enhancement of high threshold calcium currents in rat primary afferent neurons by constitutively active protein kinase C. J Neurosci. 1995;15:6069–6076. doi: 10.1523/JNEUROSCI.15-09-06069.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa E, Yanagisawa M, Sakurai T, Mieda M. Orexin neurons suppress narcolepsy via 2 distinct efferent pathways. J Clin Invest. 2014;124:604–616. doi: 10.1172/JCI71017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassani OK, Krause MR, Mainville L, Cordova CA, Jones BE. Orexin neurons respond differentially to auditory cues associated with appetitive versus aversive outcomes. J Neurosci. 2016;36:1747–1757. doi: 10.1523/JNEUROSCI.3903-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Kushikata T, Kudo M, Kudo T, Lambert DG, Matsuki A. Orexin A and B evoke noradrenaline release from rat cerebrocortical slices. Br J Pharmacol. 2001;134:1461–1466. doi: 10.1038/sj.bjp.0704409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmqvist T, Akerman KE, Kukkonen JP. Orexin signaling in recombinant neuron-like cells. FEBS Lett. 2002;526:11–14. doi: 10.1016/S0014-5793(02)03101-0. [DOI] [PubMed] [Google Scholar]
- Hong M, Manita S, Ross WN. Calcium waves generated by uncaging IP3 or synaptic stimulation evoke an apamin-sensitive AHP in the perisomatic region of hippocampal CA1 pyramidal neurons. Soc Neurosci Abstr. 2007;33:786.6. [Google Scholar]
- Innis RB, Nestler EJ, Aghajanian GK. Evidence for G protein mediation of serotonin- and GABAB-induced hyperpolarization of rat dorsal raphe neurons. Brain Res. 1988;459:27–36. doi: 10.1016/0006-8993(88)90282-X. [DOI] [PubMed] [Google Scholar]
- Inoue T, Kato K, Kohda K, Mikoshiba K. Type 1 inositol 1,4,5-trisphosphate receptor is required for induction of long-term depression in cerebellar Purkinje neurons. J Neurosci. 1998;18:5366–5373. doi: 10.1523/JNEUROSCI.18-14-05366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AC, Bean BP. State-dependent enhancement of subthreshold A-type potassium current by 4-aminopyridine in tuberomammillary nucleus neurons. J Neurosci. 2007;27:10785–10796. doi: 10.1523/JNEUROSCI.0935-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs BL, Fornal CA. Activity of serotonergic neurons in behaving animals. Neuropsychopharmacology. 1999;21:9S–15S. doi: 10.1016/S0893-133X(99)00012-3. [DOI] [PubMed] [Google Scholar]
- Jerng HH, Pfaffinger PJ, Covarrubias M. Molecular physiology and modulation of somatodendritic A-type potassium channels. Mol Cell Neurosci. 2004;27:343–369. doi: 10.1016/j.mcn.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Kelly E, Bailey CP, Henderson G. Agonist-selective mechanisms of GPCR desensitization. Br J Pharmacol. 2008;153(Suppl 1):S379–S388. doi: 10.1038/sj.bjp.0707604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keros S, McBain CJ. Arachidonic acid inhibits transient potassium currents and broadens action potentials during electrographic seizures in hippocampal pyramidal and inhibitory interneurons. J Neurosci. 1997;17:3476–3487. doi: 10.1523/JNEUROSCI.17-10-03476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Wei DS, Hoffman DA. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J Physiol. 2005;569:41–57. doi: 10.1113/jphysiol.2005.095042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klink R, Robichaud M, Debonnel G. Gender and gonadal status modulation of dorsal raphe nucleus serotonergic neurons: I. Effects of gender and pregnancy. Neuropharmacology. 2002;43:1119–1128. doi: 10.1016/S0028-3908(02)00219-8. [DOI] [PubMed] [Google Scholar]
- Kohlmeier KA, Inoue T, Leonard CS. Hypocretin/orexin peptide signaling in the ascending arousal system: elevation of intracellular calcium in the mouse dorsal raphe and laterodorsal tegmentum. J Neurophysiol. 2004;92:221–235. doi: 10.1152/jn.00076.2004. [DOI] [PubMed] [Google Scholar]
- Kohlmeier KA, Watanabe S, Tyler CJ, Burlet S, Leonard CS. Dual orexin actions on dorsal raphe and laterodorsal tegmentum neurons: noisy cation current activation and selective enhancement of Ca transients mediated by L-type calcium channels. J Neurophysiol. 2008;100:2265–2281. doi: 10.1152/jn.01388.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmeier KA, Tyler CJ, Kalogiannis M, Ishibashi M, Kristensen MP, Gumenchuk I, Chemelli RM, Kisanuki YY, Yanagisawa M, Leonard CS. Differential actions of orexin receptors in brainstem cholinergic and monoaminergic neurons revealed by receptor knockouts: implications for orexinergic signaling in arousal and narcolepsy. Front Neurosci. 2013;7:246. doi: 10.3389/fnins.2013.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M, Offermanns S, Stocker M, Pedarzani P. Functional specificity of G alpha q and G alpha 11 in the cholinergic and glutamatergic modulation of potassium currents and excitability in hippocampal neurons. J Neurosci. 2002;22:666–673. doi: 10.1523/JNEUROSCI.22-03-00666.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukkonen JP, Leonard CS. Orexin/hypocretin receptor signalling cascades. Br J Pharmacol. 2014;171:314–331. doi: 10.1111/bph.12324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Callaway JC, Foehring RC. Effects of temperature on calcium transients and Ca2+-dependent afterhyperpolarizations in neocortical pyramidal neurons. J Neurophysiol. 2005a;93:2012–2020. doi: 10.1152/jn.01017.2004. [DOI] [PubMed] [Google Scholar]
- Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J Neurosci. 2005b;25:6716–6720. doi: 10.1523/JNEUROSCI.1887-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard CS, Kukkonen JP. Orexin/hypocretin receptor signalling: a functional perspective. Br J Pharmacol. 2014;171:294–313. doi: 10.1111/bph.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhong W, Wang D, Feng Q, Liu Z, Zhou J, Jia C, Hu F, Zeng J, Guo Q, Fu L, Luo M. Serotonin neurons in the dorsal raphe nucleus encode reward signals. Nat Commun. 2016;7:10503. doi: 10.1038/ncomms10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–376. doi: 10.1016/S0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- Liu RJ, van den Pol AN, Aghajanian GK. Hypocretins (orexins) regulate serotonin neurons in the dorsal raphe nucleus by excitatory direct and inhibitory indirect actions. J Neurosci. 2002;22:9453–9464. doi: 10.1523/JNEUROSCI.22-21-09453.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Zhou J, Li Y, Hu F, Lu Y, Ma M, Feng Q, Zhang JE, Wang D, Zeng J, Bao J, Kim JY, Chen ZF, El Mestikawy S, Luo M. Dorsal raphe neurons signal reward through 5-HT and glutamate. Neuron. 2014;81:1360–1374. doi: 10.1016/j.neuron.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry CA, Johnson PL, Hay-Schmidt A, Mikkelsen J, Shekhar A. Modulation of anxiety circuits by serotonergic systems. Stress. 2005;8:233–246. doi: 10.1080/10253890500492787. [DOI] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Cyclic adenosine 3′,5′-monophosphate mediates B-receptor actions of noradrenaline in rat hippocampal pyramidal cells. J Physiol. 1986;372:245–259. doi: 10.1113/jphysiol.1986.sp016007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Madison DV, Andrade R, Nicoll RA. Phorbol esters mimic some cholinergic actions in hippocampal pyramidal neurons. J Neurosci. 1986;6:475–480. doi: 10.1523/JNEUROSCI.06-02-00475.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus JN, Aschkenasi CJ, Lee CE, Chemelli RM, Saper CB, Yanagisawa M, Elmquist JK. Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol. 2001;435:6–25. doi: 10.1002/cne.1190. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Sugiyama K. A modulatory action of divalent cations on transient outward current in cultured rat sensory neurones. J Physiol. 1988;396:417–433. doi: 10.1113/jphysiol.1988.sp016970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieda M, Hasegawa E, Kisanuki YY, Sinton CM, Yanagisawa M, Sakurai T. Differential roles of orexin receptor-1 and -2 in the regulation of non-REM and REM sleep. J Neurosci. 2011;31:6518–6526. doi: 10.1523/JNEUROSCI.6506-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milescu LS, Yamanishi T, Ptak K, Mogri MZ, Smith JC. Real-time kinetic modeling of voltage-gated ion channels using dynamic clamp. Biophys J. 2008;95:66–87. doi: 10.1529/biophysj.107.118190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron. 2005;46:787–798. doi: 10.1016/j.neuron.2005.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti JM. Serotonin control of sleep–wake behavior. Sleep Med Rev. 2011;15:269–281. doi: 10.1016/j.smrv.2010.11.003. [DOI] [PubMed] [Google Scholar]
- Murai Y, Akaike T. Orexins cause depolarization via nonselective cationic and K+ channels in isolated locus coeruleus neurons. Neurosci Res. 2005;51:55–65. doi: 10.1016/j.neures.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Muroya S, Funahashi H, Yamanaka A, Kohno D, Uramura K, Nambu T, Shibahara M, Kuramochi M, Takigawa M, Yanagisawa M, Sakurai T, Shioda S, Yada T. Orexins (hypocretins) directly interact with neuropeptide Y, POMC and glucose-responsive neurons to regulate Ca 2+ signaling in a reciprocal manner to leptin: orexigenic neuronal pathways in the mediobasal hypothalamus. Eur J Neurosci. 2004;19:1524–1534. doi: 10.1111/j.1460-9568.2004.03255.x. [DOI] [PubMed] [Google Scholar]
- Pan ZZ, Grudt TJ, Williams JT. Alpha 1-adrenoceptors in rat dorsal raphe neurons: regulation of two potassium conductances. J Physiol. 1994;478:437–447. doi: 10.1113/jphysiol.1994.sp020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF. PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron. 1993;11:1023–1035. doi: 10.1016/0896-6273(93)90216-E. [DOI] [PubMed] [Google Scholar]
- Penington NJ, Kelly JS, Fox AP. Whole-cell recordings of inwardly rectifying K+ currents activated by 5-HT1A receptors on dorsal raphe neurones of the adult rat. J Physiol. 1993;469:387–405. doi: 10.1113/jphysiol.1993.sp019819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, Nevsimalova S, Aldrich M, Reynolds D, Albin R, Li R, Hungs M, Pedrazzoli M, Padigaru M, Kucherlapati M, Fan J, Maki R, Lammers GJ, Bouras C, Kucherlapati R, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6:991–997. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper DC, Upton N, Smith MI, Hunter AJ. The novel brain neuropeptide, orexin-A, modulates the sleep–wake cycle of rats. Eur J Neurosci. 2000;12:726–730. doi: 10.1046/j.1460-9568.2000.00919.x. [DOI] [PubMed] [Google Scholar]
- Pulver SR, Griffith LC. Spike integration and cellular memory in a rhythmic network from Na+/K+ pump current dynamics. Nat Neurosci. 2010;13:53–59. doi: 10.1038/nn.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranade SP, Mainen ZF. Transient firing of dorsal raphe neurons encodes diverse and specific sensory, motor, and reward events. J Neurophysiol. 2009;102:3026–3037. doi: 10.1152/jn.00507.2009. [DOI] [PubMed] [Google Scholar]
- Rood BD, Calizo LH, Piel D, Spangler ZP, Campbell K, Beck SG. Dorsal raphe serotonin neurons in mice: immature hyperexcitability transitions to adult state during first three postnatal weeks suggesting sensitive period for environmental perturbation. J Neurosci. 2014;34:4809–4821. doi: 10.1523/JNEUROSCI.1498-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouchet N, Waroux O, Lamy C, Massotte L, Scuvée-Moreau J, Liégeois JF, Seutin V. SK channel blockade promotes burst firing in dorsal raphe serotonergic neurons. Eur J Neurosci. 2008;28:1108–1115. doi: 10.1111/j.1460-9568.2008.06430.x. [DOI] [PubMed] [Google Scholar]
- Sailer CA, Kaufmann WA, Marksteiner J, Knaus HG. Comparative immunohistochemical distribution of three small-conductance Ca2+-activated potassium channel subunits, SK1, SK2, and SK3 in mouse brain. Mol Cell Neurosci. 2004;26:458–469. doi: 10.1016/j.mcn.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Sakai K. Sleep-waking discharge profiles of dorsal raphe nucleus neurons in mice. Neuroscience. 2011;197:200–224. doi: 10.1016/j.neuroscience.2011.09.024. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, et al. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/S0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- Schubert R, Noack T, Serebryakov VN. Protein kinase C reduces the KCa current of rat tail artery smooth muscle cells. Am J Physiol. 1999;276:C648–C658. doi: 10.1152/ajpcell.1999.276.3.C648. [DOI] [PubMed] [Google Scholar]
- Scuvée-Moreau J, Boland A, Graulich A, Van Overmeire L, D'hoedt D, Graulich-Lorge F, Thomas E, Abras A, Stocker M, Liégeois JF, Seutin V. Electrophysiological characterization of the SK channel blockers methyl-laudanosine and methyl-noscapine in cell lines and rat brain slices. Br J Pharmacol. 2004;143:753–764. doi: 10.1038/sj.bjp.0705979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serôdio P, Rudy B, Nakamura TY, Coetzee WA, Vega-Saenz De Miera E, Artman M, Sekirnjak C, Martone ME, Weiser M, Deerinck T, Bueno E, Ellisman M, de Miera EVS, Sugimori M, Llinas R, Du J, Zhang L, McBain CJ. Differential expression of Kv4 K+ channel subunits mediating subthreshold transient K+ (A-type) currents in rat brain. J Neurophysiol. 1998;79:1081–1091. doi: 10.1152/jn.1998.79.2.1081. [DOI] [PubMed] [Google Scholar]
- Shah MM, Javadzadeh-Tabatabaie M, Benton DC, Ganellin CR, Haylett DG. Enhancement of hippocampal pyramidal cell excitability by the novel selective slow-afterhyperpolarization channel blocker 3-(triphenylmethylaminomethyl)pyridine (UCL2077) Mol Pharmacol. 2006;70:1494–1502. doi: 10.1124/mol.106.026625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh H, Tzingounis AV. The specific slow afterhyperpolarization inhibitor UCL2077 is a subtype-selective blocker of the epilepsy associated KCNQ channels. Mol Pharmacol. 2010;78:1088–1095. doi: 10.1124/mol.110.066100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker M, Pedarzani P. Differential distribution of three Ca(2+)-activated K(+) channel subunits, SK1, SK2, and SK3, in the adult rat central nervous system. Mol Cell Neurosci. 2000;15:476–493. doi: 10.1006/mcne.2000.0842. [DOI] [PubMed] [Google Scholar]
- Stocker M, Hirzel K, D'hoedt D, Pedarzani P. Matching molecules to function: neuronal Ca2+-activated K+ channels and afterhyperpolarizations. Toxicon. 2004;43:933–949. doi: 10.1016/j.toxicon.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Taylor CW, Broad LM. Pharmacological analysis of intracellular Ca2+ signalling: problems and pitfalls. Trends Pharmacol Sci. 1998;19:370–375. doi: 10.1016/S0165-6147(98)01243-7. [DOI] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel JM. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469–474. doi: 10.1016/S0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi P, Yu H, MacNeil DJ, Van der Ploeg LH, Guan XM. Distribution of orexin receptor mRNA in the rat brain. FEBS Lett. 1998;438:71–75. doi: 10.1016/S0014-5793(98)01266-6. [DOI] [PubMed] [Google Scholar]
- Turunen PM, Jäntti MH, Kukkonen JP. OX1 orexin/hypocretin receptor signaling through arachidonic acid and endocannabinoid release. Mol Pharmacol. 2012;82:156–167. doi: 10.1124/mol.112.078063. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, Nicoll RA. Contribution of KCNQ2 and KCNQ3 to the medium and slow afterhyperpolarization currents. Proc Natl Acad Sci U S A. 2008;105:19974–19979. doi: 10.1073/pnas.0810535105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzingounis AV, Heidenreich M, Kharkovets T, Spitzmaul G, Jensen HS, Nicoll RA, Jentsch TJ. The KCNQ5 potassium channel mediates a component of the afterhyperpolarization current in mouse hippocampus. Proc Natl Acad Sci U S A. 2010;107:10232–10237. doi: 10.1073/pnas.1004644107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalobos C, Foehring RC, Lee JC, Andrade R. Essential role for phosphatidylinositol 4,5-bisphosphate in the expression, regulation, and gating of the slow afterhyperpolarization current in the cerebral cortex. J Neurosci. 2011;31:18303–18312. doi: 10.1523/JNEUROSCI.3203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogalis F, Storm JF, Lancaster B. SK channels and the varieties of slow after-hyperpolarizations in neurons. Eur J Neurosci. 2003;18:3155–3166. doi: 10.1111/j.1460-9568.2003.03040.x. [DOI] [PubMed] [Google Scholar]
- White JA, Rubinstein JT, Kay AR. Channel noise in neurons. Trends Neurosci. 2000;23:131–137. doi: 10.1016/S0166-2236(99)01521-0. [DOI] [PubMed] [Google Scholar]
- Williams JT, Colmers WF, Pan ZZ. Voltage- and ligand-activated inwardly rectifying currents in dorsal raphe neurons in vitro. J Neurosci. 1988;8:3499–3506. doi: 10.1523/JNEUROSCI.08-09-03499.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willie JT, Chemelli RM, Sinton CM, Tokita S, Williams SC, Kisanuki YY, Marcus JN, Lee C, Elmquist JK, Kohlmeier KA, Leonard CS, Richardson JA, Hammer RE, Yanagisawa M. Distinct narcolepsy syndromes in orexin receptor-2 and orexin null mice: molecular genetic dissection of non-REM and REM sleep regulatory processes. Neuron. 2003;38:715–730. doi: 10.1016/S0896-6273(03)00330-1. [DOI] [PubMed] [Google Scholar]
- Yamasaki-Mann M, Demuro A, Parker I. Modulation of endoplasmic reticulum Ca2+ store filling by cyclic ADP-ribose promotes inositol trisphosphate (IP3)-evoked Ca2+ signals. J Biol Chem. 2010;285:25053–25061. doi: 10.1074/jbc.M109.095257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Ferguson AV. Orexin-A depolarizes nucleus tractus solitarius neurons through effects on nonselective cationic and K+ conductances. J Neurophysiol. 2003;89:2167–2175. doi: 10.1152/jn.01088.2002. [DOI] [PubMed] [Google Scholar]
- Yuan W, Burkhalter A, Nerbonne JM. Functional role of the fast transient outward K+ current IA in pyramidal neurons in (rat) primary visual cortex. J Neurosci. 2005;25:9185–9194. doi: 10.1523/JNEUROSCI.2858-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Kolaj M, Renaud LP. Ca2+-dependent and Na+-dependent K+ conductances contribute to a slow AHP in thalamic paraventricular nucleus neurons: a novel target for orexin receptors. J Neurophysiol. 2010;104:2052–2062. doi: 10.1152/jn.00320.2010. [DOI] [PubMed] [Google Scholar]