

Abstract
Brain-derived neurotrophic factor (BDNF) signaling in the dorsolateral striatum (DLS) keeps alcohol intake in moderation. For example, activation of the BDNF receptor tropomyosin receptor kinase B (TrkB) in the DLS reduces intake in rats that consume moderate amounts of alcohol. Here, we tested whether long-term excessive consumption of alcohol produces neuroadaptations in BDNF signaling in the rat DLS. We found that BDNF was no longer able to gate alcohol self-administration after a history of repeated cycles of binge alcohol drinking and withdrawal. We then elucidated the possible neuroadaptations that could block the ability of BDNF to keep consumption of alcohol in moderation. We report that intermittent access to 20% alcohol in a two-bottle choice paradigm that models excessive alcohol drinking produces a mobilization of DLS p75 neurotrophin receptor (p75NTR), whose activities oppose those of the Trk receptors, including TrkB. These neuroadaptations were not observed in the DLS of rats exposed to continuous access to 10% alcohol or in rats consuming sucrose. Furthermore, short hairpin RNA (shRNA)-mediated knockdown of the p75NTR gene in the DLS, as well as intra-DLS infusion or systemic administration of the p75NTR modulator, LM11A-31, significantly reduced binge drinking of alcohol. Together, our results suggest that excessive alcohol consumption produces a change in BDNF signaling in the DLS, which is mediated by the recruitment of p75NTR. Our data also imply that modulators of p75NTR signaling could be developed as medications for alcohol abuse disorders.
SIGNIFICANCE STATEMENT Neuroadaptations gate or drive excessive, compulsive alcohol drinking. We previously showed that brain-derived neurotrophic factor and its receptor, TrkB, in the dorsolateral striatum (DLS), are part of an endogenous system that keeps alcohol drinking in moderation. Here, we show that a history of excessive alcohol intake produces neuroadaptations in the DLS that preclude BDNF's ability to gate alcohol self-administration in rats by the recruitment of the low-affinity neurotrophin receptor, p75NTR, whose activities opposes those of the Trk receptors. Finally, we show that the administration of the p75NTR modulator, LM11A-31, significantly reduces excessive alcohol intake suggesting that the drug may be developed as a new treatment for alcohol abuse disorders.
Keywords: addiction, alcohol, BDNF, dorsal striatum, neurotrophic factor, p75NTR
Introduction
Brain-derived neurotrophic factor (BDNF) is a member of the neurotrophins family of growth factors that plays an important role in the survival of neurons as well as in the formation and maturation of synapses (Park and Poo, 2013). BDNF also contributes to long-term potentiation (LTP), enhancement of neurotransmitter release, and in alterations of channel function and spine morphology (Park and Poo, 2013).
BDNF ligation with the receptor tyrosine kinase tropomyosin receptor kinase B (TrkB) results in the activation of the receptor and in the corresponding activation of the phosphatidylinositol-3-kinase, and/or phospholipase C and/or the mitogen activated protein kinase (MAPK) pathways that in turn initiate transcription and/or translation events (Patapoutian and Reichardt, 2001; Yoshii and Constantine-Paton, 2010; Andero et al., 2014). In addition to binding to TrkB, BDNF, like other neurotrophins, can also interact, albeit with lower affinity, with the p75 neurotrophin receptor (p75NTR), a member of the tumor necrosis factor receptor superfamily (Dechant and Barde, 2002; Kraemer et al., 2014a). Interestingly, activation of TrkB and p75NTR produce opposite outcomes (Lu et al., 2005; Kraemer et al., 2014a). For example, activation of TrkB induces spine formation and maturation (Orefice et al., 2013; Deinhardt and Chao, 2014), whereas p75NTR contributes to a reduction in dendritic complexity and spine density (Zagrebelsky et al., 2005). Activation of TrkB in the hippocampus contributes to late phase long-term potentiation (Messaoudi et al., 2002; Lu et al., 2008; Minichiello, 2009; Panja and Bramham, 2014), whereas hippocampal p75NTR enhances long-term depression (Woo et al., 2005). In addition, TrkB mediates neuronal survival (Andero et al., 2014; Deinhardt and Chao, 2014), whereas p75NTR was reported to induce apoptosis (Woo et al., 2005). Finally, TrkB and p75NTR play opposing roles in fear acquisition and anxiety in mice (Olsen et al., 2013).
Whereas the BDNF/TrkB signaling plays a pivotal role on neuronal functions, dysregulation of the pathway have been associated with various psychiatric disorders (Autry and Monteggia, 2012), including depression (Kerman, 2012), schizophrenia (Buckley et al., 2007), and anxiety disorders (Andero et al., 2014). Evidence also suggests a role for BDNF in mediating critical features underlying cocaine use disorder (Li and Wolf, 2015). Interestingly, malfunctioning of the BDNF signaling pathway has also been linked to alcohol abuse disorders (AUDs). Serum BDNF levels are negatively correlated with the severity of withdrawal signs in alcohol-dependent subjects (Heberlein et al., 2010; Costa et al., 2011), and DNA methylation of the BDNF gene is increased in the serum of alcoholics compared to healthy controls (Heberlein et al., 2015). Furthermore, human carriers of the Val66Met polymorphism show earlier onset of alcohol addiction (Matsushita et al., 2004), reduced gray matter during abstinence in alcohol-dependent subjects (Mon et al., 2013), increased alcohol consumption (Colzato et al., 2011), increased propensity to relapse in alcoholics seeking treatment (Wojnar et al., 2009), and increased alcohol abuse phenotypes in adolescence (Nees et al., 2015). In line with these finding, we found that mice carrying the polymorphism exhibit a compulsive alcohol intake phenotype (Warnault et al., 2016). Together, these data suggest that dysfunction of the BDNF pathway produces adverse phenotypes that are associated with alcohol abuse disorder.
Since malfunctioning of the BDNF signaling pathway is associated with AUD, then the normal function of BDNF may be to delay or prevent the development of alcohol addiction. In line with this possibility, we demonstrated that moderate consumption of alcohol increases BDNF, but not nerve growth factor (NGF) expression in the dorsal striatum (McGough et al., 2004), and specifically in the dorsolateral striatum (DLS) of rodents (Jeanblanc et al., 2009; Logrip et al., 2009). Short hairpin RNA (shRNA)-mediated knockdown of the BDNF gene increases mice and rats intake of moderate (10%) amounts of alcohol (Jeanblanc et al., 2009; Logrip et al., 2015). In contrast, intra-DLS infusion of BDNF produces a dose-dependent decrease of rat operant self-administration of moderate (10%) levels of alcohol (Jeanblanc et al., 2009, 2013). BDNF gates the level of alcohol self-administration through the activation of TrkB (McGough et al., 2004; Logrip et al., 2008; Jeanblanc et al., 2013), resulting in the activation of MAPK (Logrip et al., 2008; Jeanblanc et al., 2013) and in the transcription of preprodynorphin (Logrip et al., 2008) and dopamine D3 receptors (Jeanblanc et al., 2006). Together, these results suggest that BDNF is a pivotal molecular contributor to mechanisms that prevent pathological use of alcohol. Here, we tested the hypothesis that the transition from moderate to excessive drinking occurs, in part, when the BDNF signaling pathway in the DLS ceases to function properly.
Materials and Methods
Materials
Rabbit anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:2000), goat anti-actin (1:2000), rabbit anti-phospho-ERK (pY204; 1:2000) and mouse anti-ERK (1:5000) antibodies were purchased from Santa Cruz Biotechnologies. The donkey horseradish peroxidase (HRP)-conjugated secondary antibodies (1:1000) were purchased from Jackson ImmunoResearch. Rabbit anti-p75NTR (1:500), anti-TrkB (1:1000), and mouse anti-NeuN (1:500) antibodies were purchased from EMD Millipore. Rabbit anti-phospho-TrkB (pY490; 1:200) antibodies were purchased from Cell Signaling Technology. Mouse anti-glial fibrillary acidic protein (GFAP; 1:1000) antibodies, phosphatase inhibitor cocktails 1 and 2, and primers for the PCR analysis were purchased from Sigma-Aldrich. Enhanced chemiluminescence (ECL) was purchased from GE Healthcare. The secondary antibodies Alexa Fluor 488-labeled donkey anti-rabbit and Alexa Fluor 594-labeled donkey anti-mouse and NuPAGE Bis-Tris pre-casted gels were purchased from Invitrogen. Rabbit (1:5000) anti-green fluorescence protein (GFP) antibodies were purchased from Abcam. The bicinchoninic acid (BCA) protein assay kit was obtained from Pierce. Human BDNF was purchased from Sigma-Aldrich. LM11A-31 was custom synthesized by Ricerca Biosciences.
Animals
Male Long–Evans rats (250 g at the time of purchase) were obtained from Harlan. Animals were housed individually under a light/dark cycle of 12 h, with lights on at 7:00 A.M. and food and water available ad libitum. All animal procedures were approved by the University of California, San Francisco (UCSF) Gallo Center and the UCSF Institutional Animal Care and Use Committee, and were conducted in agreement with the Guide for the Care and Use of Laboratory Animals (National Research Council), Gallo Center and Association for Assessment and Accreditation of Laboratory Animal Care (UCSF) guidelines.
Preparation of solutions
Alcohol solution for the drinking experiments was prepared from absolute anhydrous alcohol (190 proof) diluted to 10% or 20% alcohol (v/v) in tap water. For systemic administration, alcohol was diluted to 20% alcohol (v/v) in saline (0.9% NaCl; Hospira). Sucrose solution was diluted to 1% sucrose (v/v) in tap water.
Production of lentivirus expressing shRNA targeting p75NTR
The 21 nucleotide shRNA sequence targeting p75NTR (shp75NTR) 5′-GGA CCT ATC TGA GCT GAA AGC-3′ was selected based on a previous study (Vilar et al., 2009). A scrambled sequence (shSCR), 5′-GAA GCA ACT CGT CTG GAC AGT-3′ was designed using a public siDesign website (http://www.sirnawizard.com/scrambled.php) and was used as a nontargeting shRNA control. Both sequences were incorporated into a previously described stem-loop structure (Rubinson et al., 2003). Synthesized oligonucleotides were annealed and subcloned into the HpaI and XhoI restriction sites in the recombinant lentiviral vector pLL3.7, which also expresses enhanced GFP (EGFP). The expression of shRNA was driven by the U6 promoter and the expression of EGFP by the CMV promoter. The pLL3.7-shp75NTR or pLL3.7-shSCR plasmid was transfected into HEK293T (Clonetech) cells along with the packaging plasmids, psPAX2 and pMD2.G, using Lipofectamine 2000. Forty-eight hours after transfection, the supernatant was collected, and lentiviral particles were purified by ultracentrifugation (26000 rpm, 90 min at 4°C) as described previously (Lasek et al., 2007). Titers were determined using the HIV-1 p24 antigen ELISA kit (ZeptoMetrix) per the manufacturer's instructions. The titer of viruses used in the studies was 2 × 107 pg/ml.
Preparation of synaptosomal fraction
The DLS and the dorsomedial striatum (DMS) were dissected, and crude synaptosomal fractions were prepared as described by Gibb et al. (2011). Immediately after being collected, tissue was homogenized in a glass homogenizer containing 500 μl of ice-cold Krebs-sucrose buffer [containing (in mm) 125 NaCl, 1.2 KCl, 1.2 MgSO4 1.2 CaCl2, 22 Na2CO3, 1.2 NaH2PO4, 10 glucose, and 320 sucrose, pH 7.4] in the presence of protease and phosphatase inhibitors. The homogenate was centrifuged at 1000 × g at 4°C for 10 min to pellet heavy membranes and debris (P1). The supernatant (S1) was collected and was centrifuged at 16,000 × g at 4°C for 20 min to pellet the crude synaptosomal fraction (P2). P2 was resuspended in 100 μl RIPA buffer. Total protein concentration was determined using BCA protein assay kit.
Western blot analyses
Equal amounts of homogenates from individual rats (25 μg) were resolved on NuPAGE 4–12% Bis-Tris gels and transferred onto nitrocellulose membranes. Blots were incubated for 1 h at room temperature in blocking solution [5% (w/v) nonfat milk in Tris-buffered saline containing 0.1% (v/v) Tween 20] and then incubated overnight at 4°C in a blocking solution including the primary antibodies. Membranes were then washed and probed with HRP-conjugated secondary antibodies for 2 h at room temperature. Bands were visualized using ECL and quantified by ImageJ (NIH).
Immunochemistry
Four weeks after virus infusion, rats were deeply anesthetized with Euthasol (Virbac) and perfused with 0.9% NaCl, followed by 4% paraformaldehyde (PFA) in phosphate buffer, pH 7.4. Brains were removed and fixed in the same fixative for 2 h, and then transferred to PBS at 4°C. The following day, brains were transferred into 30% sucrose and stored at 4°C until the brain sank to the bottom of the tube. Frozen 50-μm-thick coronal sections were cut on a cryostat (Microm; Thermo Fisher Scientific) and collected into 24-well dishes. Free-floating sections containing the injection site in the DLS were selected. Coronal sections were blocked with 5% normal donkey serum in PBS for 1 h and then incubated for 24 h at 4°C on an orbital shaker with antibodies for either a neuronal marker (anti-NeuN antibody) or a glial marker (anti-GFAP antibody) in combination with the anti-GFP antibody, diluted in PBS plus 3% bovine serum albumin and 0.05% Triton X-100. The sections were then washed three times for 5 min each in PBS followed by incubation for 4 h with the secondary antibodies Alexa Fluor 488-labeled donkey anti-rabbit and Alexa Fluor 594-labeled donkey anti-mouse (both at 1:500). After staining, the sections were washed three times for 5 min each in PBS and mounted in Vectashield mounting medium. For each subject, the infected area was verified in 50 μm coronal sections using Zeiss LSM 510 META laser confocal microscope.
Systemic administration of alcohol
Rats were habituated to the intraperitoneal administration procedure with a daily injection of saline (0.9% NaCl) for 3 d. Rats then received an intraperitoneal administration of alcohol (1.5 g/kg) or saline 30 min before brain tissue collection.
Excessive alcohol intake: intermittent access to 20% alcohol
The two-bottle choice (2BC) drinking paradigm is described in detail in previous reports (Carnicella et al., 2009, 2014). Briefly, at 8 weeks of age, animals were given 24 h of access to one bottle of 20% alcohol (v/v) and one bottle of tap water concurrently for a total of 21 sessions of 24 h during a period of 7 weeks. Drinking sessions started at 12:00 P.M. on Monday, Wednesday, and Friday, with 24 or 48 h (weekend) of alcohol deprivation between the drinking sessions. The water and alcohol bottles were weighed before the beginning of each drinking session, immediately after the first 30 min of access to alcohol and at the end of the session. Rats with escalation of alcohol consumption from session 1 to session 18 reaching a baseline of drinking that was higher than 3.5 g/kg/24 h were selected for the studies. Between 65% to 70% of the rats showed escalation of drinking. Rats that did not meet the latter criteria were excluded from the study.
Moderate alcohol intake: continuous access (CA) to 10% alcohol
The paradigm was conducted as described by He et al. (2005). Briefly, rats underwent continuous access to one bottle of 10% alcohol (v/v) and one bottle of water in their home cage for 21 consecutive days (3 weeks). Alcohol and water consumption were recorded daily. CA10%-2BC does not lead to escalation of alcohol intake; i.e., there is no difference in the amount of consumed alcohol between day 1 and day 18 (N.M. and D.R., unpublished observation) and all rats were included in the study.
Operant self-administration of alcohol
After 5 weeks of the intermittent access (IA) to 20% alcohol, 2BC paradigm (IA20%-2BC), rats were trained to self-administer alcohol. The self-administration chambers (Med-Associates) contain two levers: an active lever (the alcohol lever), for which presses result in the delivery of 0.1 ml of the alcohol solution (v/v), and an inactive lever, for which presses are counted but no alcohol solution is delivered. Rats were trained under a fixed ratio 3 schedule; i.e., rats had to press three times to receive one reward. Sessions lasted for the duration of 1 h at the beginning of the training and 30 min after the first 2 weeks. The number of presses on the levers and the number of alcohol deliveries were recorded using MED-PC IV software (Med-Associates).
Intermittent access to sucrose
The procedure was similar to the IA20%-2BC paradigm described above, except that a sucrose (1%) solution was substituted for alcohol. Rats were tested for sucrose intake in response to intra-DLS administration of LM11A-31 or vehicle 1 week after the conclusion of the alcohol-drinking study. Animals were trained to drink sucrose for 2 weeks before the test day. An independent cohort of rats underwent IA-2BC sucrose-drinking paradigm for 7 weeks before systemic administration of LM11A-31.
Surgical procedures
Rats with a stable baseline level of drinking in the alcohol self-administration or IA-20%-2BC paradigms underwent surgeries under isoflurane (Baxter) anesthesia. Rats were weighed and monitored daily after the surgery to ensure the health and well being of each animal.
Infusion of virus.
The procedure was conducted as described previously by Jeanblanc et al. (2009). Two holes were drilled above the sites of injection to allow the introduction of injector cannula (acute internal cannula, C315IA; diameter, 33G; PlasticsOne). The injectors were connected to 25 μl Hamilton syringes (#1702), and infusions were controlled by an automatic pump (Harvard Apparatus). Rats received three injections of 1 μl of solution containing the virus per side at a rate of 0.2 μl/min. Injectors remained in place for 10 additional min. The coordinates used for the injections into the DLS were all relative to Bregma and were as follows: injection 1, +1.2 mm anterior, 3.5 mm lateral to the medial suture, −4.5 mm ventral to the skull surface; injection 2, +0.2 mm anterior, 3.5 mm lateral to the medial suture, −5.5 mm ventral to the skull surface; injection 3, +0.2 mm anterior, 3.5 mm lateral to the medial suture, −4.5 mm ventral to the skull surface. After 1 week of recovery, alcohol-drinking procedure was resumed.
Cannula implantation and microinfusions of BDNF and LM11A-31.
The procedure was conducted as described previously by Jeanblanc et al. (2009). Four holes were drilled for screws, and two holes were drilled for cannulae implantation (single cannula, C315GA; PlasticsOne). The coordinates for the DLS were +1.2 mm anterior to Bregma and 3.5 mm lateral to the medial suture. The cannulae were implanted into the lateral part of the dorsal striatum (-4.2 mm from the skull surface) and fixed with dental cement. After 1 week of recovery, the alcohol-drinking procedure resumed, and subjects were habituated to the microinjection procedure with two sham injections (injectors were lowered but no infusion took place). The experimental microinfusions began after each rat reacquired a stable level of drinking. BDNF, LM11A-31, or PBS was infused via a 25 μl Hamilton syringe (#1702), 3 h (BDNF) or 2 h (LM11A-31) before the beginning of the alcohol-drinking session. BDNF (0.75 μg/1 μl) or LM11A-31 (30 μg/1 μl) was infused over 2 min in each side of the brain. The injectors extended 0.5 mm below the tip of the cannula and remained in position for an additional 2 min to allow for full diffusion from the injector tip.
In the experiment testing alcohol self-administration after intra-DLS administration of BDNF (Fig. 1), each concentration of alcohol was self-administered for a total period of 2 weeks, and microinfusions were performed 4 d after the change in alcohol concentration. All subjects received each treatment in a counterbalanced manner within each alcohol concentration.
Figure 1.
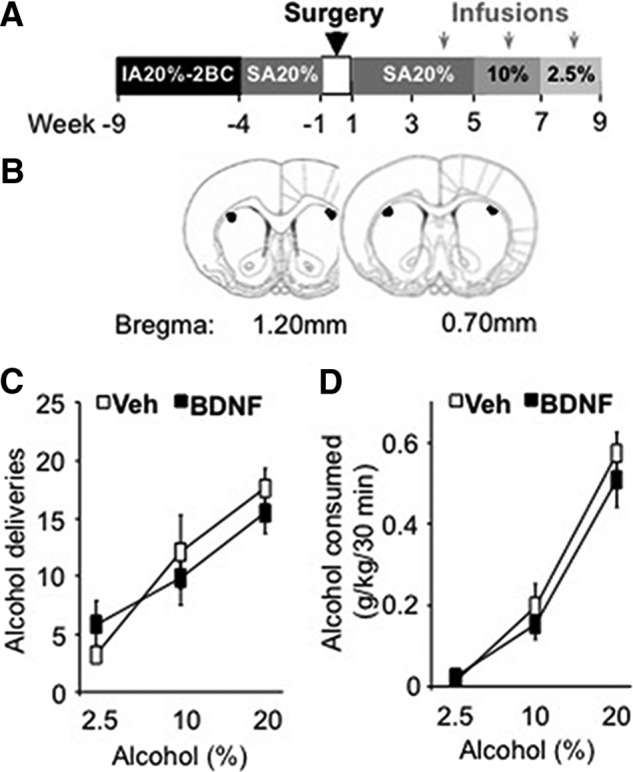
Intra-DLS infusion of BDNF does not reduce alcohol self-administration in rats with a history of excessive alcohol drinking. A, Rats underwent 5 weeks of an IA20%-2BC paradigm and were then trained to lever press for a 20% alcohol solution during 30 min sessions. BDNF (0.75 μg/μl) or vehicle (Veh; PBS) was infused into the DLS 3 h before the beginning of the operant self-administration sessions. The concentration of alcohol was decreased (from 20 to 10% and from 10 to 2.5%) every 2 weeks to allow the rats to reach a new baseline of consumption before BDNF administration. B, Schematic drawings of coronal sections of the rat brain showing the placement of bilateral infusion sites in the DLS (Paxinos and Watson, 2007). C, D, Data are expressed as the mean ± SEM of the number of alcohol deliveries (C) and alcohol consumed in grams per kilogram (D). n = 7 per treatment.
In the experiments testing intra-DLS administration of LM11A-31, all subjects received each treatment in a counterbalanced manner, with one microinfusion per week. Two alcohol-drinking sessions without treatment were carried out between drug administration to allow intake to return to baseline.
Systemic administration of LM11A-31
Before the beginning of experiments, rats were habituated for 3 d to intraperitoneal administration by daily injections of saline (0.9% NaCl). LM11A-31 (50–150 mg/kg) or vehicle (0.9% saline) was administered intraperitoneally 2 h before the beginning of the drinking session. Doses used in the study were based on a previous report (Tep et al., 2013). Test days occurred once a week, with two alcohol-drinking sessions without treatment that were performed between injections to allow intake to return to baseline. For the highest dose of LM11A-31, vehicle and drug were tested in different animals.
Histology
Rats implanted with cannulae were perfused transcardially with 4% PFA. Fixed brains were frozen, and 50 μm coronal slices were cut and examined for cannulae placement using a bright-field microscope.
Data analysis
Data are expressed as mean ± SEM. Biochemical data were analyzed with a two-tailed unpaired t test. Behavioral data were analyzed with a two-tailed paired or unpaired t test or by one- or two-way ANOVA with or without repeated measures (RM), depending on the experiment, followed by the Student–Newman–Keuls (SNK) test when indicated by significant effects of treatments or interactions. Statistical significance was set at p < 0.05.
Results
BDNF in the DLS does not alter alcohol consumption in rats with a history of excessive alcohol drinking and withdrawal
We showed previously that the administration of exogenous BDNF into the DLS reduces operant alcohol self-administration in rats with history of moderate (10%) alcohol drinking, whereas knockdown of BDNF in the DLS increases 10% alcohol self-administration and intake (Jeanblanc et al., 2009, 2013; Logrip et al., 2015). These data suggest that the BDNF signaling pathway in the DLS gates the level of alcohol intake. To test the possibility that BDNF signaling in the DLS is disrupted after chronic excessive alcohol drinking, we first tested the ability of the neurotrophic factor to control alcohol self-administration in rats with a history of excessive alcohol drinking. We used the IA20%-2BC drinking paradigm to induce escalation to excessive alcohol consumption (Carnicella et al., 2014), and after 5 weeks of binge and withdrawal, rats were trained to self-administer alcohol in an operant self-administration paradigm (Carnicella et al., 2014; Fig. 1A). After achieving a stable baseline, BDNF (0.75 μg/side) was infused into the DLS (Fig. 1B) 3 h before the session and the self-administration of three alcohol concentrations was measured. We found that intra-DLS administration of BDNF did not decrease the number of alcohol deliveries (Fig. 1C; two-way RM-ANOVA, treatment effect, F(1,12) = 0.11, p = 0.74) or the amount of alcohol consumed (Fig. 1D; two-way RM-ANOVA, treatment effect, F(1,12) = 0.53, p = 0.48), independently of the concentrations of alcohol (2.5, 10, and 20%). These findings suggest that repeated access to high amounts of alcohol together with repeated episodes of withdrawal preclude the ability of BDNF in the DLS to gate the level of alcohol self-administration.
p75NTR localization in the DLS is altered in rats with a history of excessive alcohol drinking and withdrawal
As described above, the beneficial actions of BDNF to reduce alcohol self-administration are abolished after repeated cycles of binge drinking and withdrawal. It is well established that chronic alcohol intake produces molecular adaptations that contribute to the development or maintenance of alcohol-drinking behaviors (Ron and Barak, 2016). We therefore hypothesized that long-term exposure to alcohol alters BDNF/TrkB signaling in the DLS.
We first examined the levels of TrkB in the DLS of rats that were subjected to repeated cycles of binge drinking and withdrawal. Specifically, after 7 weeks of IA20%-2BC, the DLS (Fig. 2A) was dissected after the first 30 min of alcohol access (binge), immediately at the end of the alcohol-drinking session (end) or after 24 h of alcohol deprivation (withdrawal; Fig. 2B). Individual alcohol consumption values before dissection are reported in Table 1. No differences were observed in total homogenate levels for TrkB after alcohol binge drinking (Fig. 2C; t(22) = −0.57, p = 0.58), at the end of the last 24 h of alcohol-drinking session (Fig. 2E; t(14) = 1.14, p = 0.28), or after 24 h of withdrawal (Fig. 2G; t(14) = 1.57, p = 0.14). The levels of TrkB in the synaptosomal fraction were also unaltered in the DLS of alcohol-drinking animals following binge (Fig. 2D; t(22) = −1.50, p = 0.15), 24 h of consumption (Fig. 2F; t(14) = 1.96, p = 0.07), and after 24 h of withdrawal (Fig. 2H; t(14) = 0.56, p = 0.58) compared to control animals that consumed water only. Finally, we measured the levels of TrkB phosphorylation as well as the phosphorylation of ERK1/2 in total homogenate and found that neither TrkB nor ERK1/2 were phosphorylated, and thus activated, in response to binge alcohol drinking (p-TrkB, t(7) = 1.23, p = 0.26, n = 4–5/group; p-ERK1/2, t(15) = 1.20, p = 0.24, n = 8–9) or after 24 h of withdrawal (p-TrkB, t(6) = 0.42, p = 0.21, n = 4/group; p-ERK1/2, t(12) = 1.65, p = 0.13, n = 7/group; data not shown). Together, these data suggest that long-term excessive drinking and withdrawal do not alter the expression or activity of TrkB.
Figure 2.

Excessive alcohol drinking alters the synaptosomal localization of p75NTR but not TrkB in the DLS. A, Schematic drawings of coronal sections of the rat brain showing the DLS slices in black (Paxinos and Watson, 2007). B, Schematic representation of dissection timeline. Alc, Alcohol; W, water. C–H, Rats received IA20%-2BC (black) or water only (white) for 7 weeks. DLS of high-drinking rats (alcohol intake equal to or >3.5 g/kg/24 h) were dissected immediately after the last 30 min (binge; C, D), at the end of the last 24 h drinking session (end; E, F), or after 24 h of withdrawal (WD; G, H). TrkB and p75NTR levels in total homogenates (C, E, G) and in synaptosomal fractions (D, F, H) were measured by Western blot analysis. Histograms depict the mean ratio of TrkB or p75NTR to actin ± SEM, and values are expressed as percentages of water controls. ***p < 0.0001 versus water (unpaired t test). n = 10–11 (C, D) and 8 per time point of dissection (E–H).
Table 1.
Individual alcohol intake data for the biochemical experiments
| Rat number | Last drinking session (g/kg/24 h) | Binge (g/kg/30 min) | ||
|---|---|---|---|---|
| Figs. 2C,D and 4B,C | Drinking procedure, IA20%-2BC; harvest time point, 30 min binge | 1 (Figs. 2, 4) | 11.17 | 0.30 |
| 2 (Figs. 2, 4) | 7.25 | 0.07 | ||
| 3 (Figs. 2, 4) | 4.79 | 0.43 | ||
| 4 (Figs. 2, 4) | 5.19 | 0.31 | ||
| 5 (Figs. 2, 4) | 9.70 | 3.06 | ||
| 6 (Figs. 2, 4) | 6.35 | 2.54 | ||
| 7 (Figs. 2, 4) | 5.35 | 1.24 | ||
| 8 (Figs. 2, 4) | 4.05 | 0.30 | ||
| 9 (Fig. 2) | 6.05 | 1.22 | ||
| 10 (Fig. 2) | 9.69 | 1.59 | ||
| Mean ± SEM | 6.96 ± 0.77 | 1.11 ± 0.33 |
| Rat number | Last drinking session (g/kg/24 h) | |||
|---|---|---|---|---|
| Figs. 2E,F and 4D,E | Drinking procedure, IA20%-2BC; harvest time point, end of drinking | 1 | 9.05 | |
| 2 | 5.24 | |||
| 3 | 4.91 | |||
| 4 | 4.29 | |||
| 5 | 7.40 | |||
| 6 | 3.81 | |||
| 7 | 5.87 | |||
| 8 | 4.00 | |||
| Mean ± SEM | 5.57 ± 0.64 |
| Rat number | Last drinking session (g/kg/24 h) | |||
|---|---|---|---|---|
| Fig. 2G,H | Drinking procedure, IA20%-2BC; harvest time point, 24 h withdrawal | 1 | 5.28 | |
| 2 | 7.18 | |||
| 3 | 3.53 | |||
| 4 | 3.75 | |||
| 5 | 9.06 | |||
| 6 | 6.50 | |||
| 7 | 5.35 | |||
| 8 | 4.81 | |||
| Mean ± SEM | 5.68 ± 0.65 |
| Rat number | Last drinking session (g/kg/24 h) | |||
|---|---|---|---|---|
| Fig. 3C,D | Drinking procedure, CA10%-2BC; harvest time point, end of drinking | 1 | 0.98 | |
| 2 | 1.85 | |||
| 3 | 1.28 | |||
| 4 | 1.07 | |||
| 5 | 1.06 | |||
| 6 | 0.28 | |||
| 7 | 0.68 | |||
| 8 | 0.22 | |||
| Mean ± SEM | 0.93 ± 0.19 |
| Rat number | Last drinking session (g/kg/24 h) | Binge (g/kg/30 min) | ||
|---|---|---|---|---|
| Fig. 3E,F | Drinking procedure, IA1% sucrose-2BC; harvest time point, 30 min binge | 1 | 45.99 | 1.45 ml/kg/30 min |
| 2 | 47.77 | 4.58 ml/kg/30 min | ||
| 3 | 54.48 | 6.72 ml/kg/30 min | ||
| 4 | 44.71 | 8.75 ml/kg/30 min | ||
| Mean ± SEM | 48.24 ± 2.17 | 5.38 ± 1.56 ml/kg/30 min |
Next, we examined whether a history of excessive alcohol intake alters the levels and or localization of the low-affinity neurotrophin receptor p75NTR. The protein levels of p75NTR in total homogenate of DLS were unaltered in response to a binge drinking session (Fig. 2C; t(21) = 1.99, p = 0.06), at the end of the last 24 h of alcohol-drinking session (Fig. 2E; t(14) = 1.81, p = 0.09), or after 24 h of withdrawal (Fig. 2G; t(14) = −1.87, p = 0.08). In contrast, a 30 min binge alcohol-drinking session decreased the synaptosomal localization of p75NTR in the DLS (Fig. 2D; t(21) = 12.18, p < 0.0001), whereas the membranal level of p75NTR was significantly increased at the end of the final 24 h drinking session compared to water-consuming rats (Fig. 2F; t(14) = −5.18, p < 0.0001), which was maintained even after 24 h of withdrawal (Fig. 2H; t(14) = −10.10, p < 0.0001). These results suggest that the synaptosomal localization of p75NTR is dynamic, changing over the course of a 24 h drinking session and withdrawal.
Expression levels and localization of p75NTR are unaltered in the DLS after acute alcohol administration and in response to moderate alcohol consumption or sucrose intake
Next, we examined whether the alterations in p75NTR localization are a consequence of the history of high levels of voluntary alcohol intake or are due to the acute pharmacological actions of alcohol per se. To do so, we tested p75NTR levels in the rat DLS after an acute systemic administration of a nonhypnotic dose of alcohol (1.5 g/kg). Acute alcohol exposure did not alter the total protein levels of p75NTR (Fig. 3A; t(14) = −0.42, p = 0.68), and the synaptosomal levels of the receptor were also unaltered (Fig. 3B; t(14) = 0.99, p = 0.34). TrkB levels in both the total homogenate (Fig. 3A; t(14) = 1.08, p = 0.30) and the synaptosomal fraction (Fig. 3B; t(14) = −0.29, p = 0.78) were also unaffected in response to the systemic administration of alcohol.
Figure 3.

Systemic administration of alcohol, moderate alcohol intake, or sucrose consumption do not alter BDNF receptors expression or localization in the DLS. A, B, Rats received a systemic administration of alcohol (Alc; 1.5 g/kg, i.p.; black) or saline (Sal; white), and the DLS was dissected 30 min later. C, D, Rats received continuous access to 10% alcohol (CA10%; black) or water only (white) for 21 d, and the DLS was dissected immediately after the last drinking session. E, F, Rats received 7 weeks of IA of 1% sucrose (Suc) or water only, and the DLS was dissected immediately after the last 30 min drinking session. TrkB and p75NTR levels in total homogenate (A, C, E) or in the synaptosomal fraction (B, D, F) were measured by Western blot analysis. Histograms show mean ratio of TrkB or p75NTR to actin ± SEM, and values are expressed as percentages of water controls. n = 8 per treatment (A, B); n = 8 (C, D) and 4 per drinking regimen (E, F).
To examine whether the alterations in the synaptosomal p75NTR levels are specific to excessive alcohol drinking and are not a result of prolonged alcohol exposure per se, we measured the levels of both receptors in the DLS of rats that underwent a paradigm of moderate alcohol intake. Rats experienced CA10%-2BC for 21 d (i.e., for the same number of alcohol access sessions as the rats that underwent an IA-20%-2BC paradigm), and the DLS was dissected at the end of the last 24 h drinking session. The individual alcohol consumption values before dissection are reported in Table 1. The levels of both receptors in the total homogenate (Fig. 3C; TrkB, t(14) = 1.39, p = 0.19; p75NTR, t(14) = 1.62, p = 0.13) and in the synaptosomal fraction (Fig. 3D; TrkB, t(14) = 0.20, p = 0.84; p75NTR, t(14) = −1.35, p = 0.20) were unaltered in the DLS of rats consuming moderate levels of alcohol compared to control water-only-consuming rats.
Finally, we determined whether the consumption of another rewarding substance (e.g., sucrose) attenuates the expression and/or localization of the BDNF receptors. Rats underwent 7 weeks of IA1% sucrose-2BC, and the DLS was dissected immediately after the last 30 min of drinking. Individual sucrose consumption values before dissection are reported in Table 1. No changes were observed in the total (Fig. 3E; TrkB, t(6) = 0.54, p = 0.61; p75NTR, t(6) = −0.47, p = 0.65) or synaptosomal (Fig. 3F; TrkB, t(6) = −1.45, p = 0.20; p75NTR, t(6) = −1.37, p = 0.22) homogenates of the DLS of sucrose-drinking rats compared to water-only-consuming animals. Together, these results demonstrate that the changes in p75NTR localization are specific to a regime of alcohol consumption, i.e., a history of high, binge-like levels of intake with intermittent access and withdrawal periods.
TrkB and p75NTR expression and localization are unaltered in the DMS in response to alcohol consumption
To examine whether altered p75NTR localization was specific to the DLS, we tested whether the expression and/or localization of TrkB and p75NTR are altered in response to alcohol in the DMS (Fig. 4A), a striatal region where endogenous BDNF does not control alcohol self-administration (Jeanblanc et al., 2009). Individual alcohol consumption values before dissection are reported in Table 1. The total levels of TrkB (Fig. 4B; t(14) = −0.07, p = 0.94) and p75NTR (Fig. 4B; t(14) = −0.70, p = 0.49) and the synaptosomal localization of both receptors (Fig. 4C; TrkB, t(14) = −2. 092, p = 0.06; p75NTR; t(14) = −0.46, p = 0.65) were unchanged in the DMS of rats after a 30 min binge alcohol intake session. Similarly, no change was observed after a 24 h alcohol-drinking session in the total homogenate (Fig. 4D; TrkB, t(14) = 2.2, p = 0.06; p75NTR, t(14) = −0.05, p = 0.96) and synaptosomal fraction (Fig. 4E; TrkB, t(14) = −0.91, p = 0.38; p75NTR, t(14) = −0.06, p = 0.95). Together, these results suggest that the changes in p75NTR localization in response to binge drinking of, and withdrawal from, excessive alcohol intake are specific to the DLS.
Figure 4.

Excessive alcohol intake does not alter BDNF receptors expression or localization in the DMS. A, Schematic drawings of coronal sections of the rat brain showing the DMS in black (Paxinos and Watson, 2007). B–E, Rats received IA20%-2BC for 7 weeks (black) or water only (white), and the DMS was dissected immediately after the last 30 min drinking session (binge; B, C) or at the end of the 24 h drinking session (end; D, E). TrkB and p75NTR levels in total homogenates (B, D) or in the synaptosomal fraction (C, E) were measured by Western blot analysis. Histograms show the mean ratio of TrkB or p75NTR to actin ± SEM, and values are expressed as percentages of water controls. n = 8 per time point of dissection.
Knockdown of p75NTR expression attenuates excessive alcohol drinking
The BDNF/TrkB signaling pathway in the DLS limits the levels of moderate alcohol drinking (Jeanblanc et al., 2009, 2013), whereas, as shown above, BDNF ceases to function properly after repeated cycle of excessive alcohol drinking and withdrawal. Since a history of alcohol drinking and withdrawal produced dynamic changes in the synaptosomal localization of p75NTR in the DLS, and because p75NTR has opposing actions to TrkB (Dechant and Barde, 2002; Messaoudi et al., 2002; Lu et al., 2005, 2008; Orefice et al., 2013; Deinhardt and Chao, 2014; Panja and Bramham, 2014), we hypothesized that the breakdown in BDNF signaling upon excessive alcohol consumption may be due to the increased functional contribution of p75NTR. To test this possibility, we generated a lentivirus expressing EGFP and a shRNA sequence targeting p75NTR (Ltv-shp75NTR), as well as a virus expressing a scrambled p75NTR shRNA sequence (Ltv-shSCR). Infusion of Ltv-shp75NTR into the DLS of rats led to a high level of virus infection in DLS neurons but not in glia, as shown by colocalization of GFP and NeuN but not GFAP staining (Fig. 5A). The protein levels of p75NTR were significantly reduced in the DLS of rats infected with the Ltv-shp75NTR compared with rats infected with Ltv-shSCR control, as measured 4 weeks after virus infusion (Fig. 5B; t(6) = 5.40, p < 0.001). Next, we tested the consequences of p75NTR knockdown on alcohol intake. After 7 weeks of the IA20%-2BC paradigm, rats received a bilateral infusion of either Ltv-shSCR or Ltv-shp75NTR into the DLS, and alcohol intake was determined 4 weeks after virus infection (Fig. 5C). As shown in Figure 5, D and E, knockdown of p75NTR in the DLS significantly reduced binge-like alcohol drinking (Fig. 5D; t(12) = 3.62, p < 0.01), without affecting concurrent water intake (Fig. 5E; t(12) = 0.52, p = 0.62). Thus, p75NTR in the DLS contributes to the maintenance of excessive alcohol intake.
Figure 5.
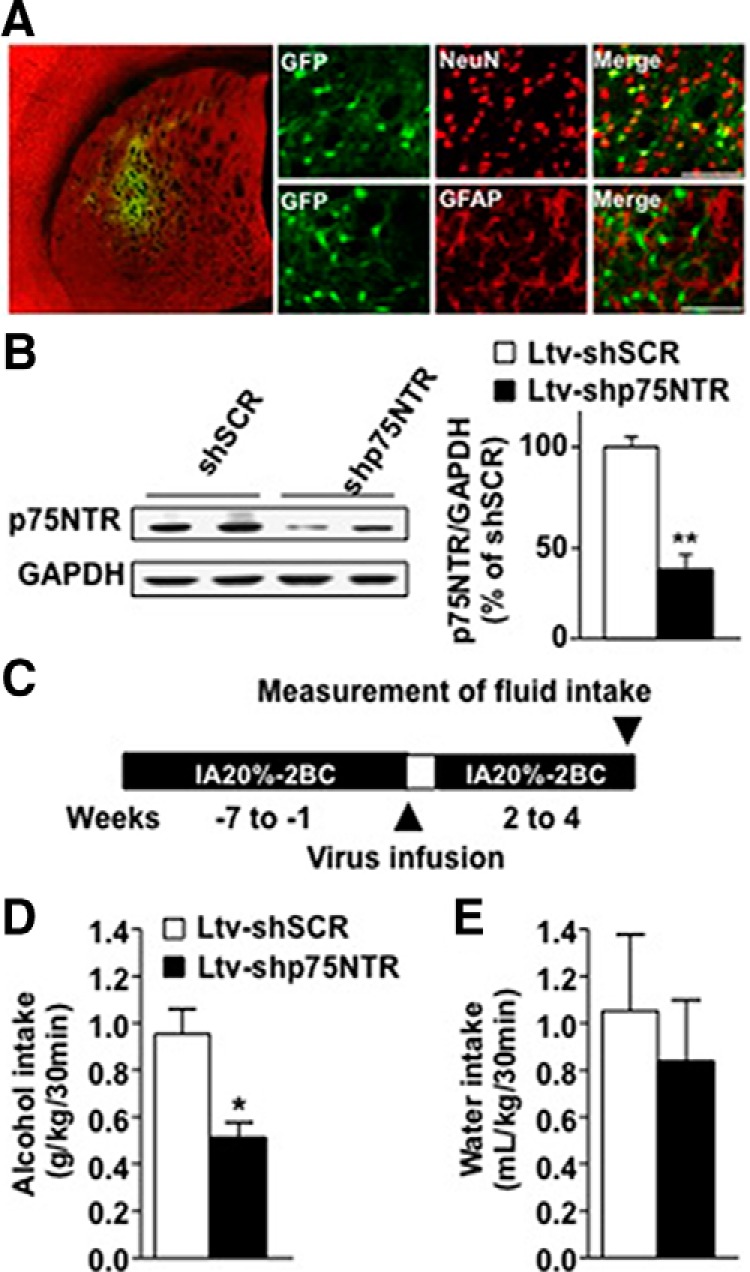
Knockdown of p75NTR in the DLS reduces binge drinking of alcohol. A, B, Ltv-shp75NTR or Ltv-shSCR was bilaterally infused at a titer of 2 × 107 pg/ml into the DLS. A, Ltv-shp75NTR infects DLS neurons. Four weeks after virus infusion, slices containing the DLS were costained with anti-GFP (green) and anti-NeuN (red) or costained with anti-GFP and anti-GFAP antibodies. The left image depicts the specificity of the site of infection. The right images depict Ltv-shp75NTR infection of neurons (costaining of GFP with NeuN; top) but not glia (costaining GFP with GFAP; bottom). B, Ltv-shp75NTR-mediated knockdown of p75NTR. The DLS was dissected 4 weeks after virus infusion, and p75NTR and GAPDH protein levels were measured by Western blot analysis. Data are expressed as mean ratio of p75NTR to GAPDH ± SEM and presented as percentages of Ltv-shSCR control. C, Schematic representation of the behavioral experiment. Rats underwent IA20%-2BC alcohol for 7 weeks. High-drinking rats (baseline level of alcohol intake equal or higher than 3.5 g/kg/24 h) received a bilateral infusion of Ltv-shSCR or Ltv-shp75NTR into the DLS, and after 1 week of recovery, the alcohol-drinking procedure resumed. D, E, Alcohol intake (grams per kilogram per 30 min; D) and water intake (milliliters per kilogram per 30 min; E) were recorded 4 weeks after viral infection. Results are expressed as mean ± SEM. *p < 0.01; **p < 0.001 versus Ltv-shSCR (unpaired t test). n = 4 (B) or 7 per treatment (D, E).
The p75NTR modulator, LM11A-31, reduces excessive alcohol consumption
The small nonpeptide p75NTR ligand LM11A-31 modulates p75NTR signaling (Massa et al., 2006; Longo and Massa, 2013; Tep et al., 2013) by disrupting the binding between p75NTR and neurotrophins including BDNF and by modulating its intrinsic signaling actions (Massa et al., 2006; Longo and Massa, 2013). LM11A-31 blocks p75NTR-mediated neuronal death (Knowles et al., 2013; Tep et al., 2013) and decreases the phosphorylation of Jun kinase, a downstream effector of p75NTR (Shi et al., 2013). We therefore tested whether LM11A-31 reduces excessive alcohol drinking. To test this possibility, LM11A-31 was infused into the DLS (Fig. 6A) 2 h before the beginning of a 30 min alcohol binge-drinking session. As shown in Figure 6, B and C, intra-DLS administration of LM11A-31 significantly reduced binge-like alcohol intake (Fig. 6B; t(10) = 2.35, p < 0.05) without changing concurrent water intake (Fig. 6C; t(10) = 0.76, p = 0.47). Importantly, the effect of LM11A-31 on alcohol intake was specific, as sucrose intake was unaltered upon infusion of the drug into the DLS (Fig. 6D; sucrose, t(9) = 0.85, p = 0.41; E, water, t(9) = −0.73, p = 0.45). These results indicate that the reduction of binge alcohol drinking by LM11A-31 was unlikely to be due to nonspecific motor or reward sensitivity changes.
Figure 6.
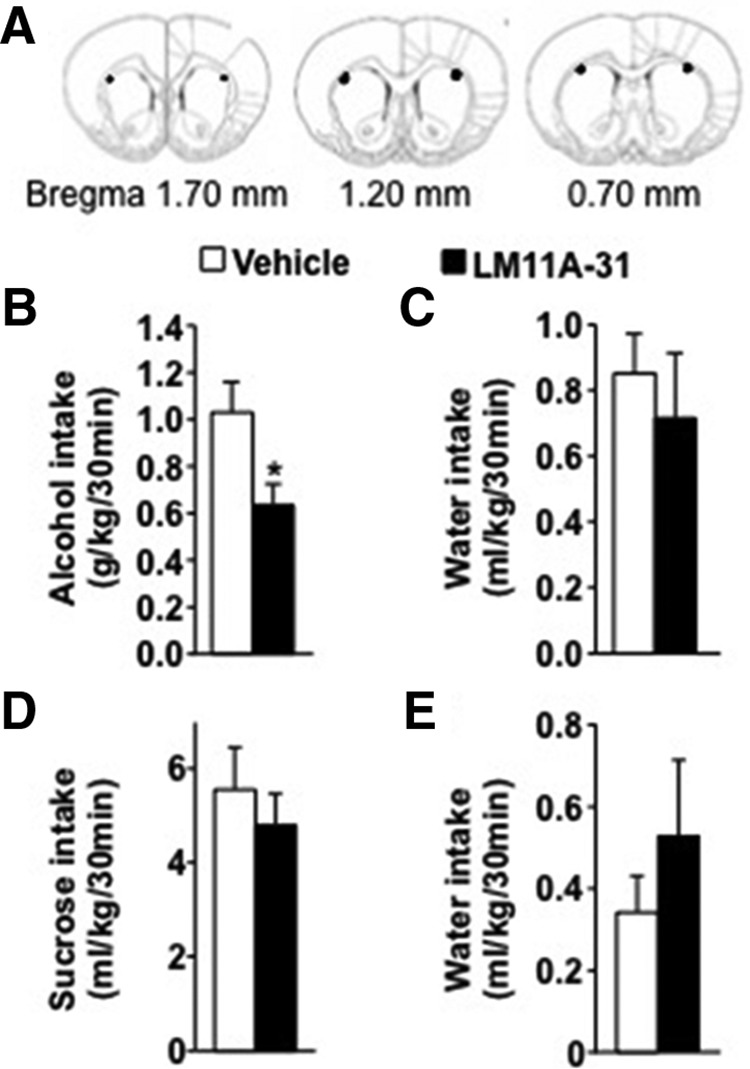
Intra-DLS infusion of LM11A-31 reduces binge drinking of alcohol but not sucrose. A–E, Rats underwent IA20%-2BC for 7 weeks, and bilateral guide cannula were then surgically implanted in the DLS. After 1 week of recovery, rats were administered with LM11A-31 (30 μg/μl; Tep et al., 2013) or vehicle (PBS) into the DLS 2 h before the beginning of the drinking session and consumption was recorded. A, Schematic drawings of coronal sections of the rat brain showing the placement of bilateral infusion sites in the DLS (Paxinos and Watson, 2007). B, C, Alcohol (grams per kilogram per 30 min; B) and water (milliliters per kilogram per 30 min; C) intake after LM11A-31 or vehicle infusion. D, E, After 1 week of withdrawal from alcohol consumption, rats underwent 2 weeks of IA-2BC of 1% sucrose and then received intra-DLS administration of LM11A-31 (30 μg/μl) or vehicle. Sucrose consumption is presented as milliliters per kilogram per min; D and water (milliliters per kilogram per 30 min; E). Results are expressed as mean ± SEM. *p < 0.05 versus vehicle (paired t test). n = 11 (B, C) and 10 per treatment (D, E).
Finally, we examined the possibility that LM11A-31 could potentially be developed for the treatment of alcohol abuse disorders by testing the effects of systemic administration of LM11A-31 on alcohol intake. As shown in Figure 7, modulation of p75NTR-mediated signaling significantly attenuated binge drinking of alcohol (Fig. 7A; one-way ANOVA, F(3,56) = 7.12, p < 0.001) at the dose of 150 mg/kg (p < 0.01), without altering water intake (Fig. 7B; one-way ANOVA, F(3,56) = 2.07, p = 0.11). We further tested whether systemic administration of LM11A-31 attenuates binge drinking of sucrose. To do so, rats had IA to 1% sucrose in a 2BC paradigm for 7 weeks, and sucrose consumption was measured after 30 min access following LM11A-31 injection. As shown in Figure 7C, LM11A-31 (150 mg/kg) did not alter binge consumption of sucrose, and the corresponding water intake was also unaltered (Fig. 7D; sucrose, t(13) = 0.93, p = 0.37; water, t(13) = 0.39, p = 0.71). Together these data suggest that pharmacological targeting of p75NTR is a potential treatment of alcohol abuse disorder by attenuating alcohol intake without affecting the consumption of a natural rewarding substance.
Figure 7.
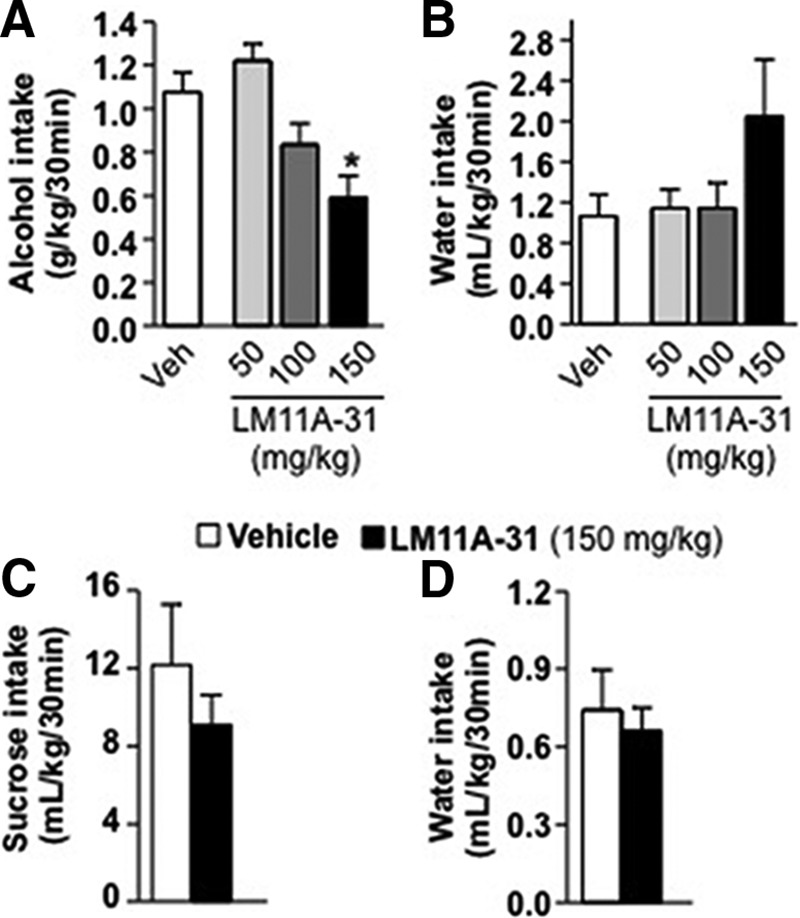
Systemic administration of LM11A-31 reduces binge drinking of alcohol but not sucrose. A–D, Rats underwent an IA-2BC paradigm with 20% alcohol (A, B) or 1% sucrose (C, D) for 7 weeks. LM11A-31 (50–150 mg/kg) or vehicle (0.9% NaCl) was administered intraperitoneally 2 h before the beginning of the drinking session. Alcohol (grams per kilogram per 30 min; A) and water (milliliters per kilogram per 30 min; B) or sucrose (C) and water (both milliliters per kilogram per 30 min; D) intake were recorded. Results are expressed as mean ± SEM. *p < 0.01 versus vehicle (one-way ANOVA with SNK post hoc test). A, B, Vehicle, n = 24 per dose; LM11A-31, n = 12 per dose; C, D, n = 7–8 per treatment.
Discussion
Here, we report that BDNF in the DLS is no longer able to gate the levels of alcohol intake in rats with a history of long-term excessive drinking, an effect that is concomitant with a dynamic change in the synaptosomal localization of p75NTR, which interacts with the NGF family of growth factors, including BDNF (Lu et al., 2005). We further demonstrate that blocking p75NTR signaling within the DLS with shRNA-mediated knockdown of the receptor, or by using the p75NTR modulator LM11A-31, dampened alcohol intake. Our findings suggest that the BDNF/TrkB pathway in the DLS ceases to participate in mechanisms that gate alcohol self-administration in rats with a history of excessive drinking. Our data further suggest that the neurotrophin receptor p75NTR, which signals in part through BDNF, contributes to mechanisms that drive excessive alcohol drinking.
We found that synaptosomal p75NTR levels were increased in the DLS at the end of a 24 h alcohol access or after 24 h of withdrawal compared to water-only-consuming rats. In contrast, a 30 min binge alcohol-drinking session reduced p75NTR levels in the synaptosomal fraction. As p75NTR levels were not altered in the total DLS homogenates, these data suggest that the receptors cycle in and out of the synaptosomal membrane in response to excessive alcohol intake and withdrawal. Activation of the p75NTR-mediated signaling cascade causes receptor internalization (Bronfman et al., 2003). Thus, it is plausible that the forward movement of p75NTR at the end of a 24 h withdrawal session (e.g., at the beginning of a binge alcohol-drinking session) allows the activation of the p75NTR signaling pathway, which then leads to a rapid internalization and increased intracellular content of the receptor.
How does p75NTR contribute to the maintenance of excessive alcohol drinking? One possibility is that the secreted BDNF interacts with p75NTR. However, another intriguing possibility is that p75NTR mediates its actions by binding to proBDNF, the BDNF precursor, which under certain conditions is released from neurons (Teng et al., 2005; Woo et al., 2005; Hempstead, 2014). Thus, it is of interest to determine whether repeated cycles of binge drinking and withdrawal alter the processing of the growth factor to its mature form. p75NTR signals through the other members of the NGF family of neurotrophic factors (Kraemer et al., 2014a), and thus we cannot exclude the possibility that p75NTR's actions to drive excessive alcohol intake are mediated via other neurotrophic factors. p75NTR signals through downstream cytoplasmic effectors (Kraemer et al., 2014b), activating several pathways, including nuclear factor-κB (Carter et al., 1996), Jun kinase (Casaccia-Bonnefil et al., 1996), the small G-proteins RhoA (Sun et al., 2012; Orefice et al., 2016) and RAC (Tian et al., 2014), as well as via ceramide signaling (Dobrowsky et al., 1994). Thus, it is likely that in response to excessive alcohol intake, membranal p75NTR recruits these effectors or others, and the involvement of downstream mediators of p75NTR signaling merits further investigation. Interestingly, p75NTR signaling was shown to counteract TrkB signaling (Song et al., 2010; Chapleau and Pozzo-Miller, 2012); thus, it is plausible that the recruitment of p75NTR in the DLS in response to excessive alcohol intake attenuates TrkB signaling.
Lentivirus-mediated knockdown of p75NTR specifically in DLS neurons produced a reduction of binge drinking, suggesting a role for neuronal p75NTR in mechanisms underlying excessive alcohol intake. However, synaptosomal fractions contain glial plasma membranes, and p75NTR is moderately expressed in glia cells including astrocytes (Cragnolini and Friedman, 2008). Astrocytic signaling has been shown to contribute to adverse phenotypes associated with alcohol exposure (Lee et al., 2013; Bull et al., 2014; Robinson et al., 2014). Since the fractionation method used herein cannot differentiate between possible changes in p75NTR localization in neurons versus glia, we cannot exclude the possibility that p75NTR in nonneuronal cells such as astrocytes contributes to the maintenance of excessive alcohol drinking. Furthermore, our protocol does not differentiate between presynaptosomal and postsynaptosomal membranes, and further studies will be aimed to address this point.
The underlying behavioral mechanism by which p75NTR signaling contributes to excessive alcohol drinking is unknown. One possibility is that p75NTR promotes the reinforcing effects of alcohol itself. Alternatively, it is plausible that p75NTR exaggerates the negative affective-like states associated with alcohol withdrawal (Koob, 2013). Furthermore, the DLS plays an essential role in habitual alcohol seeking (Corbit et al., 2012) since the molecular mechanisms underlying habit formation of alcohol taking are not well understood. Thus, it would be of interest to determine whether the recruitment of p75NTR in the DLS by alcohol participates in the formation of habit of alcohol seeking.
We show that the administration of the p75NTR modulator LM11A-31 decreased excessive alcohol drinking to levels of intake of ∼0.6 g/kg/30 min, that corresponds to blood alcohol concentration of <30 mg% which is equivalent to one or two drinks of alcohol in humans (Carnicella et al., 2014). Furthermore, we show that knockdown of p75NTR levels in the DLS as well as intra-DLS and systemic administration of LM11A-31 reduced alcohol drinking without affecting the consumption of water or sucrose solution, ruling out nonspecific effects of the manipulations on locomotion or general reward sensitivity. LM11A-31 has therapeutic potential for the treatment of Alzheimer's disease and spinal cord injury (Zeng et al., 2011; Longo and Massa, 2013; Tep et al., 2013); thus, our findings suggest that LM11A-31 may also be a potential new treatment for alcohol abuse disorders and specifically for harmful excessive alcohol use (Enoch and Goldman, 2002).
The raw data can be found at https://portal.g-node.org/data/. Readers can access the data by creating an account and searching for “Darcq.”
Footnotes
This work was supported by NIAAA Grants R01 AA016848, R37 AA01684, and P50 AA017072 (D.R.), the Jean Perkins Foundation (F.L.), and VA Merit Grant I01BX000267 (S.M.M.). We thank Samuel Sakhai for reading this manuscript.
F.M.L. and S.M.M. are listed as inventors on patents relating to a compound in this report, which are assigned to the University of North Carolina, University of California, San Francisco, and the Department of Veterans Affairs. They are entitled to royalties distributed by the assigned universities per their standard agreements. F.M.L. is a principal of, and has financial interest in, PharmatrophiX, a company focused on the development of small molecule ligands for neurotrophin receptors, which has licensed several of these patents.
References
- Andero R, Choi DC, Ressler KJ. BDNF-TrkB receptor regulation of distributed adult neural plasticity, memory formation, and psychiatric disorders. Prog Mol Biol Trans Sci. 2014;122:169–192. doi: 10.1016/B978-0-12-420170-5.00006-4. [DOI] [PubMed] [Google Scholar]
- Autry AE, Monteggia LM. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev. 2012;64:238–258. doi: 10.1124/pr.111.005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronfman FC, Tcherpakov M, Jovin TM, Fainzilber M. Ligand-induced internalization of the p75 neurotrophin receptor: a slow route to the signaling endosome. J Neurosci. 2003;23:3209–3220. doi: 10.1523/JNEUROSCI.23-08-03209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley PF, Mahadik S, Pillai A, Terry A., Jr Neurotrophins and schizophrenia. Schizophr Res. 2007;94:1–11. doi: 10.1016/j.schres.2007.01.025. [DOI] [PubMed] [Google Scholar]
- Bull C, Freitas KC, Zou S, Poland RS, Syed WA, Urban DJ, Minter SC, Shelton KL, Hauser KF, Negus SS, Knapp PE, Bowers MS. Rat nucleus accumbens core astrocytes modulate reward and the motivation to self-administer ethanol after abstinence. Neuropsychopharmacology. 2014;39:2835–2845. doi: 10.1038/npp.2014.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnicella S, Amamoto R, Ron D. Excessive alcohol consumption is blocked by glial cell line-derived neurotrophic factor. Alcohol. 2009;43:35–43. doi: 10.1016/j.alcohol.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnicella S, Ron D, Barak S. Intermittent ethanol access schedule in rats as a preclinical model of alcohol abuse. Alcohol. 2014;48:243–252. doi: 10.1016/j.alcohol.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhäuser N, Böhm-Matthaei R, Baeuerle PA, Barde YA. Selective activation of NF-kappa B by nerve growth factor through the neurotrophin receptor p75. Science. 1996;272:542–545. doi: 10.1126/science.272.5261.542. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature. 1996;383:716–719. doi: 10.1038/383716a0. [DOI] [PubMed] [Google Scholar]
- Chapleau CA, Pozzo-Miller L. Divergent roles of p75NTR and Trk receptors in BDNF's effects on dendritic spine density and morphology. Neural Plast. 2012;2012:578057. doi: 10.1155/2012/578057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colzato LS, Van der Does AJ, Kouwenhoven C, Elzinga BM, Hommel B. BDNF Val66Met polymorphism is associated with higher anticipatory cortisol stress response, anxiety, and alcohol consumption in healthy adults. Psychoneuroendocrinology. 2011;36:1562–1569. doi: 10.1016/j.psyneuen.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Corbit LH, Nie H, Janak PH. Habitual alcohol seeking: time course and the contribution of subregions of the dorsal striatum. Biol Psychiatry. 2012;72:389–395. doi: 10.1016/j.biopsych.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa MA, Girard M, Dalmay F, Malauzat D. Brain-derived neurotrophic factor serum levels in alcohol-dependent subjects 6 months after alcohol withdrawal. Alcohol Clin Exp Res. 2011;35:1966–1973. doi: 10.1111/j.1530-0277.2011.01548.x. [DOI] [PubMed] [Google Scholar]
- Cragnolini AB, Friedman WJ. The function of p75NTR in glia. Trends Neurosci. 2008;31:99–104. doi: 10.1016/j.tins.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Dechant G, Barde YA. The neurotrophin receptor p75(NTR): novel functions and implications for diseases of the nervous system. Nat Neurosci. 2002;5:1131–1136. doi: 10.1038/nn1102-1131. [DOI] [PubMed] [Google Scholar]
- Deinhardt K, Chao MV. Trk receptors. Handb Exp Pharmacol. 2014;220:103–119. doi: 10.1007/978-3-642-45106-5_5. [DOI] [PubMed] [Google Scholar]
- Dobrowsky RT, Werner MH, Castellino AM, Chao MV, Hannun YA. Activation of the sphingomyelin cycle through the low-affinity neurotrophin receptor. Science. 1994;265:1596–1599. doi: 10.1126/science.8079174. [DOI] [PubMed] [Google Scholar]
- Enoch MA, Goldman D. Problem drinking and alcoholism: diagnosis and treatment. Am Fam Physician. 2002;65:441–448. [PubMed] [Google Scholar]
- Gibb SL, Hamida SB, Lanfranco MF, Ron D. Ethanol-induced increase in Fyn kinase activity in the dorsomedial striatum is associated with subcellular redistribution of protein tyrosine phosphatase alpha. J Neurochem. 2011;119:879–889. doi: 10.1111/j.1471-4159.2011.07485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He DY, McGough NN, Ravindranathan A, Jeanblanc J, Logrip ML, Phamluong K, Janak PH, Ron D. Glial cell line-derived neurotrophic factor mediates the desirable actions of the anti-addiction drug ibogaine against alcohol consumption. J Neurosci. 2005;25:619–628. doi: 10.1523/JNEUROSCI.3959-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heberlein A, Muschler M, Wilhelm J, Frieling H, Lenz B, Gröschl M, Kornhuber J, Bleich S, Hillemacher T. BDNF and GDNF serum levels in alcohol-dependent patients during withdrawal. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:1060–1064. doi: 10.1016/j.pnpbp.2010.05.025. [DOI] [PubMed] [Google Scholar]
- Heberlein A, Büscher P, Schuster R, Kleimann A, Lichtinghagen R, Rhein M, Kornhuber J, Bleich S, Frieling H, Hillemacher T. Do changes in the BDNF promoter methylation indicate the risk of alcohol relapse? Eur Neuropsychopharmacol. 2015;25:1892–1897. doi: 10.1016/j.euroneuro.2015.08.018. [DOI] [PubMed] [Google Scholar]
- Hempstead BL. Deciphering proneurotrophin actions. Handb Exp Pharmacol. 2014;220:17–32. doi: 10.1007/978-3-642-45106-5_2. [DOI] [PubMed] [Google Scholar]
- Jeanblanc J, He DY, McGough NN, Logrip ML, Phamluong K, Janak PH, Ron D. The dopamine D3 receptor is part of a homeostatic pathway regulating ethanol consumption. J Neurosci. 2006;26:1457–1464. doi: 10.1523/JNEUROSCI.3786-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanblanc J, He DY, Carnicella S, Kharazia V, Janak PH, Ron D. Endogenous BDNF in the dorsolateral striatum gates alcohol drinking. J Neurosci. 2009;29:13494–13502. doi: 10.1523/JNEUROSCI.2243-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanblanc J, Logrip ML, Janak PH, Ron D. BDNF-mediated regulation of ethanol consumption requires the activation of the MAP kinase pathway and protein synthesis. Eur J Neurosci. 2013;37:607–612. doi: 10.1111/ejn.12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerman IA. New insights into BDNF signaling: relevance to major depression and antidepressant action. Am J Psychiatry. 2012;169:1137–1140. doi: 10.1176/appi.ajp.2012.12081053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles JK, Simmons DA, Nguyen TV, Vander Griend L, Xie Y, Zhang H, Yang T, Pollak J, Chang T, Arancio O, Buckwalter MS, Wyss-Coray T, Massa SM, Longo FM. Small molecule p75NTR ligand prevents cognitive deficits and neurite degeneration in an Alzheimer's mouse model. Neurobiol Aging. 2013;34:2052–2063. doi: 10.1016/j.neurobiolaging.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. Theoretical frameworks and mechanistic aspects of alcohol addiction: alcohol addiction as a reward deficit disorder. Curr Top Behav Neurosci. 2013;13:3–30. doi: 10.1007/7854_2011_129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer BR, Yoon SO, Carter BD. The biological functions and signaling mechanisms of the p75 neurotrophin receptor. Handb Exp Pharmacol. 2014a;220:121–164. doi: 10.1007/978-3-642-45106-5_6. [DOI] [PubMed] [Google Scholar]
- Kraemer BR, Snow JP, Vollbrecht P, Pathak A, Valentine WM, Deutch AY, Carter BD. A role for the p75 neurotrophin receptor in axonal degeneration and apoptosis induced by oxidative stress. J Biol Chem. 2014b;289:21205–21216. doi: 10.1074/jbc.M114.563403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasek AW, Janak PH, He L, Whistler JL, Heberlein U. Downregulation of mu opioid receptor by RNA interference in the ventral tegmental area reduces ethanol consumption in mice. Genes Brain Behav. 2007;6:728–735. doi: 10.1111/j.1601-183X.2007.00303.x. [DOI] [PubMed] [Google Scholar]
- Lee MR, Ruby CL, Hinton DJ, Choi S, Adams CA, Young Kang N, Choi DS. Striatal adenosine signaling regulates EAAT2 and astrocytic AQP4 expression and alcohol drinking in mice. Neuropsychopharmacology. 2013;38:437–445. doi: 10.1038/npp.2012.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wolf ME. Multiple faces of BDNF in cocaine addiction. Behav Brain Res. 2015;279:240–254. doi: 10.1016/j.bbr.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logrip ML, Janak PH, Ron D. Dynorphin is a downstream effector of striatal BDNF regulation of ethanol intake. FASEB J. 2008;22:2393–2404. doi: 10.1096/fj.07-099135. [DOI] [PubMed] [Google Scholar]
- Logrip ML, Janak PH, Ron D. Escalating ethanol intake is associated with altered corticostriatal BDNF expression. J Neurochem. 2009;109:1459–1468. doi: 10.1111/j.1471-4159.2009.06073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logrip ML, Barak S, Warnault V, Ron D. Corticostriatal BDNF and alcohol addiction. Brain Res. 2015;1628:60–67. doi: 10.1016/j.brainres.2015.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo FM, Massa SM. Small-molecule modulation of neurotrophin receptors: a strategy for the treatment of neurological disease. Nat Rev Drug Discov. 2013;12:507–525. doi: 10.1038/nrd4024. [DOI] [PubMed] [Google Scholar]
- Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nat Rev Neurosci. 2005;6:603–614. doi: 10.1038/nrn1726. [DOI] [PubMed] [Google Scholar]
- Lu Y, Christian K, Lu B. BDNF: a key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol Learn Mem. 2008;89:312–323. doi: 10.1016/j.nlm.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa SM, Xie Y, Yang T, Harrington AW, Kim ML, Yoon SO, Kraemer R, Moore LA, Hempstead BL, Longo FM. Small, nonpeptide p75NTR ligands induce survival signaling and inhibit proNGF-induced death. J Neurosci. 2006;26:5288–5300. doi: 10.1523/JNEUROSCI.3547-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita S, Kimura M, Miyakawa T, Yoshino A, Murayama M, Masaki T, Higuchi S. Association study of brain-derived neurotrophic factor gene polymorphism and alcoholism. Alcohol Clin Exp Res. 2004;28:1609–1612. doi: 10.1097/01.ALC.0000145697.81741.D2. [DOI] [PubMed] [Google Scholar]
- McGough NN, He DY, Logrip ML, Jeanblanc J, Phamluong K, Luong K, Kharazia V, Janak PH, Ron D. RACK1 and brain-derived neurotrophic factor: a homeostatic pathway that regulates alcohol addiction. J Neurosci. 2004;24:10542–10552. doi: 10.1523/JNEUROSCI.3714-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messaoudi E, Ying SW, Kanhema T, Croll SD, Bramham CR. Brain-derived neurotrophic factor triggers transcription-dependent, late phase long-term potentiation in vivo. J Neurosci. 2002;22:7453–7461. doi: 10.1523/JNEUROSCI.22-17-07453.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci. 2009;10:850–860. doi: 10.1038/nrn2738. [DOI] [PubMed] [Google Scholar]
- Mon A, Durazzo TC, Gazdzinski S, Hutchison KE, Pennington D, Meyerhoff DJ. Brain-derived neurotrophic factor genotype is associated with brain gray and white matter tissue volumes recovery in abstinent alcohol-dependent individuals. Genes Brain Behav. 2013;12:98–107. doi: 10.1111/j.1601-183X.2012.00854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nees F, Witt SH, Dinu-Biringer R, Lourdusamy A, Tzschoppe J, Vollstädt-Klein S, Millenet S, Bach C, Poustka L, Banaschewski T, Barker GJ, Bokde AL, Bromberg U, Büchel C, Conrod PJ, Frank J, Frouin V, Gallinat J, Garavan H, Gowland P, et al. BDNF Val66Met and reward-related brain function in adolescents: role for early alcohol consumption. Alcohol. 2015;49:103–110. doi: 10.1016/j.alcohol.2014.12.004. [DOI] [PubMed] [Google Scholar]
- Olsen D, Kaas M, Schwartz O, Nykjaer A, Glerup S. Loss of BDNF or its receptors in three mouse models has unpredictable consequences for anxiety and fear acquisition. Learn Memory. 2013;20:499–504. doi: 10.1101/lm.032045.113. [DOI] [PubMed] [Google Scholar]
- Orefice LL, Waterhouse EG, Partridge JG, Lalchandani RR, Vicini S, Xu B. Distinct roles for somatically and dendritically synthesized brain-derived neurotrophic factor in morphogenesis of dendritic spines. J Neurosci. 2013;33:11618–11632. doi: 10.1523/JNEUROSCI.0012-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orefice LL, Shih CC, Xu H, Waterhouse EG, Xu B. Control of spine maturation and pruning through proBDNF synthesized and released in dendrites. Mol Cell Neurosci. 2016;71:66–79. doi: 10.1016/j.mcn.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panja D, Bramham CR. BDNF mechanisms in late LTP formation: a synthesis and breakdown. Neuropharmacology. 2014;76:664–676. doi: 10.1016/j.neuropharm.2013.06.024. [DOI] [PubMed] [Google Scholar]
- Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013;14:7–23. doi: 10.1038/nrc3653. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol. 2001;11:272–280. doi: 10.1016/S0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Amsterdam: Elsevier Academic; 2007. [Google Scholar]
- Robinson G, Most D, Ferguson LB, Mayfield J, Harris RA, Blednov YA. Neuroimmune pathways in alcohol consumption: evidence from behavioral and genetic studies in rodents and humans. Int Rev Neurobiol. 2014;118:13–39. doi: 10.1016/B978-0-12-801284-0.00002-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Barak S. Molecular mechanisms underlying alcohol-drinking behaviours. Nat Rev Neurosci. 2016;17:576–591. doi: 10.1038/nrn.2016.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Zhang M, Ihrig MM, McManus MT, Gertler FB, Scott ML, Van Parijs L. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- Shi J, Longo FM, Massa SM. A small molecule p75(NTR) ligand protects neurogenesis after traumatic brain injury. Stem Cells. 2013;31:2561–2574. doi: 10.1002/stem.1516. [DOI] [PubMed] [Google Scholar]
- Song W, Volosin M, Cragnolini AB, Hempstead BL, Friedman WJ. ProNGF induces PTEN via p75NTR to suppress Trk-mediated survival signaling in brain neurons. J Neurosci. 2010;30:15608–15615. doi: 10.1523/JNEUROSCI.2581-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Lim Y, Li F, Liu S, Lu JJ, Haberberger R, Zhong JH, Zhou XF. ProBDNF collapses neurite outgrowth of primary neurons by activating RhoA. PLoS One. 2012;7:e35883. doi: 10.1371/journal.pone.0035883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng HK, Teng KK, Lee R, Wright S, Tevar S, Almeida RD, Kermani P, Torkin R, Chen ZY, Lee FS, Kraemer RT, Nykjaer A, Hempstead BL. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci. 2005;25:5455–5463. doi: 10.1523/JNEUROSCI.5123-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tep C, Lim TH, Ko PO, Getahun S, Ryu JC, Goettl VM, Massa SM, Basso M, Longo FM, Yoon SO. Oral administration of a small molecule targeted to block proNGF binding to p75 promotes myelin sparing and functional recovery after spinal cord injury. J Neurosci. 2013;33:397–410. doi: 10.1523/JNEUROSCI.0399-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Tep C, Benedick A, Saidi N, Ryu JC, Kim ML, Sadasivan S, Oberdick J, Smeyne R, Zhu MX, Yoon SO. p75 regulates Purkinje cell firing by modulating SK channel activity through Rac1. J Biol Chem. 2014;289:31458–31472. doi: 10.1074/jbc.M114.589937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilar M, Charalampopoulos I, Kenchappa RS, Simi A, Karaca E, Reversi A, Choi S, Bothwell M, Mingarro I, Friedman WJ, Schiavo G, Bastiaens PI, Verveer PJ, Carter BD, Ibáñez CF. Activation of the p75 neurotrophin receptor through conformational rearrangement of disulphide-linked receptor dimers. Neuron. 2009;62:72–83. doi: 10.1016/j.neuron.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnault V, Darcq E, Morisot N, Phamluong K, Wilbrecht L, Massa SM, Longo FM, Ron D. The BDNF valine 68 to methionine polymorphism increases compulsive alcohol drinking in mice that is reversed by tropomyosin receptor kinase B activation. Biol Psychiatry. 2016;79:463–473. doi: 10.1016/j.biopsych.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojnar M, Brower KJ, Strobbe S, Ilgen M, Matsumoto H, Nowosad I, Sliwerska E, Burmeister M. Association between Val66Met brain-derived neurotrophic factor (BDNF) gene polymorphism and post-treatment relapse in alcohol dependence. Alcohol Clin Exp Res. 2009;33:693–702. doi: 10.1111/j.1530-0277.2008.00886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo NH, Teng HK, Siao CJ, Chiaruttini C, Pang PT, Milner TA, Hempstead BL, Lu B. Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat Neurosci. 2005;8:1069–1077. doi: 10.1038/nn1510. [DOI] [PubMed] [Google Scholar]
- Yoshii A, Constantine-Paton M. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev Neurobiol. 2010;70:304–322. doi: 10.1002/dneu.20765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagrebelsky M, Holz A, Dechant G, Barde YA, Bonhoeffer T, Korte M. The p75 neurotrophin receptor negatively modulates dendrite complexity and spine density in hippocampal neurons. J Neurosci. 2005;25:9989–9999. doi: 10.1523/JNEUROSCI.2492-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng F, Lu JJ, Zhou XF, Wang YJ. Roles of p75NTR in the pathogenesis of Alzheimer's disease: a novel therapeutic target. Biochem Pharmacol. 2011;82:1500–1509. doi: 10.1016/j.bcp.2011.06.040. [DOI] [PubMed] [Google Scholar]