

Abstract
Alzheimer's disease (AD) is the most common form of dementia in individuals over the age of 65 years. The most prevalent genetic risk factor for AD is the ε4 allele of apolipoprotein E (ApoE4), and novel AD treatments that target ApoE are being considered. One unresolved question in ApoE biology is whether ApoE is necessary for healthy brain function. ApoE knock-out (KO) mice have synaptic loss and cognitive dysfunction; however, these findings are complicated by the fact that ApoE knock-out mice have highly elevated plasma lipid levels, which may independently affect brain function. To bypass the effect of ApoE loss on plasma lipids, we generated a novel mouse model that expresses ApoE normally in peripheral tissues, but has severely reduced ApoE in the brain, allowing us to study brain ApoE loss in the context of a normal plasma lipid profile. We found that these brain ApoE knock-out (bEKO) mice had synaptic loss and dysfunction similar to that of ApoE KO mice; however, the bEKO mice did not have the learning and memory impairment observed in ApoE KO mice. Moreover, we found that the memory deficit in the ApoE KO mice was specific to female mice and was fully rescued in female bEKO mice. Furthermore, while the AMPA/NMDA ratio was reduced in ApoE KO mice, it was unchanged in bEKO mice compared with controls. These findings suggest that plasma lipid levels can influence cognition and synaptic function independent of ApoE expression in the brain.
SIGNIFICANCE STATEMENT One proposed treatment strategy for Alzheimer's disease (AD) is the reduction of ApoE, whose ε4 isoform is the most common genetic risk factor for the disease. A major concern of this strategy is that an animal model of ApoE deficiency, the ApoE knock-out (KO) mouse, has reduced synapses and cognitive impairment; however, these mice also develop dyslipidemia and severe atherosclerosis. Here, we have shown that genetic restoration of plasma ApoE to wild-type levels normalizes plasma lipids in ApoE KO mice. While this does not rescue synaptic loss, it does completely restore learning and memory in the mice, suggesting that both CNS and plasma ApoE are independent parameters that affect brain health.
Keywords: Alzheimer's, ApoE, ApoE4, cognition, dementia
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder characterized by progressive memory loss and is the most common form of dementia. The primary genetic risk factor for late-onset AD is the ε4 allele of apolipoprotein E (ApoE4), which is present in 15–20% of the population, but has a prevalence of 50–80% in AD patients (Corder et al., 1993). The more common ApoE3 allele is considered to be risk neutral, while the relatively rare ApoE2 allele appears to protect against AD (Corder et al., 1994). Two decades of research into the mechanisms by which ApoE4 contributes to disease pathogenesis has led to a large body of literature on the role of ApoE in the CNS; however, the field remains divided on whether ApoE-directed therapeutics should reduce or increase ApoE levels.
AD is characterized by the buildup of plaques of amyloid-β (Aβ) followed by neurofibrillary tangles of hyperphosphorylated tau. ApoE4 carriers and ApoE4 targeted replacement mice have increased amyloid deposition, which is a major contributor to accelerated disease development in ApoE4 carriers (Holtzman et al., 2000; Scherzer et al., 2004). ApoE mediates this effect by impeding the clearance of Aβ from the CNS, leading to increased deposition of amyloid-laden plaques (Castellano et al., 2011; Verghese et al., 2013). All forms of ApoE impede Aβ clearance; however, ApoE4 has the strongest effect, while ApoE2 has the weakest (Castellano et al., 2011). Haploinsufficiency of ApoE3 or ApoE4 in an Aβ-overproducing mouse model leads to reduced levels of Aβ, indicating that lowering ApoE could be a promising therapeutic target (Kim et al., 2011). Moreover, peripheral administration of an anti-ApoE antibody reduces amyloid deposition and improves cognitive deficits in an AD mouse model (Liao et al., 2014).
Despite the promise of ApoE-lowering therapeutics, two key concerns remain. First, ApoE knock-out (KO) mice exhibit an age-dependent synaptic loss and learning deficit, suggesting a requirement for ApoE for healthy brain aging (Masliah et al., 1995). The current understanding of what causes this synaptic loss is limited. Some studies have suggested that ApoE deficiency leaves neurons susceptible to oxidative damage (Veinbergs et al., 2000; Shea et al., 2002), while others have suggested that loss of ApoE causes impaired blood–brain barrier (BBB) function (Fullerton et al., 2001). Additionally, ApoE knock-out mice exhibit cholinergic dysfunction and tau hyperphosphorylation, which are key findings in AD (Gordon et al., 1995, 1996). Finally, cholesterol supply to neurons via ApoE-containing lipoproteins is an important stimulant of synaptogenesis, and cholesterol depletion may limit synapse development (Mauch et al., 2001). It is likely that a combination of these factors contributes to synaptic dysfunction and loss. A second concern for ApoE-reducing drugs is that ApoE knock-out mice have highly increased plasma lipid levels (Plump et al., 1992; Zhang et al., 1992), which may independently cause synaptic dysfunction and cognitive deficits (Anstey et al., 2008).
Given the potential of reducing brain ApoE levels for treating AD, it is important to determine the cause of synaptic loss in the ApoE knock-out mice and to evaluate any impact this may have on the development of ApoE-reducing therapeutics. To address one aspect of this question, we have used a novel mouse model that fortuitously arose in our colony and in which ApoE is expressed normally in the plasma, but is virtually absent in the brain. This mouse has normal plasma lipid levels, thus allowing us to differentiate the contribution of the separate pools of ApoE to synaptic loss in the ApoE knock-out mice (Fig. 1). We found that brain ApoE KO (bEKO) mice exhibit a synaptic loss and dysfunction similar to that in ApoE KO mice; however, bEKO mice do not exhibit cognitive impairment. Together, these findings suggest that both CNS and plasma ApoE are important for brain function.
Figure 1.

Peripheral and central sources of ApoE. In peripheral tissues, ApoE is expressed primarily by the liver and released into the blood. In the brain, ApoE is expressed primarily by astrocytes, but can also be expressed by neurons and microglia during cell stress or injury (Pitas et al., 1987; Stone et al., 1997; Xu et al., 2008). Typically, ApoE cannot pass through the BBB (Björkhem et al., 1998); thus, the two pools of ApoE are largely separate. In ApoE knock-out mice, the lack of peripheral ApoE leads to a catastrophic increase of lipoprotein particles (Sheng et al., 1998). To study the effect of ApoE loss on synaptic loss in the presence of normal plasma lipids, we have developed an ApoE “brain knockout” (bEKO mice), which has normalized the peripheral ApoE level.
Materials and Methods
Mice.
Animals were group housed in a standard 12 h light/dark cycle and were fed standard chow ad libitum. All animal care protocols were followed in accordance with the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center.
The bEKO mouse was generated as the result of the random integration of a human ApoE expression cassette containing an inadvertent mutation (R224Q; Sullivan et al., 1997). The site of transgenic integration has an expression profile that is restricted to peripheral tissues. To generate the bEKO mice, the transgenic mouse line was bred to homozygosity and then backcrossed to ApoE knock-outs (Zhang et al., 1992). ApoE3-targeted replacement mice were used as a control (Sullivan et al., 1997). Male mice were used for performing immunohistochemistry, electrophysiology, and biochemistry; both sexes were used for behavioral experiments.
Antibodies.
The following primary antibodies were used: goat polyclonal anti-ApoE (1:1000 for both Western blotting and immunohistochemistry; Calbiochem), mouse anti-synaptophysin (1:1000; Millipore), rabbit polyclonal anti-RAP (1:1000; 692), and rabbit polyclonal anti-Transferrin (1:1000; ab1223, Abcam). Anti-rabbit 680 and anti-goat 800 secondary antibodies (1:3000; LI-COR) were used for Western blotting. Donkey anti-goat Alexa Fluor 594 and goat anti-mouse Alexa Fluor 594 secondary antibodies (1:200; Invitrogen) were used for immunohistochemistry.
Tissue preparation and Western blotting.
The 2- to 3-month-old bEKO, ApoE KO, and ApoE3/E3 mice were fasted overnight. The following morning, mice were anesthetized, and blood was collected retro-orbitally using heparinized hematocrit tubes (Drummond). Samples were centrifuged for 10 min at 4000 rpm at 4°C, and the supernatant (plasma) was retained. Mice were then deeply anesthetized by isoflurane and transcardially perfused with ice-cold PBS. The hippocampi were dissected out and flash frozen in liquid nitrogen. A portion of the right lobe of the liver was also removed and flash frozen. Samples were homogenized in RIPA buffer with protease and phosphatase inhibitors (Sigma-Aldrich). Proteins were resolved on polyacrylamide gels (Bio-Rad) and transferred to nitrocellulose membranes. Membranes were blocked in blocking buffer (LI-COR) and then incubated with primary antibody. Membranes were then treated with secondary antibody, and proteins were visualized using Odyssey CLx (LI-COR).
Immunohistochemistry.
The 7- to 8-month-old bEKO, ApoEKO, and ApoE3/E3 nonfasted mice were deeply anesthetized by isoflurane, killed, and then transcardially perfused with ice-cold PBS followed by 4% paraformaldehyde (PFA) in PBS. The brains were postfixed in 4% PFA overnight, then stored in PBS containing 0.01% sodium azide. Brains were mounted in agarose and 40 μm coronal sections were obtained using a Leica VT1000 S Vibratome. Sections were blocked for 1 h in 10% donkey serum with 0.1% Triton-X in PBS and incubated overnight at 4°C with primary antibody in blocking buffer. Sections were rinsed three times with PBS-T, and incubated for 1 h in secondary antibody. Sections were rinsed in PBS-T, incubated briefly in DAPI, rinsed again with PBS-T, and mounted with Prolong DAPI antifade (Life Technologies). Images were obtained using a fluorescent microscope.
To evaluate neurodegeneration, sections were blocked for 1 h in 10% goat serum with 0.1% Triton-X in PBS. Sections were incubated overnight in blocking buffer with mouse anti-synaptophysin antibody overnight at 4°C. Sections were rinsed three times with PBS-T and then incubated for 1 h in secondary antibody. Sections were rinsed in PBS-T, incubated briefly in DAPI, rinsed again with PBS-T, and then mounted with Fluorescence Mounting Medium (Dako).
Lipid profile measurement.
Plasma was obtained from 2- to 3-month-old bEKO, ApoEKO, and ApoE3/E3 mice that had been fasted overnight. Two microliters of plasma was incubated with 200 μl of Infinity Cholesterol or Infinity Triglyceride (Thermo Scientific) for 20 min at 37°C, and the reaction was read using a plate reader. The concentration of cholesterol or triglyceride was determined using a standard curve.
Semiquantitative assessment of synaptic loss.
Sections prepared as described were stained with anti-synaptophysin and imaged using a Leica TCS SP5 confocal microscope. Four sections per mouse and two visual fields per section were obtained for both hippocampus and neocortex. As described by Masliah et al. (1995), images were analyzed in ImageJ. Briefly, a threshold was set, and the area occupied was measured for each section and averaged.
Long-term potentiation.
Acute hippocampal slices from 6-month-old bEKO, ApoE KO, and ApoE3/E3 mice were used for field recordings to measure long-term potentiation (LTP). Theta-burst LTP was performed as previously described (Chen et al., 2010). Briefly, mice were deeply anesthetized with isoflurane, and their brains were quickly removed and placed in ice-cold high-sucrose slicing solution, as follows (in mm): 110 sucrose, 60 NaCl, 3 KCl, 1.25 NaH2PO4, 28 NaHCO3, 0.5 CaCl2, 5 glucose, 0.6 ascorbic acid, and 7 MgSO4. The 350 μm transverse slices were cut using a Leica VT1000S Vibratome. Slices were allowed to recover in artificial CSF (ACSF), consisting of the following (in mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 d-glucose, 2 CaCl2, and 1 MgSO4, for 1 h at room temperature before experiments. For recording, slices were transferred to an interface chamber perfused with ACSF at 31°C. Slices were stimulated within the stratum radiatum using concentric bipolar electrodes (FHC) and an isolated pulse stimulator. The stimulus intensity was set to 30–50% of the maximum peak amplitude, as determined by measuring the input–output curve. After stabilization of the baseline, theta-burst stimulation was applied with four pulses at 100 Hz repeated 10 times with 200 ms intervals, and the train was repeated five times at 10 s intervals. After induction, LTP was followed for an hour and averaged between 40 and 60 min for analysis.
Input–output analysis.
fEPSP traces were exported from LabView as text files and imported into Clampfit 10. The fiber volley amplitude was calculated at the minimum of the first derivative of the average of all traces. The fEPSC slope was calculated along the linear portion of the rising phase just after the fiber volley. The fiber volley amplitude and fEPSP slope were binned by fiber volley amplitude in Microsoft Excel 2010. The binned fEPSP slopes for each genotype were fit in GraphPad Prism version 6.07 (best fit of either a one-phase association or straight line), and fits were output up to 16 data points. These fits were analyzed as a grouped table using an ordinary two-way ANOVA with a Sidak post hoc multiple comparison between genotypes of the fEPSC slopes at each fiber volley amplitude interval.
Whole-cell voltage-clamp recordings on CA1 pyramidal neurons.
Hippocampal slices were prepared from 6- to 9-month-old mice. After perfusion with an ice-cold high-sucrose cutting solution (solutions as for LTP, per above), brains were quickly removed and placed into the same solution. Transverse 350 μm sections were cut using a vibratome. Slices were kept in a chamber containing 50% ACSF and 50% cutting solution until the experiment started. Slices were then transferred into an interface recording chamber and kept at room temperature (26°C) at a flow rate of 2–3 ml/min with ACSF. Patch electrodes (3–6 MΩ) were filled with patch solution containing 110 mm K-gluconate, 20 mm KCl, 10 mm NaCl, 10 mm HEPES, 0.6 mm EGTA, 4 mm Mg-ATP, 0.3 mm GTP, and 10 mm N-ethyl bromide quaternary salt, pH 7.2. Series resistance (Rs) ranged from 7 to 20 MΩ. Experiments in which Rs varied ≥20% within a recording session were discarded. Recordings were obtained with an Axopatch-200B Patch-Clamp Amplifier (Molecular Devices) digitized at 10 kHz through a Digidata 1322A Digitizer (Molecular Devices). EPSCs were evoked by stimulation (duration, 200 μs; amplitude, 100–600 μA) of Schaeffer collateral commissural afferents (30–50% of maximum response, usually ∼100 pA) using concentric bipolar electrodes (catalog #CBBRC75, FHC) through an isolated pulse stimulator (Model 2100, A-M Systems). AMPA and NMDA currents were plotted in the presence of the GABAA receptor antagonist picrotoxin (50 μm; Sigma-Aldrich) at −70 and +50 mV, respectively. Paired pulses are recorded at −60 mV with the same stimulus intensities.
Behavior.
Mice were trained for 7 d to find a hidden platform in a 120-cm-diameter tube of cloudy water at 21°C. The platform was 10 cm in diameter and 1 cm below the water surface. Each day, mice underwent three trials, during which they were placed into the water at a pseudorandom start point and given 60 s to find the platform, guided by large cues on the wall. Mice that did not find the platform within 60 s were guided to the platform by hand. Mice were then given 5 s on the platform before being returned to their holding cage. On the eighth day, mice were tested with a probe trial in which the platform was removed and they were allowed to swim for 60 s. On the ninth day, mice were tested on a visual version on the task, in which the platform was moved to the opposite side of the pool and made visible with a flag. One male ApoE KO mouse failed the visual probe task and was removed from the analysis. Mouse movements were recorded and analyzed using HVS water maze software (HVS Image).
Statistical analysis.
Data were analyzed with Prism software version 6.0 (GraphPad Software). All data are shown as the mean ± SEM, and statistical significance was set at p < 0.05.
Results
Plasma ApoE levels are normal in the bEKO mouse
Loss of ApoE results in age-dependent synaptic loss in mice (Masliah et al., 1995); however, the loss of ApoE causes extremely elevated plasma lipid levels (Schaefer et al., 1986; Plump et al., 1992; Zhang et al., 1992; Mak et al., 2014), which may independently contribute to neurodegenerative processes or affect cognition. To differentiate between these two effects of ApoE loss on brain function, we took advantage of a novel mouse that has a random integration of a functional human ApoE expression cassette, with expression restricted to peripheral tissues (primarily liver; Fig. 2A,B) and virtually complete absence of ApoE in the brain (Fig. 3A), which will be referred to here as the bEKO mouse.
Figure 2.
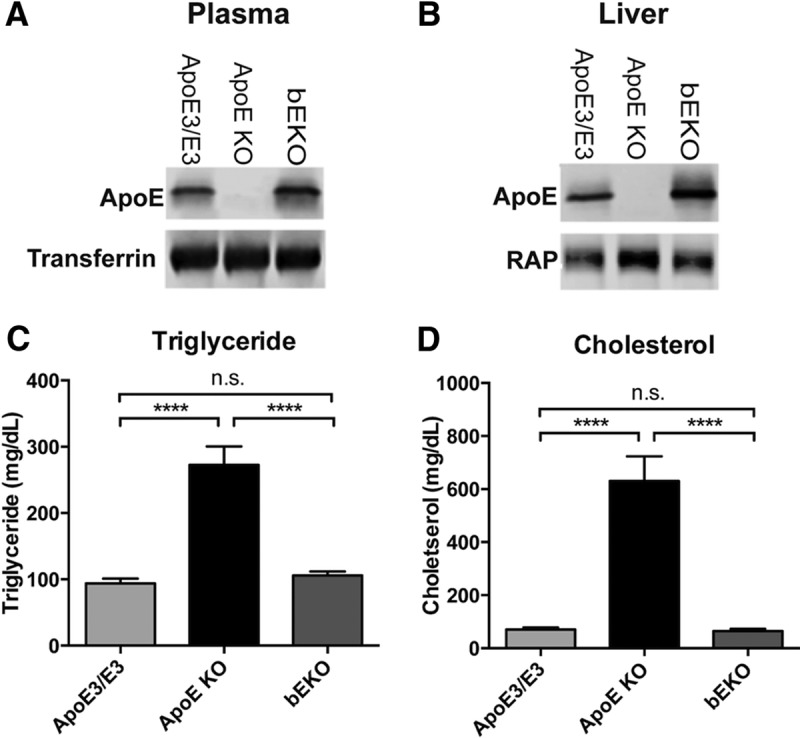
Tissue distribution of ApoE in the bEKO mouse and restoration of normal lipid profile in bEKO mouse. A, Plasma ApoE levels (Kruskal Wallis nonparametric one-way ANOVA, p < 0.01). B, Liver ApoE levels (Kruskal Wallis nonparametric one-way ANOVA, p < 0.0001). C, Plasma triglyceride levels. ApoE KO mice had highly elevated plasma triglyceride levels (one-way ANOVA, ****p < 0.0001), while bEKO mice had levels that were similar to ApoE3/E3 mice (Tukey's post hoc test, p = n.s.) D, Plasma cholesterol levels. ApoE KO mice had highly elevated plasma cholesterol (one-way ANOVA, ****p < 0.0001), while bEKO mice had levels that were similar to ApoE3/E3 mice (post hoc test, p = n.s.). ApoE3/E3 (n = 5), ApoE KO (n = 4), and bEKO (n = 5) mice.
Figure 3.

Minimal brain ApoE presence in bEKO mice and reduced synapse numbers in ApoE KO and bEKO mice. A, Hippocampal ApoE levels in ApoE3/E3 (n = 5), ApoE KO (n = 3), and bEKO (n = 5) mice (Kruskal Wallis nonparametric one-way ANOVA, **p < 0.0001). B, Anti-ApoE-stained (red) sections from 7- to 8-month-old mice (n = 3 mice per genotype). C, Sections from 9-month-old mice were immunostained for synaptophysin and analyzed for the area of the neuropil. No significant difference was observed in the hippocampus (one-way ANOVA, p = 0.5912). In the neocortex, a reduction in area occupied by synaptophysin was observed in both ApoE KO and bEKO mice (one-way ANOVA, **p = 0.0011; n = 3 mice/genotype, n = 3–4 sections/brain region per mouse, n = 2 fields of view/section).
bEKO mice have a normal plasma lipid profile
First, we determined whether the restoration of peripheral ApoE in the bEKO mouse normalized the lipid profile. The 6-month-old mice were fasted overnight to prevent dietary effects on plasma lipid levels, and plasma levels of cholesterol and triglyceride were measured. As has been previously shown, the ApoE KO mice showed a remarkable elevation in plasma cholesterol and triglyceride levels (Plump et al., 1992; Zhang et al., 1992). The restoration of plasma ApoE completely normalized plasma cholesterol and triglyceride to levels that were similar to the ApoE3/E3 mice (Fig. 2C,D).
bEKO mice have greatly reduced brain ApoE
In contrast to peripheral tissues, ApoE expression in the hippocampus was barely detectable in bEKO lysates (Fig. 3A). Since there was some residual ApoE in the hippocampus of bEKO mice, we wanted to confirm the distribution of brain ApoE in the bEKO mice. In the ApoE3/E3 mice, immunohistochemical staining with an anti-ApoE antibody revealed distinct cells, whose distribution and shape matched astrocytes, as well as a diffuse light staining, which matched previous findings for ApoE TR mice (Hamanaka et al., 2000). In the ApoE KO sections, no ApoE staining was observed. Sections from the bEKO mice showed none of the cellular staining, but did show a small amount of the diffuse interstitial staining observed in ApoE3/E3 sections (Fig. 3B). It is likely that this staining is the residual ApoE observed in the Western blots (Fig. 3A).
bEKO mice have a loss of synaptic markers similar to that of ApoE KO mice
We next determined whether the bEKO mice showed a loss of synaptic markers similar to that previously observed in ApoE KO mice (Masliah et al., 1995). Sections from 7- to 8-month-old male ApoE3/E3, ApoE KO, and bEKO mice were stained with an anti-synaptophysin antibody and semiquantitatively analyzed for the area of the neuropil covered by synaptophysin (Fig. 3C). Similar to previously published results, we observed a reduction in synaptophysin immunoreactivity in the neocortex of the ApoE KO mice (Masliah et al., 1995), and this was also observed in the bEKO mice (Fig. 3C). However, contrary to what was previously reported, we did not see a significant reduction in synaptophysin staining in the hippocampus in either the ApoE KO or bEKO mice (Fig. 3C). This may be an age-dependent effect, as previous studies indicated delayed loss of synaptic markers in the ApoE KO hippocampus (Masliah et al., 1995).
bEKO mice have similar LTP deficits as ApoE KO mice
Having shown that bEKO mice have synaptic loss similar to that of ApoE KO mice, we then investigated whether they also exhibited impaired LTP, as had been observed in the total ApoE knockouts (Veinbergs and Masliah, 1999). Acute hippocampal slices from male 7- to 8-month-old ApoE3/E3, ApoE KO, and bEKO mice were evaluated for deficits in synaptic plasticity. After achieving a stable baseline, LTP was induced with theta-burst stimulation of CA1 Schaffer collaterals and recorded for 1 h. Slices from bEKO mice showed a reduction in LTP that was similar to ApoE KO slices (Fig. 4A,B). To further compare synaptic strength in these mice, input–output curves were calculated. Similar to the LTP findings, the bEKO and ApoE KO mice had reduced input–output responses compared with the ApoE3/E3 mice (Fig. 4C).
Figure 4.

Reduced theta-burst LTP and synaptic function in both ApoE KO and bEKO mice. A, Theta-burst LTP. B, LTP was reduced at 40–60 min in both ApoE KO and bEKO mice [one-way ANOVA, p = 0.0040, Tukey's post hoc ApoE3/E3 vs ApoE KO; *p = 0.0142, ApoE3/E3 vs bEKO; **p = 0.0078, ApoE KO vs bEKO; p = 0.9372 (n.s.); ApoE3/E3, n = 23; ApoE KO, n = 25; bEKO, n = 21]. C, Input–output plot of the average fEPSP slopes vs the fiber volley amplitude depicting reduced synaptic strength in both ApoE KO and bEKO mice [two-way ANOVA, pfEPSP slope < 0.0001, pGenotype < 0.0001, pInteraction = 0.0067; fEPSP slope post hoc analyses: ApoE3 KO vs bEKO, p > 0.9999 (n.s.); ApoE3/E3 vs ApoE KO, *p = 0.0268; ApoE3/E3 vs bEKO, *p = 0.0279; ApoE3/E3, n = 61; ApoE KO, n = 14; bEKO, n = 26].
bEKO mice are not impaired in spatial learning, in contrast to ApoE KO mice
Together, the immunohistochemistry and LTP data suggest that the bEKO mice have synaptic loss and dysfunction similar to that of ApoE KO mice. ApoE KO mice have an age-dependent deficit on the Morris water maze (MWM; Masliah et al., 1995, 1997); thus, we also tested 6.5- to 7.5-month-old mice on the MWM task. No differences in acquisition were observed between the genotypes (Fig. 5A). The ApoE KO mice showed impairment on the probe trial, which was not observed in the bEKO mice (Fig. 5B). These results suggest that while bEKO mice exhibit signs of synaptic changes, they are able to compensate behaviorally.
Figure 5.

Spatial learning impairment is observed only in female ApoE KO mice. A, Latency to find the platform for each day of MWM training [two-way, repeated measures (rm) ANOVA: pTime < 0.0001; pGenotype = 0.6561; pInteraction = 0.0750]. B, Preference for the target quadrant is reduced in ApoE KO mice on the probe day, but not in bEKO mice [two-way ANOVA: pQuadrant < 0.0001; pGenotype > 0.9999; pInteraction = 0.0293; target quadrant post hoc analyses: ApoE KO compared with bEKO, *p < 0.05; ApoE3/E3 compared with ApoE KO, p = n.s.; ApoE3/E3 compared with bEKO, p = n.s.). A, B, n = 22 ApoE3/E3 and bEKO mice, n = 16 ApoE KO mice). C, D, No difference in MWM acquisition or probe trial in male ApoE KO and bEKO mice (acquisition two-way rmANOVA: pTime < 0.0001; pGenotype and pInteraction = n.s; probe trial two-way ANOVA: pQuadrant < 0.0001; pInteraction and pGenotye = n.s.) E, Impaired MWM acquisition in female ApoE KO mice only (two-way, rmANOVA: pTime < 0.0001; pGenotype and pInteraction = n.s.; post hoc day 6: ApoE3/3 female vs ApoE KO female, **p < 0.01; post hoc day 7: ApoE3/E3 vs ApoE KO, *p < 0.05). F, Time spent in each quadrant by female mice on the probe trial, day 8 (two-way ANOVA: pQuadrant < 0.0001; pGenotype > 0.9999; pInteraction = 0.0042; target quadrant post hoc test: ApoE KO vs bEKO, ***p < 0.001; ApoE KO vs ApoE3/E3, *p < 0.05). C–F: ApoE3/E3 male, n = 12; ApoE3/E3 female, n = 10; ApoE KO male, n = 8; ApoE KO female, n = 8; bEKO male, n = 9; bEKO female, n = 13. Dashed line indicates 25% of time spent in quadrant.
Learning impairment in ApoE KO mice is sex dependent
The behavioral results shown in Figure 5, A and B, reflect male and female mice. When the data were broken down by sex, differences emerged. In the wild-type mice, there was no difference in performance between the sexes, and the male ApoE KO mice performed similarly to wild-type mice. In stark contrast to the males, the female ApoE KO mice had impaired results in the MWM test, showing both increased latency to reach the platform and a reduced preference for the target quadrant on the probe trial. The impairment seen in the probe trial is not present in the bEKO mice, for either sex (Fig. 5C–F).
ApoE KO mice have synaptic properties different from those of bEKO and ApoE3/E3 mice
Since the bEKO mice did not exhibit the same behavioral impairment as the ApoE KO mice, despite having a similar deficit in LTP, we decided to investigate synaptic function in more detail. Neurons from 6-month-old ApoE3/E3, ApoE KO, and bEKO mice were evaluated using patch-clamp techniques. First, we examined paired-pulse facilitation and found no difference between the genotypes, indicating no effect of ApoE deficiency on presynaptic function (Fig. 6A,B). We then evaluated AMPA and NMDA currents. In contrast to our LTP findings, we observed a reduction of AMPA/NMDA ratios compared with wild-type controls only in the ApoE KO slices, but not in the bEKO slices (Fig. 6C–E).
Figure 6.

Normal paired-pulse facilitation and reduced AMPA/NMDA ratio in ApoE KO mice. A, B, Paired-pulse facilitation (ApoE3/E3, n = 6; ApoE KO, n = 9; bEKO n = 7). A, Representative traces of EPSCs. B, Normal facilitation of EPSC amplitudes evoked at 20, 50, 100, and 200 ms intervals (two-way ANOVA: pInteraction = 0.9995; pInterval < 0.0001; pgenotype = 0.9096). C–E, AMPA/NMDA ratio in ApoE3/E3, ApoE KO, and bEKO neurons (ApoE3/E3, n = 6; ApoE KO, n = 9; bEKO n = 7). C, Representative traces of EPSCs evoked at either +50 mV (top) or −70 mV (bottom). D, Average EPSC amplitude at −70 and +50 mV (two-way RM ANOVA: pInteraction = 0.6232; pmV < 0.0001; pGenotype = 0.0027; genotype post hoc test, p = n.s. at either holding potential). E, Average AMPA/NMDA ratio calculated by dividing the EPSC amplitudes evoked at −70 mV by those at +50 mV [one-way ANOVA, p = 0.0002; Tukey's post hoc test: ApoE3/E3 vs ApoE KO, *p = 0.0142; ApoE3/E3 vs bEKO, p = 0.1907 (n.s.); ApoE KO vs bEKO, **p = 0.0002].
Discussion
We investigated the effect of genetically restoring plasma ApoE in an ApoE knock-out mouse (bEKO mouse) on lipid profile, synaptic function, and behavior. Genetic restoration of peripheral ApoE normalized plasma cholesterol and triglyceride levels in ApoE KO mice. Despite this normalization of plasma lipids, bEKO mice exhibited reduced levels of synaptic markers and reduced LTP, which were comparable to those of ApoE KO mice. In contrast, the bEKO mice did not share the modest behavioral impairment of the ApoE KO mice. Additionally, this impairment was present only in the female ApoE KO mice and was completely rescued in the female bEKO mice. At a synaptic level, we found that while paired-pulse facilitation was unaltered by ApoE deficiency, AMPA/NMDA ratios were reduced only in the ApoE KO mice, but not in the bEKO mice. Together, these results suggest distinct contributions of peripheral and central ApoE, as well as plasma lipids, to brain function, which appear to have a greater impact in female brains.
How does complete loss of ApoE lead to loss of synaptic markers, electrophysiological deficits, and behavioral impairments, while brain-specific loss of ApoE leads only to the loss of synaptic markers and more selective electrophysiological changes? ApoE receptors have a variety of roles in synaptic function and plasticity that may be altered by the loss of ApoE (Lane-Donovan et al., 2014), and the shared synaptic changes observed in ApoE KO and bEKO point to an effect of ApoE loss on synapses that is independent of peripheral ApoE levels. Astrocyte-derived, ApoE-containing lipoproteins are required for the development of mature synapses (Mauch et al., 2001). This is consistent with our findings that both the bEKO and ApoE KO mice—which lack astrocyte-derived ApoE—had reduced synaptophysin staining and reduced input–output curves, indicating reduced synapse numbers. However, while the bEKO mice were able to adapt to the reduced synapse numbers and exhibited normal behavior and AMPA/NMDA ratios, the ApoE KO mice had spatial learning deficits and reduced AMPA/NMDA ratios. These data suggest that loss of peripheral ApoE has a detrimental effect on CNS function and network activity, for which several potential mechanisms could be posited (Fig. 7).
Figure 7.

Possible mechanisms by which peripheral ApoE loss impacts CNS function. In the CNS, astrocyte-derived ApoE-rich lipoprotein particles are required for synapse maturation, a function that is lost in both the bEKO and ApoE KO mice. CNS and plasma ApoE may both contribute to the maintenance of the BBB. When both pools are lost, leakage of metabolic products or inflammatory cytokines associated with atherosclerosis and hyperlipidemia that can affect synapse function across the BBB may occur. Finally, the elevated levels of cholesterol and triglycerides resulting from the loss of plasma ApoE may lead to the generation of excessive free fatty acid (FFA) and sterol transport across the BBB.
One mechanism by which peripheral ApoE loss could lead to cognitive impairment is its effect on the vasculature. Vascular dysfunction and atherosclerosis occur early in ApoE KO mice, which leads to reduced cerebral blood flow and dysfunctional autonomic regulation of the cerebrovasculature (Plump et al., 1992; Zhang et al., 1992; Ayata et al., 2013). This reduction in brain perfusion is accompanied by impaired functional connectivity in ApoE KO mice (Zerbi et al., 2014), supporting an intersection of cerebrovascular dysfunction and cognitive impairment. In the bEKO mice, the level of peripheral ApoE is returned to control levels, and plasma lipid levels are completely normalized, thus potentially averting much of the vascular dysfunction.
In addition to vascular changes, ApoE knock-out mice exhibit an age-dependent breakdown of the BBB, which correlates with behavioral impairments (Mulder et al., 2001; Bell et al., 2012). This breakdown can be partially restored with a bone marrow transplant from wild-type mice, which introduces ApoE-expressing leukocytes and restores a small amount (∼3%) of ApoE to the plasma (Hafezi-Moghadam et al., 2007). A mechanism by which ApoE maintains BBB strength was recently proposed. In ApoE KO and ApoE4/E4 mice, cyclophilin A is dysregulated, leading to increased matrix metalloproteinase 9 activity and breakdown of the BBB (Bell et al., 2012). A breakdown in the BBB could conceivably permit easier flow of neurotoxic agents, including inflammatory cytokines which are elevated in hyperlipidemia and atherosclerosis, from the plasma into the CNS.
The ApoE KO mice have highly elevated levels of triglycerides and cholesterol as a result of inefficient uptake of particles by lipoprotein receptors (Zhang et al., 1992). Hypertriglyceridemia in mice causes cognitive dysfunction (Farr et al., 2008), and numerous studies have investigated the effect of high-fat diets on cognition in animal and human models (Freeman et al., 2014). These lipids have various metabolites that could be detrimental to CNS function. For example, cholesterol can be metabolized to 27-hydroxycholesterol, which can more easily cross the BBB, and negatively impacts cognitive function in mice (Heverin et al., 2015). Also, mechanisms to prevent the processing of triglyceride-rich lipoproteins exist in the brain. Briefly, lipoprotein lipase (LpL) in the lumen of capillaries hydrolyzes triglycerides into free fatty acids (Goldberg, 1996). Although LpL is present in the CNS (Vilaró et al., 1990), it requires glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPIHBP1) on the endothelial surface for efficient lipolytic processing (Wang and Eckel, 2012). GPIHBP1 is notably absent from brain capillary lumens (Beigneux et al., 2007), which suggests a need to avoid excessive exposure of the CNS to free fatty acids. These studies have largely not evaluated the effect of these lipid species on neuronal function.
In the bEKO mice, there is a small amount of residual ApoE in the CNS observed in Western blotting and immunohistochemistry. The source of this residual ApoE is unclear. Since there is no ApoE staining present in cell bodies, this suggests that the ApoE is not astrocyte derived, but is likely derived from peripheral sources. This may simply reflect incomplete perfusion of the CNS with PBS. Alternatively, a recent study showed that ApoA-I is able to enter the CNS via the choroid plexus (Stukas et al., 2014). If a similar mechanism exists for ApoE, it could account for the residual amounts observed in the CNS. Recent sophisticated studies have confirmed that ApoE does not normally cross the blood–CSF barrier (Liu et al., 2012); however, if there is some leakage of the BBB in the bEKO mice, this could permit entry of ApoE into the CNS. It is important to note that it is therefore possible that this residual amount of ApoE, however small, may be sufficient to rescue the behavioral deficits caused by ApoE.
It has been shown that female ApoE KO mice display greater cognitive impairment than male ApoE KO mice (Raber et al., 1998), a result we have replicated here. Importantly, the impairment in the female ApoE KO mice was completely rescued in the female bEKO mice. This finding supports the hypothesis that atherosclerosis in the ApoE KO mice contributes to cognitive dysfunction, because female ApoE KO mice develop more severe atherosclerosis and vascular impairment earlier (Caligiuri et al., 1999).
An effect of sex on AD pathology has previously been noted. In AD mice, a difference in cognitive function by sex is observed both at baseline and in response to treatment targeting ApoE receptor signaling (Hinrich et al., 2016). Moreover, women are disproportionately more likely than men to develop the disease, primarily due to longer lifespan (Bachman et al., 1992), and ApoE4 has a stronger effect on AD risk in women compared with men (Altmann et al., 2014), but only because of the greater life expectancy that females enjoy. Since plasma and brain ApoE levels are slightly reduced in ApoE4 individuals (Slooter et al., 1998; Beffert et al., 1999), the mechanism by which ApoE4 causes enhanced risk in women may include a greater susceptibility of women to reduced ApoE. Additionally, our finding that normalizing lipid levels restores normal learning in the bEKO mouse may suggest that lipid-lowering interventions would be more effective at reducing AD risk in women.
The answer to whether ApoE is required for cognitive function in humans was seemingly answered by a recent case report. While the patient, a 40-year-old male with no detectable ApoE had severe hyperlipidemia, he was cognitively unremarkable; suggesting that ApoE may not be essential for cognitive function (Lane-Donovan and Herz, 2014; Mak et al., 2014). However, in the context of our finding that male ApoE KO mice were unimpaired on the MWM task while female ApoE KO mice were moderately impaired, this conclusion may have been premature. Moving forward, it will be important for clinical trials investigating ApoE-reducing therapeutics to consider sex in their study design and analysis.
The simultaneous effects of ApoE genotype on AD pathology and plasma cholesterol level have long been appreciated in the field as independent risk factors for AD. What has been difficult thus far has been to differentiate between the importance of ApoE produced in the brain and its essential role in plasma lipid metabolism in peripheral tissues. The findings described here delineate the distinct roles played by peripheral and central ApoE in synaptic function and behavior, and suggest a dual mechanism by which ApoE deficiency causes behavioral deficits. In this hypothesis, CNS ApoE deficiency causes synaptic loss, while peripheral ApoE deficiency concomitantly leads to dyslipidemia, vascular dysfunction, and aberrant neuronal function; these disturbances together cause cognitive dysfunction. In addition, our findings highlight the fact that female sex enhances susceptibility to ApoE changes, possibly due to hyperlipidemia in combination with increased atherosclerosis. In conclusion, although ApoE-reducing drugs show promise as potential AD therapeutics, their effect on atherosclerosis risk and possible adverse CNS effects, particularly in women, need to be addressed.
Footnotes
This work was supported by National Institutes of Health Grants F30-AG-047799 (to C.L.D.), and R37-HL-63762 and R01-NS-093382 (to J.H.); the American Health Assistance Foundation; the Consortium for Frontotemporal Dementia Research; the Bright Focus Foundation; the Lupe Murchison Foundation; and the Ted Nash Long Life Foundation. We thank Rebekah Hewitt, Barsha Subba, and Charisma Desai for technical assistance.
The authors declare no competing financial interests.
References
- Altmann A, Tian L, Henderson VW, Greicius MD. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann Neurol. 2014;75:563–573. doi: 10.1002/ana.24135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstey KJ, Lipnicki DM, Low LF. Cholesterol as a risk factor for dementia and cognitive decline: a systematic review of prospective studies with meta-analysis. Am J Geriatr Psychiatry. 2008;16:343–354. doi: 10.1097/01.JGP.0000310778.20870.ae. [DOI] [PubMed] [Google Scholar]
- Ayata C, Shin HK, Dileköz E, Atochin DN, Kashiwagi S, Eikermann-Haerter K, Huang PL. Hyperlipidemia disrupts cerebrovascular reflexes and worsens ischemic perfusion defect. J Cereb Blood Flow Metab. 2013;33:954–962. doi: 10.1038/jcbfm.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman DL, Wolf PA, Linn R, Knoefel JE, Cobb J, Belanger A, D'Agostino RB, White LR. Prevalence of dementia and probable senile dementia of the Alzheimer type in the Framingham Study. Neurology. 1992;42:115–119. doi: 10.1212/WNL.42.1.115. [DOI] [PubMed] [Google Scholar]
- Beffert U, Cohn JS, Petit-Turcotte C, Tremblay M, Aumont N, Ramassamy C, Davignon J, Poirier J. Apolipoprotein E and beta-amyloid levels in the hippocampus and frontal cortex of Alzheimer's disease subjects are disease-related and apolipoprotein E genotype dependent. Brain Res. 1999;843:87–94. doi: 10.1016/S0006-8993(99)01894-6. [DOI] [PubMed] [Google Scholar]
- Beigneux AP, Davies BS, Gin P, Weinstein MM, Farber E, Qiao X, Peale F, Bunting S, Walzem RL, Wong JS, Blaner WS, Ding ZM, Melford K, Wongsiriroj N, Shu X, de Sauvage F, Ryan RO, Fong LG, Bensadoun A, Young SG. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 2007;5:279–291. doi: 10.1016/j.cmet.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk BC, Zlokovic BV. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkhem I, Lütjohann D, Diczfalusy U, Ståhle L, Ahlborg G, Wahren J. Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J Lipid Res. 1998;39:1594–1600. [PubMed] [Google Scholar]
- Caligiuri G, Nicoletti A, Zhou X, Törnberg I, Hansson GK. Effects of sex and age on atherosclerosis and autoimmunity in apoE-deficient mice. Atherosclerosis. 1999;145:301–308. doi: 10.1016/S0021-9150(99)00081-7. [DOI] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Durakoglugil MS, Xian X, Herz J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci U S A. 2010;107:12011–12016. doi: 10.1073/pnas.0914984107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, Jr, Rimmler JB, Locke PA, Conneally PM, Schmader KE. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180–184. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- Farr SA, Yamada KA, Butterfield DA, Abdul HM, Xu L, Miller NE, Banks WA, Morley JE. Obesity and hypertriglyceridemia produce cognitive impairment. Endocrinology. 2008;149:2628–2636. doi: 10.1210/en.2007-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman LR, Haley-Zitlin V, Rosenberger DS, Granholm AC. Damaging effects of a high-fat diet to the brain and cognition: a review of proposed mechanisms. Nutr Neurosci. 2014;17:241–251. doi: 10.1179/1476830513Y.0000000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton SM, Shirman GA, Strittmatter WJ, Matthew WD. Impairment of the blood-nerve and blood-brain barriers in apolipoprotein e knockout mice. Exp Neurol. 2001;169:13–22. doi: 10.1006/exnr.2001.7631. [DOI] [PubMed] [Google Scholar]
- Goldberg IJ. Lipoprotein lipase and lipolysis: central roles in lipoprotein metabolism and atherogenesis. J Lipid Res. 1996;37:693–707. [PubMed] [Google Scholar]
- Gordon I, Grauer E, Genis I, Sehayek E, Michaelson DM. Memory deficits and cholinergic impairments in apolipoprotein E-deficient mice. Neurosci Lett. 1995;199:1–4. doi: 10.1016/0304-3940(95)12006-P. [DOI] [PubMed] [Google Scholar]
- Gordon I, Genis I, Grauer E, Sehayek E, Michaelson DM. Biochemical and cognitive studies of apolipoprotein-E-deficient mice. Mol Chem Neuropathol. 1996;28:97–103. doi: 10.1007/BF02815210. [DOI] [PubMed] [Google Scholar]
- Hafezi-Moghadam A, Thomas KL, Wagner DD. ApoE deficiency leads to a progressive age-dependent blood-brain barrier leakage. Am J Physiol Cell Physiol. 2007;292:C1256–C1262. doi: 10.1152/ajpcell.00563.2005. [DOI] [PubMed] [Google Scholar]
- Hamanaka H, Katoh-Fukui Y, Suzuki K, Kobayashi M, Suzuki R, Motegi Y, Nakahara Y, Takeshita A, Kawai M, Ishiguro K, Yokoyama M, Fujita SC. Altered cholesterol metabolism in human apolipoprotein E4 knock-in mice. Hum Mol Genet. 2000;9:353–361. doi: 10.1093/hmg/9.3.353. [DOI] [PubMed] [Google Scholar]
- Heverin M, Maioli S, Pham T, Mateos L, Camporesi E, Ali Z, Winblad B, Cedazo-Minguez A, Björkhem I. 27-hydroxycholesterol mediates negative effects of dietary cholesterol on cognition in mice. Behav Brain Res. 2015;278:356–359. doi: 10.1016/j.bbr.2014.10.018. [DOI] [PubMed] [Google Scholar]
- Hinrich AJ, Jodelka FM, Chang JL, Brutman D, Bruno AM, Briggs CA, James BD, Stutzmann GE, Bennett DA, Miller SA, Rigo F, Marr RA, Hastings ML. Therapeutic correction of ApoER2 splicing in Alzheimer's disease mice using antisense oligonucleotides. EMBO Mol Med. 2016;8:328–345. doi: 10.15252/emmm.201505846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2000;97:2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, Yoon H, Basak JM, Finn MB, Holtzman DM. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-β amyloidosis. J Neurosci. 2011;31:18007–18012. doi: 10.1523/JNEUROSCI.3773-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane-Donovan C, Herz J. Is apolipoprotein e required for cognitive function in humans? Implications for Alzheimer drug development. JAMA Neurol. 2014;71:1213–1215. doi: 10.1001/jamaneurol.2014.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane-Donovan C, Philips GT, Herz J. More than cholesterol transporters: lipoprotein receptors in CNS function and neurodegeneration. Neuron. 2014;83:771–787. doi: 10.1016/j.neuron.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F, Hori Y, Hudry E, Bauer AQ, Jiang H, Mahan TE, Lefton KB, Zhang TJ, Dearborn JT, Kim J, Culver JP, Betensky R, Wozniak DF, Hyman BT, Holtzman DM. Anti-ApoE antibody given after plaque onset decreases Aβ accumulation and improves brain function in a mouse model of Aβ amyloidosis. J Neurosci. 2014;34:7281–7292. doi: 10.1523/JNEUROSCI.0646-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Kuhel DG, Shen L, Hui DY, Woods SC. Apolipoprotein E does not cross the blood-cerebrospinal fluid barrier, as revealed by an improved technique for sampling CSF from mice. Am J Physiol Regul Integr Comp Physiol. 2012;303:R903–R908. doi: 10.1152/ajpregu.00219.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak AC, Pullinger CR, Tang LF, Wong JS, Deo RC, Schwarz JM, Gugliucci A, Movsesyan I, Ishida BY, Chu C, Poon A, Kim P, Stock EO, Schaefer EJ, Asztalos BF, Castellano JM, Wyss-Coray T, Duncan JL, Miller BL, Kane JP, et al. Effects of the absence of apolipoprotein e on lipoproteins, neurocognitive function, and retinal function. JAMA Neurol. 2014;71:1228–1236. doi: 10.1001/jamaneurol.2014.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Ge N, Alford M, Veinbergs I, Roses AD. Neurodegeneration in the central nervous system of apoE-deficient mice. Exp Neurol. 1995;136:107–122. doi: 10.1006/exnr.1995.1088. [DOI] [PubMed] [Google Scholar]
- Masliah E, Samuel W, Veinbergs I, Mallory M, Mante M, Saitoh T. Neurodegeneration and cognitive impairment in apoE-deficient mice is ameliorated by infusion of recombinant apoE. Brain Res. 1997;751:307–314. doi: 10.1016/S0006-8993(96)01420-5. [DOI] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- Mulder M, Blokland A, van den Berg DJ, Schulten H, Bakker AH, Terwel D, Honig W, de Kloet ER, Havekes LM, Steinbusch HW, de Lange EC. Apolipoprotein E protects against neuropathology induced by a high-fat diet and maintains the integrity of the blood-brain barrier during aging. Lab Invest. 2001;81:953–960. doi: 10.1038/labinvest.3780307. [DOI] [PubMed] [Google Scholar]
- Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta. 1987;917:148–161. doi: 10.1016/0005-2760(87)90295-5. [DOI] [PubMed] [Google Scholar]
- Plump AS, Smith JD, Hayek T, Aalto-Setälä K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-G. [DOI] [PubMed] [Google Scholar]
- Raber J, Wong D, Buttini M, Orth M, Bellosta S, Pitas RE, Mahley RW, Mucke L. Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc Natl Acad Sci U S A. 1998;95:10914–10919. doi: 10.1073/pnas.95.18.10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer EJ, Gregg RE, Ghiselli G, Forte TM, Ordovas JM, Zech LA, Brewer HB., Jr Familial apolipoprotein E deficiency. J Clin Invest. 1986;78:1206–1219. doi: 10.1172/JCI112704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherzer CR, Offe K, Gearing M, Rees HD, Fang G, Heilman CJ, Schaller C, Bujo H, Levey AI, Lah JJ. Loss of apolipoprotein E receptor LR11 in Alzheimer disease. Arch Neurol. 2004;61:1200–1205. doi: 10.1001/archneur.61.8.1200. [DOI] [PubMed] [Google Scholar]
- Shea TB, Rogers E, Ashline D, Ortiz D, Sheu MS. Apolipoprotein E deficiency promotes increased oxidative stress and compensatory increases in antioxidants in brain tissue. Free Radic Biol Med. 2002;33:1115–1120. doi: 10.1016/S0891-5849(02)01001-8. [DOI] [PubMed] [Google Scholar]
- Sheng H, Laskowitz DT, Bennett E, Schmechel DE, Bart RD, Saunders AM, Pearlstein RD, Roses AD, Warner DS. Apolipoprotein E isoform-specific differences in outcome from focal ischemia in transgenic mice. J Cereb Blood Flow Metab. 1998;18:361–366. doi: 10.1097/00004647-199804000-00003. [DOI] [PubMed] [Google Scholar]
- Slooter AJ, de Knijff P, Hofman A, Cruts M, Breteler MM, Van Broeckhoven C, Havekes LM, van Duijn CM. Serum apolipoprotein E level is not increased in Alzheimer's disease: the Rotterdam study. Neurosci Lett. 1998;248:21–24. doi: 10.1016/S0304-3940(98)00339-5. [DOI] [PubMed] [Google Scholar]
- Stone DJ, Rozovsky I, Morgan TE, Anderson CP, Hajian H, Finch CE. Astrocytes and microglia respond to estrogen with increased apoE mRNA in vivo and in vitro. Exp Neurol. 1997;143:313–318. doi: 10.1006/exnr.1996.6360. [DOI] [PubMed] [Google Scholar]
- Stukas S, Robert J, Lee M, Kulic I, Carr M, Tourigny K, Fan J, Namjoshi D, Lemke K, DeValle N, Chan J, Wilson T, Wilkinson A, Chapanian R, Kizhakkedathu JN, Cirrito JR, Oda MN, Wellington CL. Intravenously injected human apolipoprotein A-I rapidly enters the central nervous system via the choroid plexus. J Am Heart Assoc. 2014;3:e001156. doi: 10.1161/JAHA.114.001156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PM, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick RL, Quarfordt SH, Maeda N. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J Biol Chem. 1997;272:17972–17980. doi: 10.1074/jbc.272.29.17972. [DOI] [PubMed] [Google Scholar]
- Veinbergs I, Masliah E. Synaptic alterations in apolipoprotein E knockout mice. Neuroscience. 1999;91:401–403. doi: 10.1016/S0306-4522(98)00602-2. [DOI] [PubMed] [Google Scholar]
- Veinbergs I, Mallory M, Sagara Y, Masliah E. Vitamin E supplementation prevents spatial learning deficits and dendritic alterations in aged apolipoprotein E-deficient mice. Eur J Neurosci. 2000;12:4541–4546. doi: 10.1111/j.1460-9568.2000.01308.x. [DOI] [PubMed] [Google Scholar]
- Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, Bu G, Frieden C, Holtzman DM. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc Natl Acad Sci U S A. 2013;110:E1807–E1816. doi: 10.1073/pnas.1220484110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilaró S, Camps L, Reina M, Perez-Clausell J, Llobera M, Olivecrona T. Localization of lipoprotein lipase to discrete areas of the guinea pig brain. Brain Res. 1990;506:249–253. doi: 10.1016/0006-8993(90)91258-I. [DOI] [PubMed] [Google Scholar]
- Wang H, Eckel RH. Lipoprotein lipase in the brain and nervous system. Annu Rev Nutr. 2012;32:147–160. doi: 10.1146/annurev-nutr-071811-150703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Walker D, Bernardo A, Brodbeck J, Balestra ME, Huang Y. Intron-3 retention/splicing controls neuronal expression of apolipoprotein E in the CNS. J Neurosci. 2008;28:1452–1459. doi: 10.1523/JNEUROSCI.3253-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbi V, Wiesmann M, Emmerzaal TL, Jansen D, Van Beek M, Mutsaers MP, Beckmann CF, Heerschap A, Kiliaan AJ. Resting-state functional connectivity changes in aging apoE4 and apoE-KO mice. J Neurosci. 2014;34:13963–13975. doi: 10.1523/JNEUROSCI.0684-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]