

Abstract
The impact of aging on CNS white matter (WM) is of general interest because the global effects of aging on myelinated nerve fibers are more complex and profound than those in cortical gray matter. It is important to distinguish between axonal changes created by normal aging and those caused by neurodegenerative diseases, including multiple sclerosis, stroke, glaucoma, Alzheimer's disease, and traumatic brain injury. Using three-dimensional electron microscopy, we show that in mouse optic nerve, which is a pure and fully myelinated WM tract, aging axons are larger, have thicker myelin, and are characterized by longer and thicker mitochondria, which are associated with altered levels of mitochondrial shaping proteins. These structural alterations in aging mitochondria correlate with lower ATP levels and increased generation of nitric oxide, protein nitration, and lipid peroxidation. Moreover, mitochondria–smooth endoplasmic reticulum interactions are compromised due to decreased associations and decreased levels of calnexin and calreticulin, suggesting a disruption in Ca2+ homeostasis and defective unfolded protein responses in aging axons. Despite these age-related modifications, axon function is sustained in aging WM, which suggests that age-dependent changes do not lead to irreversible functional decline under normal conditions, as is observed in neurodegenerative diseases.
SIGNIFICANCE STATEMENT Aging is a common risk factor for a number of neurodegenerative diseases, including stroke. Mitochondrial dysfunction and oxidative damage with age are hypothesized to increase risk for stroke. We compared axon–myelin–node–mitochondrion–smooth endoplasmic reticulum (SER) interactions in white matter obtained at 1 and 12 months. We show that aging axons have enlarged volume, thicker myelin, and elongated and thicker mitochondria. Furthermore, there are reduced SER connections to mitochondria that correlate with lower calnexin and calreticulin levels. Despite a prominent decrease in number, elongated aging mitochondria produce excessive stress markers with reduced ATP production. Because axons maintain function under these conditions, our study suggests that it is important to understand the process of normal brain aging to identify neurodegenerative changes.
Keywords: aging, axon, mitochondrial dynamic, myelin, neurodegeneration, three-dimensional electron microscopy
Introduction
Aging is associated with many functional and morphological changes in the CNS. The impact of aging on CNS white matter (WM) is of particular interest because it appears that the global effects of aging on myelinated nerve fibers are more complex and profound than those in cortical gray matter (Peters, 1999). Myelinated nerve fibers in WM deteriorate structurally and functionally with age, leading to impaired communication between different parts of the CNS. The underlying reasons for increased susceptibility with age to stroke, dementia, Alzheimer's disease, or other neurodegenerative diseases are just beginning to be explored; therefore, it is important to distinguish between WM structural and functional changes that are due to natural aging and pathological changes associated with neurodegenerative diseases.
The isolated optic nerve is a unique part of the CNS; it is devoid of neuronal cell bodies and comprises myelinated axons of retinal ganglion cells together with supporting glial cells (Foster et al., 1982; Ross and Pawlina, 2016). Optic nerve axons are sensitive to the aging process (Cavallotti et al., 2002, 2003), and supporting glial cells undergo age-associated changes (Sandell and Peters, 2002). Aging is associated with an increased incidence of ocular diseases, including glaucoma and macular degeneration, as well as ocular complications that are part of systemic diseases such as hypertension and diabetes mellitus (Heflin and Lum, 2008). Moreover, because the optic nerve is a pure CNS WM tract, it has been extensively used to investigate WM injury mechanisms during aglycemia, stretch injury, anoxia, and ischemia (Baltan et al., 2008; Baltan, 2012; Murphy et al., 2014). We studied axon function responses to ischemia as a function of age by exposing mouse optic nerves (MONs) to oxygen and glucose deprivation (OGD) and showed that aging, specifically at 12 months of age, marks the onset of a prominent vulnerability of axon function to ischemia (Baltan et al., 2008; Baltan, 2009, 2012). WM structure and function are integrated, and thus several age-related changes in WM structure, such as upregulation of GLT-1 and the resultant early and robust release of glutamate, contribute to the increased vulnerability of aging WM to ischemia (Baltan et al., 2008; Baltan, 2014). Consequently, approaches that promote axon functional recovery in aging WM after ischemia correlate with reducing excitotoxicity. Interestingly, agents that conserve ATP and preserve mitochondria when applied preinjury or postinjury confer protection to axons in WM (Baltan et al., 2011; Baltan, 2012). Therefore, we hypothesize that age-dependent alterations in mitochondrial structure and function play a crucial role in the increased vulnerability of axon function to ischemia. The aim of this study was to systematically quantify axonal changes, alterations in axonal mitochondrial structure, and changes in mitochondrial interactions with smooth endoplasmic reticulum (SER) in MONs obtained from 12-month-old mice, because at this age MONs show the most prominent changes in response to OGD (Baltan et al., 2008; Baltan, 2014). Our results demonstrate that aging correlates with a reduction in axonal numbers but an increase in axon diameter, axon volume, myelin thickness, and nodal and internodal lengths. Moreover, there were fewer axonal mitochondria in aging MONs, but they were longer and thicker, correlating to alterations in mitochondrial shaping proteins underlying fission and fusion. Furthermore, less mitochondria were associated with SER in aging MONs. In addition, levels of SER stress and Ca2+ binding proteins were lower with age. All of these structural changes correlated with reduced ATP production and increased oxidative injury by-products, suggesting that morphological changes in aging axonal mitochondria compromise energy status. Despite these structural changes, aging axons sustain their conduction properties and fail only when challenged with an injury such as ischemia (Baltan et al., 2008). This is an important distinction between the sustained functional capacity of an aging brain in contrast to the prominent functional decline observed during neurodegenerative diseases.
Materials and Methods
Animals and chemicals.
All experimental procedures were approved by the Institutional Animal Care and Use Committee of the Cleveland Clinic. All chemicals used for electron microscopy (EM) were purchased from Electron Microscopy Sciences except for aspartic acid (Sigma-Aldrich). The sources for chemicals for electrophysiology experiments have been described previously (Baltan et al., 2008; Uo et al., 2009). The Thy-1 mito-CFP mice (Misgeld et al., 2007) were originally received by the senior author at the University of Washington and were later brought to and bred in the Cleveland Clinic animal facility.
Preparation of optic nerves and recording techniques.
MONs were obtained from adult male C57BL/6J mice or from mice expressing mitochondrial-targeted cyan fluorescent protein (CFP) on a C57BL/6 background (Thy-1 mito-CFP; Misgeld et al. 2007) at 1 or 12 months of age. After CO2 asphyxiation, MONs were gently freed from their dural sheaths, placed in a Haas Top perfusion chamber (Harvard Apparatus) and superfused with artificial cerebrospinal fluid (ACSF) containing the following (in mmol/L): 124 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 23 NaHCO3, and 10 glucose. All experiments were performed at 37°C. Suction electrodes back-filled with ACSF were used for stimulation (Isostim 520; WPI) and for recording compound action potentials (CAPs). The recording electrode was connected to an Axoclamp 900A amplifier (Molecular Devices) and the signal was amplified 20 or 50 times, filtered at 3 kHz (SR560, Stanford Research Systems), and acquired at 20–30 kHz. Stimulus pulse (30 μs duration) strength was adjusted to evoke the maximum CAP possible and then increased another 25% for supramaximal stimulation. MONs were allowed to equilibrate for at least 15 min in the recording chamber in normal ACSF. During experiments, the supramaximal CAP was elicited every 30 s. MONs isolated from 1- or 12-month-old mice were placed in a preset grid (mesh #64-0198 NYL/Mesh, Warner Instruments) in between the two suction electrodes to keep nerve length constant (∼5.5–6 mm).
Three-dimensional electron microscopy.
Animals were anesthetized using isoflurane and perfused intracardially with 0.1 m sodium cacodylate buffer containing 4% paraformaldehyde and 2.5% glutaraldehyde, pH 7.4, with ∼80 ml per mouse. MONs were extracted and washed three times in cold 0.1 m sodium cacodylate buffer. The samples were then processed sequentially in 2% osmium tetroxide/potassium ferrocyanide for 1 h, 1% thiocarbohydrazide (freshly prepared and placed in a 60°C oven for 1 h to completely dissolve) for 20 min, and 2% osmium tetroxide solution for 30 min, with three washes in double-distilled water in between. The samples were then placed in saturated uranyl acetate solution overnight. The next day, after three washes in double-distilled water, samples were incubated in lead aspartate solution (prepared by dissolving 0.066 g of lead nitrate in 10 ml 0.03 m aspartic acid, pH 5.5) at 60°C for 30 min. After three washes in double-distilled water at room temperature, the samples were dehydrated over an ethanol gradient series of freshly prepared ice-cold solutions of 20, 60, 80, 90, and 95% ethanol once and then three times in anhydrous 100% ethanol. Samples were then incubated in anhydrous ice-cold acetone for 10 min on ice and for another 10 min at room temperature. Tissue samples were then transferred before drying to Epon resin formulated as 10 ml EMBed-812, 8 ml dodecenyl succinic anhydride, 4 ml methyl-5-norbornene-2,3-dicarboxylic anhydride, and 0.4 ml 2,4,6-tri(dimethylaminomethyl)-phenol. The samples were placed in 50% Epon (in acetone) for 2 h and switched to 100% Epon overnight. The samples were then transferred into fresh 100% Epon for 2 h. MON samples were transferred into molds, and fresh Epon resin was poured and left to polymerize at 60°C for 2 d.
From the resin-embedded tissue, a sample block of size 0.5 × 0.5 × 0.5 mm was trimmed using a razor blade and mounted on a pin, which was viewed under 3-D serial block-face scanning electron microscopy. The diamond knife was imaged by serial block-face scanning EM using a Zeiss Sigma VP scanning electron microscope equipped with a Gatan 3View in-chamber ultramicrotome and low kilovolt backscattered electron detector. Images were generated at 2.0–2.25 kV under standard vacuum conditions using an aperture set at 30 μm. The diamond knife was set to make ∼250 to 300 sections at a thickness of 100 nm at ∼8 nm/pixel resolution (4000× magnification). Image sets were registered using the Fiji (ImageJ) plugin for linear alignment with SIFT (Schindelin et al., 2012). Simple macros were written for Fiji (ImageJ) to randomly select axons for 3-D analysis. The experimenters were blinded to the conditions of the groups analyzed.
Axonal and mitochondrial 3-D reconstructions using reconstruct.
Image sets were analyzed using ImageJ and Reconstruct (Fiala and Harris, 2001) software. Areas were traced using either the wildfire selection tool or manual tracing. The edges of wildfire areas were checked and manually corrected. Each trace with the same name is considered as one object by the software, and a 3-D model can be generated for each object (axon, mitochondrion). The “Z trace” feature was used to measure 3-D lengths manually. We traced 20 axons (average length, 53 μm) per animal with their resident mitochondria from each image set. The slice thickness (100 nm) between slices and the pixel size (9.07 nm) for each image set were manually entered. Once an entire axon was traced from the image set, the object list on Reconstruct software (version 1.1.0.0; Fiala and Harris, 2001) automatically generated values for axon volume. The volume was calculated using the Cavalieri formula, where each section contributes the product of its enclosed area times the section thickness (Fiala, 2005):
The object and Z trace list generated by Reconstruct software (version 1.1.0.0; Fiala and Harris, 2001) at the end of the tracing was imported to Microsoft Excel, and the corrected volume, axon length, mitochondrial volume, and length were used to calculate corrected volume (volume/length), axon diameter, mitochondria per 10 μm, and mitochondria per unit volume for quantification. The Z trace distance list generated was sorted to acquire the distances from mitochondria to the nodes of Ranvier in the same axon.
Smooth endoplasmic reticulum: 3-D EM reconstruction and analysis.
For SER analysis, 8 bit TIFF image files were opened in Fiji and inverted for better contrast to observe the SER. After enhancing contrast, imaging parameters were set, such as pixel size and voxel depth, based on the image acquisition settings. Three stacks representing at least 10 μm of the axon of interest (randomly picked) were generated and saved in the TIFF format for axon, mitochondria, and associated SER tracing and were converted to RGB (red, green, blue) format. The paint tool macro in Fiji was used to color the SER green and the mitochondria red. The axons were flood-filled in one of the stacks and, using the paintbrush tool, coloring was completed over areas that the macro missed. The SER proximity macro was then used to generate color-coded images and to generate output text files on the information about the proximity of SER to axon, mitochondria, and the plasma membrane, which were exported to Microsoft Excel and quantified. In the proximity stack, blue represented SER portions near the plasma membrane, green represented SER portions that were cytosolic, and yellow represented SER portions near mitochondria.
Multiphoton imaging and pixel intensity analysis of Thy-1 mito-CFP mice.
Expression of CFP was imaged using a Leica TCS SP511 upright multiphoton laser-scanning microscope. Two to three adjacent sections for each MON were imaged with the wavelength set at 900 nm. A total of 10 optical sections of 1 μm thickness at 512 × 512 pixel size were collected in the z-axis from a single microscopic field using the 25× objective lens (HCX IRAPO, water immersion; numerical aperture, 0.95; free working distance, 2.5 mm) under fixed gain, laser power, pinhole, and photomultiplier tube settings. To compare pixel intensity, all sections were processed concurrently with Leica imaging software (LAS-AF version 2.7) using a single channel. The Z stacks were projected into a single plane image before analysis and assessment of pixel intensity.
Immunohistochemistry.
Perfusion-fixed (4% paraformaldehyde in PBS) MON sections were used for immunohistochemical analysis. Cryoprotection was achieved in 30% sucrose for 16–18 h. Sections, 10 to 16 μm thick, from each MON were blocked and permeabilized in 5–40% normal goat/donkey (50% by volume) serum (Sigma-Aldrich) and 0.3% Triton X-100 (Sigma-Aldrich) for 1 h at room temperature. All primary antibodies listed in Table 1 were prepared in their respective solutions. MONs were incubated in primary antibodies at 4°C overnight without agitation. After a thorough wash in PBS, the tissue was exposed to a secondary antibody prepared in 2% normal goat serum for overnight incubation. Donkey anti-rabbit Cy5, anti-mouse Cy5, anti-mouse Cy3, and anti-rabbit Cy3 (Jackson ImmunoResearch) were used at 1:100. Sections were double or triple labeled.
Table 1.
List of antibodies used
| Antigen | Host | Dilution | Source |
|---|---|---|---|
| CASPR | M | 1:100 | Neuromab |
| Nav1.6 | R | 1:100 | Alomone Labs |
| Mitofusin-1 (mfn-1) | M | 1:250 | Novus Biologicals |
| Mitofusin-2 (mfn-2) | R | 1:500 | Sigma-Aldrich |
| Optic atrophy protein 1 (Opa-1) | R | 1:500 | BD Biosciences |
| Dynamin-related protein 1 (drp-1) | R | 1:500 | BD Biosciences |
| 4-HNE (lipid peroxidation by-products) | M | 1:250 | Alpha Diagnostic |
| 3-NT (protein nitrosylation by-products) | R | 1:500 | Sigma-Aldrich |
R, Rabbit; M, mouse.
Immunoblotting.
MONs were extracted, flash frozen in liquid nitrogen, and stored at −80°C until use. MON samples (one or two pairs) were homogenized in 75 μl of RIPA buffer on ice (Millipore) with protease inhibitor and phosphatase inhibitor cocktails (Sigma-Aldrich) and sonicated five times for 1 s or until the homogenate looked smooth on ice. Following homogenization, the samples were vortexed every 2 min for a total of 10 min. The samples were centrifuged at 14,000 rpm for 10 min, the supernatant was collected, and the protein concentration was estimated using a BCA protein assay kit (Thermoscientific). Protein lysates were diluted in 4× Laemmli sample buffer (Bio-Rad) with 2-mercaptoethanol (10:1) and incubated at 95°C for 10 min. Equal amounts of protein were loaded onto each well (10–22 μg) of a stain-free 4–20% Mini-Protean TGX precast gel (Bio-Rad). After gel electrophoresis, the protein samples were transferred to a nitrocellulose membrane. The blots were blocked in 5% powdered dry milk in TBS–Tween 20 (TBST, 0.1%) for 1 h at room temperature. Table 1 shows a list of primary antibodies with their respective dilutions used. Both primary antibodies and secondary antibodies were prepared in blocking buffer. Primary antibodies were incubated overnight at 4°C. After three washes in TBST for 5 min, the blots were incubated in secondary antibodies (goat anti-mouse HRP or goat anti-rabbit HRP based on the host of the primary antibody) at room temperature for 2 h. Chemiluminescence was detected using Clarity Western ECL substrate (Bio-Rad), imaged using a Bio-Rad Chemidoc MP system (Bio-Rad), and analyzed using Imagelab software (version 4.1; Bio-Rad) for protein quantification.
Nitric oxide synthase assays.
MONs were frozen in liquid nitrogen at the end of electrophysiology experiments. A nitric oxide synthase (NOS) assay kit (catalog #NB78, Oxford Biomedical) was used. MONs were sonicated in 75 μl RIPA buffer, vortexed every 2 min for a total of 10 min, and centrifuged at 14,000 rpm for 10 min at 4°C. After estimating protein concentration of the supernatant with a BCA protein assay, supernatant with 20 μg protein was used for NOS assays. Absorbance was measured at 540 nm (SpectraMax M2, Molecular Devices). The blank values were subtracted from the raw data and the NO generated was calculated from a log–log plot of the standard curve and then normalized to protein concentration. The values are expressed as fold change relative to control.
ATP assays.
ATP extraction was performed as described previously (Khan, 2003) and modified for optic nerve (Baltan et al., 2010, 2011). Briefly, a MON was cut and placed in 75 μl of 10% perchloric acid (HClO4). The sample was sonicated five times for 1 s and centrifuged at 4500 rpm for 10 min at 4°C. The supernatant was collected, neutralized with 30 μl 2.5 m potassium hydroxide (KOH), and centrifuged at 14,000 rpm for 10 min at 4°C. The precipitate was removed and the supernatant was kept on ice for ATP measurements. Total cellular ATP was measured using a bioluminescence assay kit (catalog #11 699 709 001, Roche). Samples were diluted (1:100) in buffer and mixed with luciferase reagent provided by the manufacturer. The absorbance was measured at 560 nm on a BioTek Synergy 4 system using Gen 5 data analysis software. Absorbance of blank values was subtracted from the raw data. Samples of known ATP concentrations were used to generate a standard curve. ATP levels in experimental samples were calculated from a log–log plot of the standard curve and then normalized to protein concentration. The values are expressed as micromole of ATP per milligram of protein. Protein content was quantified in one nerve (BCA protein assay kit, Thermo Fisher Scientific), and ATP levels were measured in the other.
Data analysis.
Optic nerve function was monitored quantitatively as the area under the supramaximal CAP using Clampfit (version 10.2, Molecular Devices). The CAP area, which quantifies CAP amplitude and duration, is proportional to the total number of excited axons and represents a convenient and reliable means of monitoring optic nerve axon function (Cummins et al., 1979; Stys et al., 1991). Note that another important factor in defining the CAP area is axon diameter, as larger axons are expected to generate larger CAP areas due to influx of Na+, variation in membrane potentials, and concentrations and densities of Na+ channels (subtypes; Hodgkin and Katz, 1949). Therefore, the CAP area is a complex spatiotemporal summation of action potentials from individual axons (Stys et al., 1991). For comparing CAP areas between young and aging animals, an average of 10 CAPs at baseline was measured. Original data were normalized by setting the mean of initial baseline values (measured over 15 min) to a value of 1. Results from several nerves were pooled, averaged, and plotted against time. All data are presented as the median with the first and third quartiles (Figs. 1E, 2C,E,G, 3G,I, 5C,E,G, 6G) or mean ± SEM (Figs. 3N, 4C, 5I–L, 6C,J). Graphpad Prism (version 4.0c, Graphpad Software) was used for statistical analysis. Statistical significance was determined by unpaired two-tailed Student's t tests or one-way ANOVAs. For normally distributed 3-D EM data, Student's t tests were used for comparing two groups, and one-way ANOVAs followed by Bonferroni's post hoc tests were used for three or more groups (Fig. 6C). Nonparametric tests such as two-tailed Mann–Whitney U tests and Kruskall–Wallis tests were used for analysis for comparing the medians of nonnormally distributed data (Figs. 1E, 2C,E,G, 3G,I, 5C,E,G, 6G). The n values indicate the number of optic nerves in Figures 3N, 4C, 5I–L, and 6J, while in others they indicate the number of axons or mitochondria. The p values and significance values are indicated individually for each figure in the text.
Figure 1.

An age-related reduction in the number of optic nerve axons is associated with an increase in axonal volume. A, B, Blocks of optic nerves obtained from young (1 month; A) and aging (12 month; B) MONs imaged by 3-D EM showed fewer but thicker axons with aging. C–F, Tracing individual axons among longitudinal sections of young (yellow; C) and aging (magenta; D) optic nerve showed increased axon volume (young, median volume, 0.8 μm3/μm, n = 60 vs aging, 1.1 μm3/μm, n = 60; E) due to a majority of axons with a volume larger than 1.2 μm3 with aging (F). **p = 0.002 (two-tailed Mann–Whitney U test). Boxes (E) show median with first and third quartiles. Bars (F) indicate relative frequencies. Scale bars: (In B, D) A–D, 5 μm.
Figure 2.

An age-related increase in axonal diameter correlates with a lower g ratio due to increased thickness of the myelin sheath. A, B, Single plane cross-sectional images obtained from 3-D EM stacks show numerous round axons in young (A) and fewer but larger axons in aging (B) optic nerve. Scale bar (B), 0.5 μm. C–F, The diameter of axons increased from 0.4–1.6 μm (median, 0.8 μm; n = 1198) to 0.5–4.1 μm (median, 0.9 μm; n = 1011; ***p < 0.0001, two-tailed Mann–Whitney U test; C, D), while the number of axons decreased (E, F), reducing the number of axons per unit area with aging (median, 18.5 axons/μm2; n = 12) to 14 axons/μm2 (n = 12; ***p = 0.0002, two-tailed Mann–Whitney test). G, H, Because the myelin sheath wrapped around axons increased more compared to axon diameter, the g-ratio (diameter of axon/diameter of axon + myelin thickness) was reduced independent of axon diameter with aging. Boxes in C, E, and G show the median with the first and third quartiles. Bars in D, F, and H indicate relative frequencies.
Figure 3.
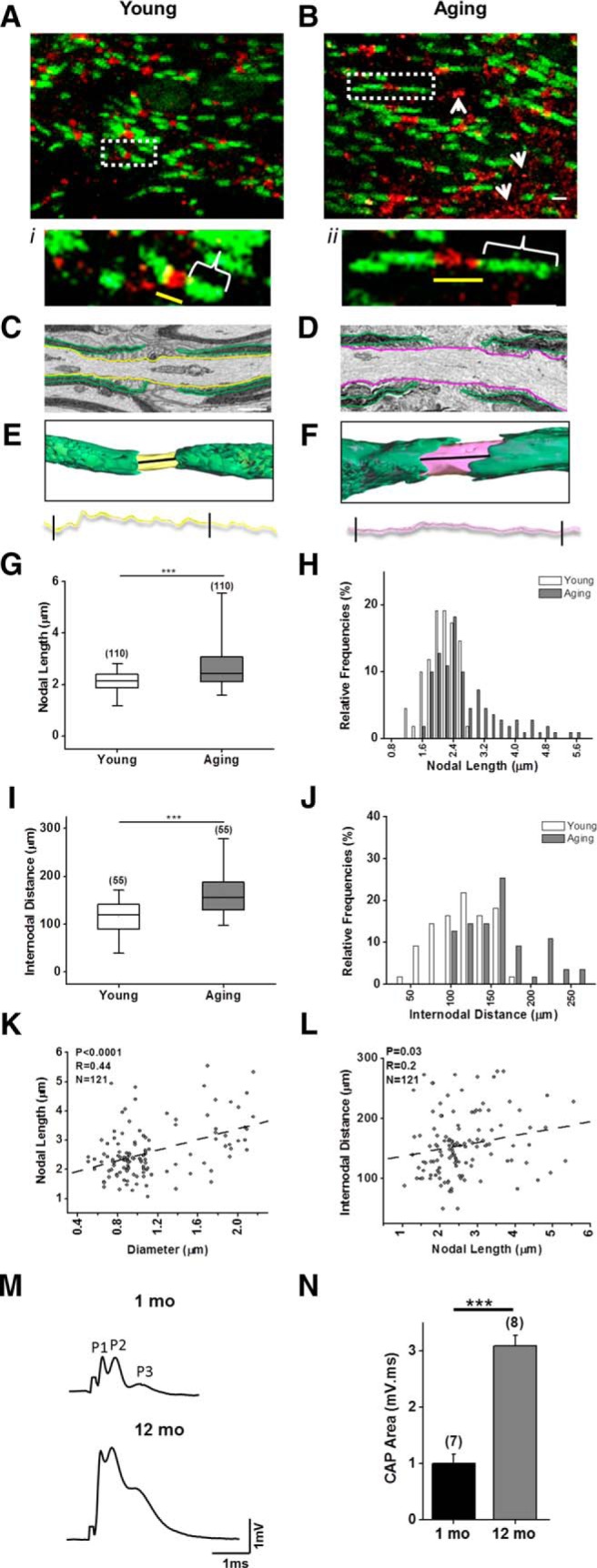
Aging results in longer nodal and paranodal lengths without affecting axon conduction properties. A, B, Immunohistochemical labeling of Nav1.6 (red) and Caspr (green) in young (1 month old; A) and aging (12 month old; B) optic nerves showed reorganization of nodal and paranodal regions with aging such that in addition to the nodal region, Nav1.6 immunoreactivity showed ectopic localization (white arrows), while Caspr was thinner and elongated (white brackets) and nodal length was longer (yellow line; i, ii). C–F, Tracing young (yellow; C) and aging (magenta; D) axons and paranodal structures (green) in longitudinal sections of 3-D EM images outlined nodal lengths in young (yellow; E) and aging (magenta; F) axons. Nodal lengths were extracted from reconstructed 3-D EM images and the interval quantified is shown between brackets (black lines). G–L, Nodal (young, median, 2.1 μm, n = 110; aging, 2.4 μm, n = 110; G, H) and internodal distances increased in aging axons (young, median 120.1 μm, n = 55; aging, 155.4 μm, n = 55; I, J), maintaining a proportional relationship such that nodal length increased with increases in axon diameter (Spearman's correlation, r = 0.44, p < 0.0001; K) and internodal distance increased with increased nodal length (Spearman's correlation, r = 0.19, p = 0.029; L). M, N, Despite age-dependent reorganization of nodal architecture (M), the conduction velocity properties of evoked CAPs remained constant, as depicted by three individual peaks (P1, P2, and P3, reflecting three subsets of axons; N) and improved spatiotemporal summation of axon function (p < 0.0001, unpaired two-tailed Student's t test). Boxes in G and I show the median with the first and third quartiles. Bars in H and J show relative frequencies, and bars in N show mean ± SEM. ***p < 0.0001 (two-tailed Mann–Whitney U test).
Figure 5.
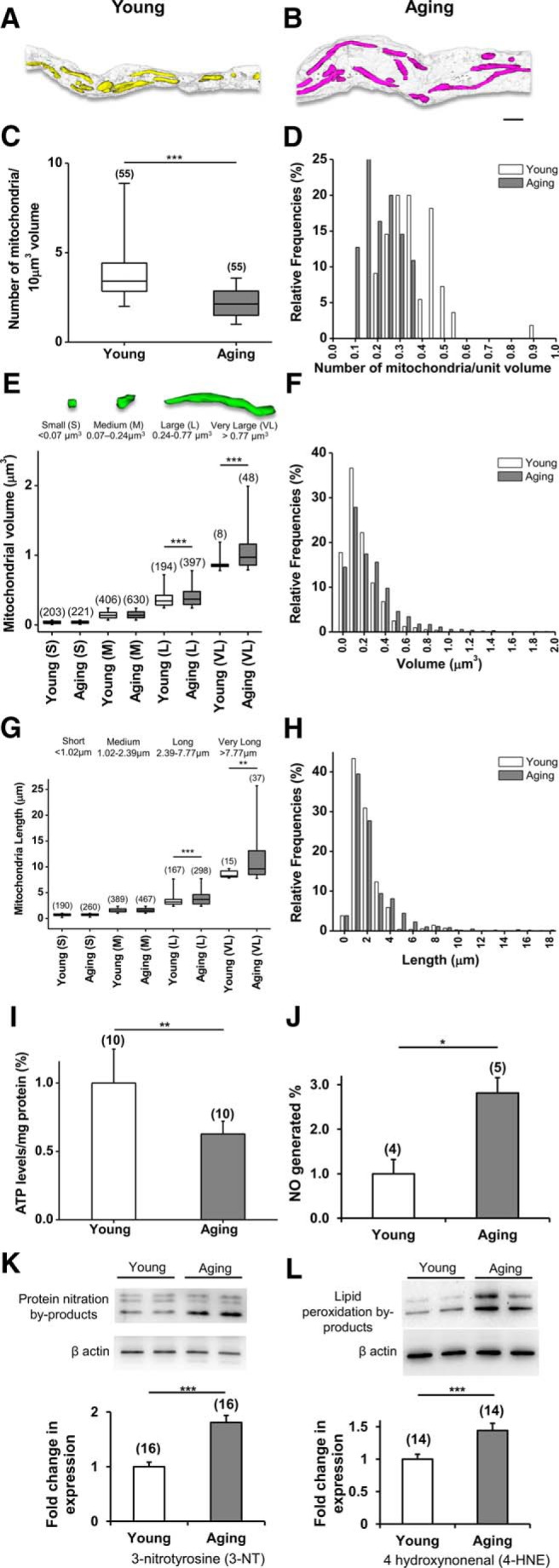
Aging axons display fewer but longer and thicker mitochondria. A, B, Tracing and reconstruction of 3-D EM images of young (1 month old; A) and aging (12 month old; B) axons revealed numerous mitochondria in young (yellow) axons and fewer mitochondria (magenta) in aging axons. Note the white-gray outline of respective axons housing the mitochondria. C–H, Together with an increase in axon volume with aging, the number of mitochondria within the axon volume was reduced (C, D), but the mitochondrial volume increased (E, F), particularly in long (L) and very long (VL) mitochondria, mainly due to an increase in mitochondrial length reaching above 10 μm with aging (G, H). I–L, Age-dependent changes in mitochondrial architecture led to lower ATP levels (young, 1.0 ± 0.1%, n = 10; old, 0.6 ± 0.1%, n = 10; **p = 0.001, unpaired two-tailed Student's test; I) and increased oxidative stress levels as measured by elevated NO levels (young, 1.0 ± 0.1%, n = 4; old, 2.8 ± 0.1, n = 5; *p = 0.03, unpaired two-tailed Student's t test; J), protein nitrosylation (young, 1.0 ± 0.1%, n = 16; old, 1.8 ± 0.1%, n = 16; ***p = 0.0001, unpaired two-tailed Student's t test; K), and lipid peroxidation (young, 1.0 ± 0.1%, n = 14; old, 1.4 ± 0.1%, n = 14; ***p = 0.004, unpaired two-tailed Student's t test; L). Boxes in C, E, and G show the median with the first and third quartiles. Bars in D, F, and H indicate relative frequencies. Bars in I–L show mean ± SEM.
Figure 6.
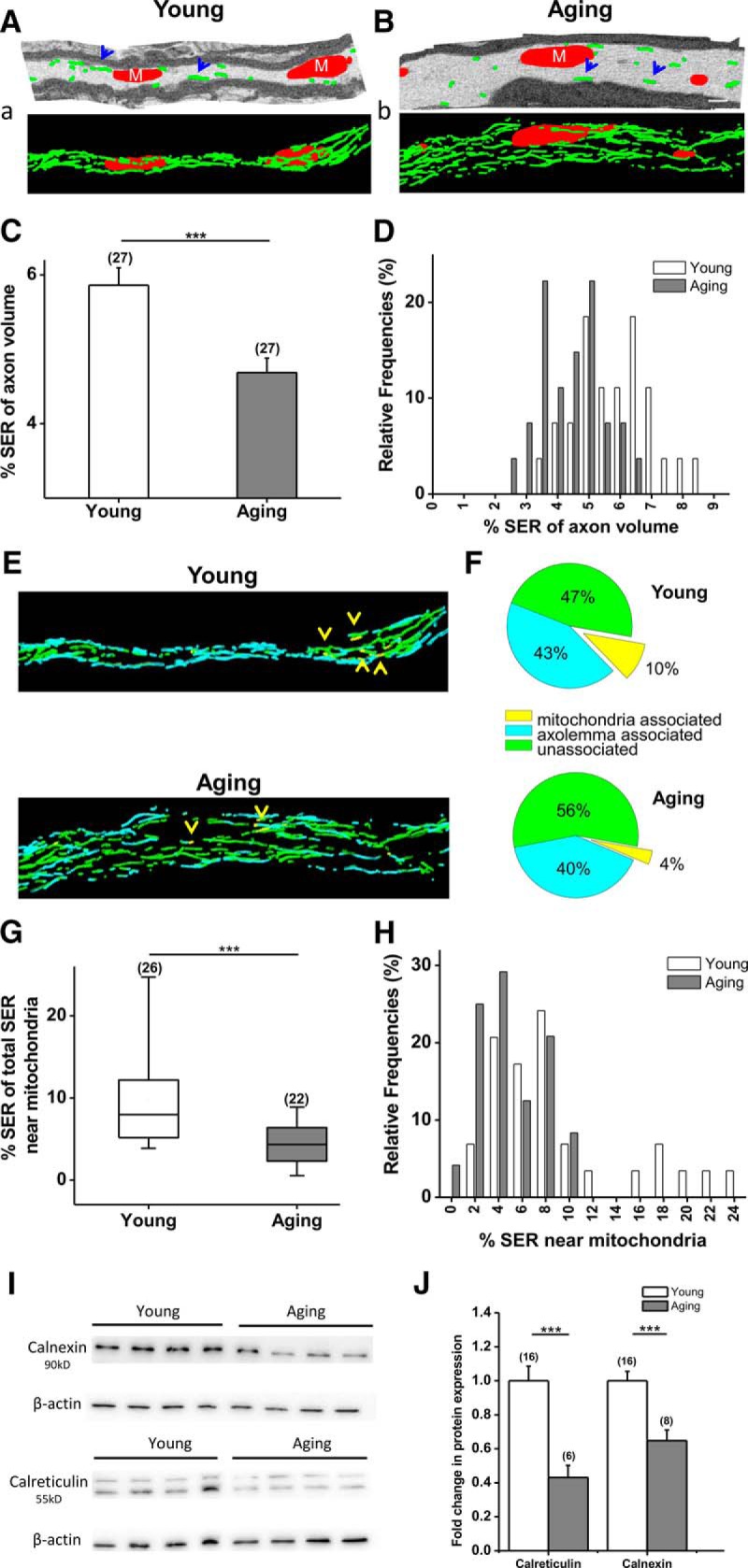
Aging impairs SER networks and mitochondria-SER interactions. A–b, Tracing mitochondria and SER in young (A) and aging (B) axons in 3-D EM reconstructed images displayed the SER network (green, blue arrowheads) and its interlacing of mitochondria (red) in young (a) and aging (b) axons. C, D, Although the SER network seemed to be expanded with aging (a, b) due to increased axon volume, the SER network covered a smaller area within aging axons (young, 5.9 ± 2.3%, n = 27; old, 4.4 ± 1.9%, n = 27). ***p < 0.0001 (unpaired two-tailed Student's t test). E, F, Consequently, portions of the SER network associated with mitochondria (yellow, yellow arrowheads) or axolemma (cyan) or unassociated (green) portions changed such that less SER network was associated with mitochondria (young, 10.0 ± 1.2%, n = 25 axons; old, 3.6 ± 0.6%, n = 25 axons; p < 0.0001, unpaired two-tailed Student's t test), while unassociated portions increased in aging axons (young, 46.8 ± 3.2%, n = 25; old, 55.8 ± 2.2%, n = 25; p = 0.03, unpaired two-tailed Student's t test). G, H, Moreover, the mitochondria-associated SER portion showed fewer contact points between the SER network and the interlaced mitochondria. I, J, Consequently, calnexin and calreticulin, two chaperone proteins located on SER–mitochondria contact points, were decreased with aging (calnexin, young, 1.0 ± 0.05, n = 16; 0.65 ± 0.06, n = 6; ***p = 0.0006, unpaired two-tailed Student's t test; calreticulin, young, 100.0 ± 8.6%, n = 16; old, 49.2 ± 7.2%, n = 8; ***p = 0.0003, unpaired two-tailed Student's t test). Boxes in G show the median with the first and third quartiles. Bars in D and H indicate relative frequencies. Bars in C and J show mean ± SEM.
Figure 4.
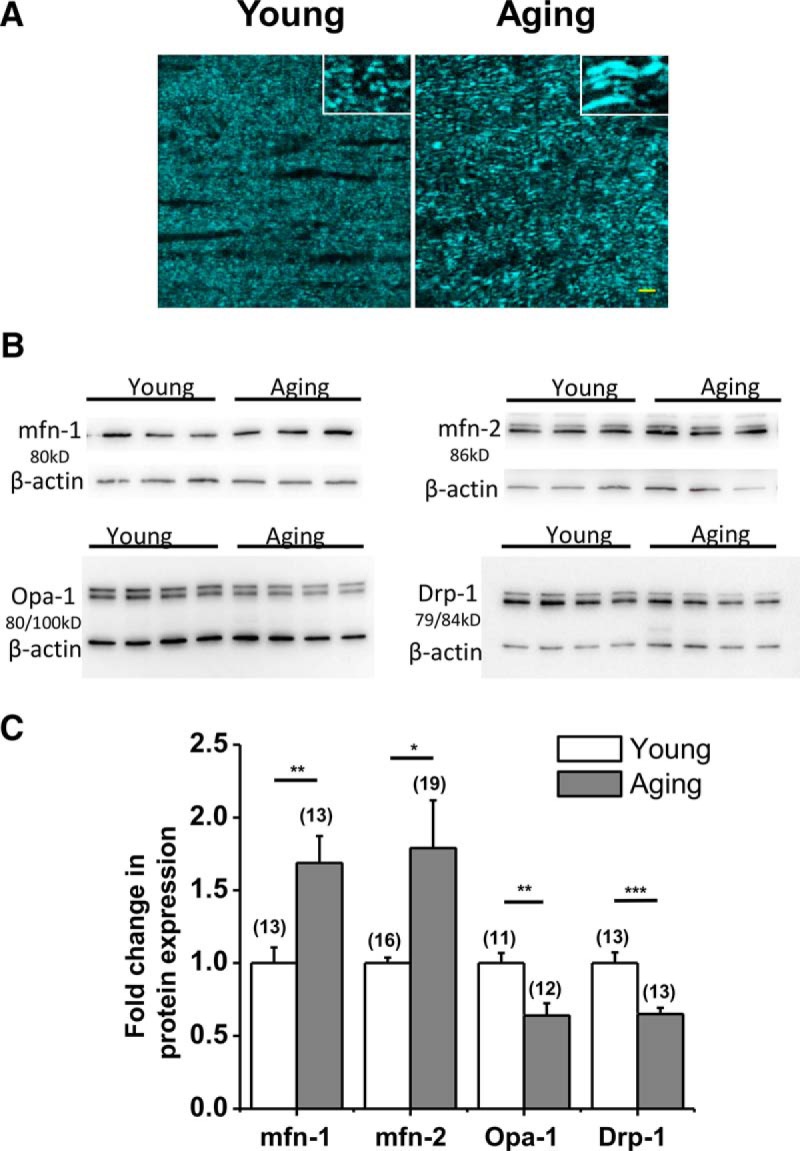
Age-related alterations in mitochondrial architecture correlate with lower ATP and higher oxidative stress levels. A, Two-photon confocal microscopy images in optic nerve obtained from Thy-1 mito-CFP mice demonstrated small round CFP(+) mitochondria in young optic nerves, as opposed to thicker longer mitochondria in aging optic nerves. Scale bar, 5 μm. Insets are enlarged magnifications. B, C, Aging (gray columns) caused a mismatch in mitochondrial shaping protein levels favoring fusion by prominent increases in mfn-1 (young, 1.0 ± 0.1%, n = 16; old, 1.7 ± 0.1%, n = 13; **p = 0.003, unpaired two-tailed Student's t test) and mfn-2 (young, 1.0 ± 0.1%, n = 16; old, 1.8 ± 0.2%, n = 19; *p = 0.02, unpaired two-tailed Student's t test), together with a reduction in Drp-1 levels (young, 1.0 ± 0.1%, n = 13; old, 0.6 ± 0.1%, n = 13; ***p = 0.0004, unpaired two-tailed Student's t test), compared to young optic nerves (white columns). Opa-1 levels also decreased with age (young, 1.0 ± 0.1%, n = 11; old, 0.6 ± 5.6%, n = 12; **p = 0.003, unpaired two-tailed Student's t test). Bars (C) show mean ± SEM. values.
Results
Aging axons are thicker and have lower g ratios
Aging axons become thicker and larger in size. Three-dimensional reconstruction blocks (dimensions, 35 × 20 × 19 μm) of serial 3-D EM images of optic nerves from mice at 1 month (Fig. 1A,C) and 12 months (B,D) of age revealed closely packed, well-myelinated nerve fibers. Aging optic nerves were characterized by a smaller number of axons having larger diameters wrapped with thicker myelin sheaths (Fig. 1B,D), compared to the more numerous thinner axons with slender myelin sheaths of young optic nerves (A,C). Three-dimensional reconstructions of individual axons showed that thin young axons weaved in and out of multiple planes (yellow), while thicker aging axons elongated in a single plane with less interlacing (magenta). Axon volume (per unit length) in aging axons was increased by 50% (Fig. 1E,F; young, median volume, 0.8 μm3/μm, n = 60 vs aging, 1.1 μm3/μm, n = 60; p = 0.002, two-tailed Mann–Whitney U test) compared to young axons due to more aging axons having larger diameters (Fig. 2C,D). Consequently, the optic nerve diameter increased modestly by 18% (data not shown), verifying that the increase in axon diameter was compensated for by a parallel reduction in the number of axons (Fig. 2E,F).
The ratio of the inner axonal diameter to the outer diameter, or g ratio, is widely used as a functional and structural index of optimal axonal myelination. In single cross-sectional slices, numerous axons with myelin sheaths were packed densely in young optic nerves (Fig. 2A), while a small number of axons with dramatically larger diameters and thicker myelin sheaths were prominent in aging optic nerves (Fig. 2B). Axon diameter [calculated as diameter = 2 × √(area/π)] increased with age; young axons had a diameter range of 0.4–1.6 μm, while aging axons had a wider range of 0.5–4.1 μm (median diameter, young, 0.8 μm, n = 1198; aged, 0.9 μm, n = 1011; p < 0.0001, two-tailed Mann–Whitney U test; Fig. 2C,D). The number of axons (per unit area of 27 μm2) was reduced with age, from a median of 18.5 axons/unit area (n = 12) down to 14 axons/unit area (n = 12; p = 0.0002, two-tailed Mann–Whitney U test; Fig. 2E,F). Subsequently, axons were grouped based on cross-sectional area (25th percentile, 0.37 μm2; 75th percentile, 0.89 μm2 for young) into three different groups (Fig. 2G,H): small axons of <0.37 μm2; medium axons of 0.37–0.89 μm2; and large axons of >0.89 μm2. All groups of axons displayed lower g ratios with age (p < 0.0001, Kruskall–Wallis test with Dunn's multiple comparison post hoc test) compared to their corresponding young groups.
Aging increases nodal length and internodal distances
Nodes of Ranvier are regular interruptions of long myelin sheaths at specialized regions of axolemma characterized by clusters of voltage-gated sodium channels. Clustering of Nav1.6, the predominant sodium channel at the nodes of Ranvier (Kaplan et al., 2001), is an indicator of proper axoglial contact dictated by myelin (Boiko et al., 2001). This architectural organization of nodal, paranodal, and juxtaparanodal structures of axons is integral for proper saltatory conduction of action potentials. As reported previously (Baltan 2014), sections (50 μm) of young optic nerves immunostained for either pan-sodium channels (Na+) or the NaV1.6 isoform (red) together with contactin-associated protein (Caspr) (green)–rich paranodes (Fig. 3A) all showed identifiable nodes in aging optic nerves (B). Caspr labeling was elongated (brackets) and became thinner (Baltan, 2014), suggesting longer internodal distance with age. Although Nav1.6 is found in unmyelinated axons (Black et al., 2002), it is not normally detected in internodal regions of myelinated axons. However, aging caused ectopic regions of Nav1.6 immunoreactivity (Fig. 3B, white arrows), which may lead to deleterious effects on axons, especially if colocalized with the Na+–Ca2+ exchanger (Craner et al., 2004). Interestingly, nodal length (Fig. 3i,ii, yellow line), measured as the distance between myelin sheaths in 3-D EM reconstructed images (Fig. 3E,F), increased from a median of 2.1 μm (n = 110) in young animals to 2.4 μm (n = 110) with age (p < 0.0001, two-tailed Mann–Whitney U test). Consequently, the internodal distance became wider, with a median of 120.1 μm (n = 55) in young MONs and a median of 155.4 μm (n = 55) in aging MONs (p < 0.0001, two-tailed Mann–Whitney U test; Fig. 3I). This was mainly due to the replacement of nodes of short and intermediate length with longer nodes (Fig. 3J). These architectural alterations in nodes correlated with an increase in axon diameter (Fig. 3K; p < 0.0001, Spearman's rank correlation coefficient, r = 0.44). Thus, internodal and nodal length maintained a proportionate relationship (Fig. 3L; Spearman's rank correlation coefficient, r = 0.19, p = 0.029). Changes in axonal dimensions seem to be proportionally compensated for by the myelin sheath to maintain proper signaling across aging axons under control conditions. Consequently, CAPs evoked by supramaximal stimulation in optic nerves from aging mice maintained the characteristic three-peak shape (Fig. 3M), but displayed a dramatically larger CAP area (Fig. 3N). These CAP peaks indicate at least three groups of axons with different axon diameters, myelin sheath thicknesses, and conduction velocities such that peak 1 (P1) consists of the fastest conducting, largest axons, while peak 2 (P2) represents medium conducting axons with medium diameter, and peak 3 (P3) represents the slowest conducting and smallest diameter axons (Fig. 3M; Baltan et al., 2010). Note that the amplitude of all three CAP peaks and the CAP area in aging MONs were increased (mean ± SEM, 3.1 ± 0.19-fold, n = 8; compared to young MONs, mean ± SEM, 1.0 ± 0.17, n = 7; p < 0.0001, two-tailed unpaired Student's t test) without any conduction delay as reported previously (Baltan et al., 2010). Because the concentration or density of Na+ channels may change the action potential amplitude (Hodgkin and Katz, 1949), prolonged nodal regions with Na+ channel clusters may underlie the increased CAP amplitude with aging. In addition, glutamate release in close proximity of axons (Sasaki et al., 2011) and changes in K+ channel expression (Bay and Butt, 2012) may contribute to the widening of CAPs, resulting in larger CAP area with age. Therefore, aging axons adapt to transmit signals faithfully under control conditions as reported previously (Baltan et al., 2008; Baltan et al., 2010).
Fewer, but longer and larger, mitochondria produce increased oxidative stress markers and less ATP in aging axons
Our observation of CFP(+) mitochondria in Thy-1 mito-CFP mice could not differentiate whether aging led individual mitochondria to become longer and thicker or whether a group of mitochondria formed clusters. For instance, the insets in Figure 4A demonstrate marked increases in length and thickness in a group of CFP(+) mitochondria in aging optic nerve axons obtained from Thy-1 mito-CFP mice compared to young optic nerves. Mitochondrial length is determined by the balance between the rates of mitochondrial fission and fusion and is important for controlling the spatiotemporal properties of mitochondrial Ca2+ responses and the physiological and pathophysiological consequences of Ca2+ signals (Szabadkai and Duchen, 2008). Mitochondrial fusion and fission processes are both mediated by large GTPases. Quantifying protein levels of GTPases such as mitochondrial fusion proteins 1 (mfn-1) and 2 (mfn-2) and optic atrophy 1 (Opa-1) and the mitochondrial fission protein dynamin-related protein 1 (Drp-1; Fig. 4B) revealed that both mfn-1 and mfn-2 were increased (mean ± SEM, mfn-1, 69 ± 18.5%, n = 13, p = 0.003; mfn-2, 79 ± 32.8%, n = 19, p = 0.02, two-tailed unpaired Student's t test), whereas Opa-1 (16 ± 8.5%, n = 12, p = 0.002, two-tailed unpaired Student's t test) and Drp-1 (30 ± 6.7%, n = 14, p = 0.005, two-tailed unpaired Student's t test) were decreased in aging MONs (Fig. 4C).
Although these results suggest that individual mitochondria become elongated with age, we further evaluated axonal ultrastructural changes using 3-D EM to determine a structure–function basis for age-related alterations in molecular architecture and response to ischemia (Baltan, 2012, 2015). Since 12 months of age marks the beginning of increased sensitivity to ischemic injury (Baltan et al., 2008; Baltan, 2009, 2014), our 3-D EM studies primarily compared MONs at 1 and 12 months (Fig. 5). Three-dimensional reconstructions of individual mitochondria (yellow) in young optic nerve axons (Fig. 5A) displayed mitochondria of various sizes and thicknesses distributed along the length of young axons. Aging axons were considerably expanded in volume, whereas their mitochondria (magenta) were mostly located closer to axolemma, leaving visibly open spaces in aging axon volume (Fig. 3B). Consequently, median mitochondrial counts per unit axon volume were reduced from 3.4 mitochondria/10 μm3 (n = 55) to 2.1 mitochondria/10 μm3 (n = 55) in aging axons (p < 0.0001, two-tailed Mann–Whitney U test; Fig. 5C,D). On the other hand, mitochondrial volume increased in aging axons. Based on axonal mitochondrial volume (young, 25th percentile, 0.07 μm3; 75th percentile, 0.24 μm3; 99th percentile, 0.77 μm3), axonal mitochondria were classified into four major groups, designated as small (<0.07 μm3), medium (0.07–0.24 μm3), large (0.24–0.77 μm3), and very large (>0.77 μm3; Fig. 5E). Mitochondrial volumes in the large and very large groups were consistently higher in aging axons (Fig. 5E,F), and this was mainly due to a greater number of elongated mitochondria reaching over 10 μm of length (Fig. 5G,H). Mitochondria were classified based on length (young, 25th percentile, 1.02 μm; 75th percentile, 2.39 μm; 98th percentile, 7.77 μm) into four major groups, designated as small (<1.02 μm), medium (1.02–2.39 μm), long (2.39–7.77 μm), and very long (>7.77 μm).
These morphological alterations in mitochondria correlated with impaired mitochondrial function such as decreased ATP production (Fig. 5I; mean ± SEM, young, 1 ± 0.02%, n = 10; aging, 0.6 ± 0.02%, n = 10; p = 0.001, two-tailed unpaired Student's t test), and augmented oxidative stress quantified as increased NO generation (Fig. 5J; mean ± SEM, young, 1 ± 0.3%, n = 4; aging, 2.8 ± 0.3 n = 5; p = 0.03 two-tailed Student's t test) and greater 3-nitrotryrosine (3-NT; Fig. 5K; mean ± SEM, young, 1 ± 0.1%, n = 16; aging, 1.8 ± 0.1%, n = 16; p = 0.0001, two-tailed unpaired Student's t test) and 4-hydroxynonenal levels with aging (4-HNE; Fig. 5L; mean ± SEM, young, 1 ± 0.1, n = 14; aging, 1.44 ± 0.1%, n = 14; p = 0.004, two-tailed unpaired Student's t test). These results suggest that aging alters structural and functional integrity within mitochondria, leading to higher oxidative stress levels and compromised local energy homeostasis, rendering axons vulnerable to increased injury following a metabolic attack such as ischemia.
Roles for interactions between mitochondria and SER are emerging in synergizing lipid synthesis, Ca2+ signaling, mitochondrial biogenesis, and intracellular trafficking. Because aging altered other mitochondrial parameters, we investigated the effects of age on mitochondria–SER junctions by first defining SER structure and distribution within axons (Figs. 6A,a,B,b, green) and then relative mitochondria to SER relationships in young and aging axons using 3-D EM (Fig. 6A,B, red). Axonal SER comprises a continuous, highly branched network of small-diameter (10–20 nm) tubules that are present in all regions of the axon (Fig. 6A,B, blue arrows). The SER was wrapped around each mitochondrion to varying degrees (Fig. 6a,b) in young and aging axons. The total amount of SER in young optic nerve axons consisted of (mean ± SEM) 5.8 ± 0.2% (n = 27) of SER area per cubic micrometer of axoplasm (Fig. 6C). Although SER networks seemed more elaborate and widely spread in aging axons (Fig. 6B,b, green), when corrected to axon volume, they were reduced to 4.3 ± 0.2% (n = 27; p = 0.0001, two-tailed unpaired Student's t test) of SER area per cubic micrometer of axoplasm. Furthermore, the mitochondria–SER interaction was defined with respect to SER such that SER near mitochondria were denoted in yellow as mitochondria associated (Fig. 6E,F, yellow arrows), SER near axon membranes in blue as axolemma associated, and SER near cytoplasm in green as unassociated (Fig. 6E,F). Aging reduced the SER area (mean ± SEM) associated with a mitochondrion to 3.6 ± 0.6% (n = 26; p = 0.001, two-tailed unpaired Student's t test; Fig. 6b,G,H) compared to young axons (10.0 ± 1.2%, n = 29). The remaining SER area (mean ± SEM) was divided equally between axolemma associated (43.2 ± 2.8%, n = 29) and unassociated (46.8 ± 3.2%, n = 29) in young axons, while the majority of SER became unassociated with aging (55.8 ± 2.2%, n = 24).
SER contains various luminal and integral membrane proteins to carry out diverse cellular functions. Calreticulin is a luminal Ca2+-binding SER chaperone implicated in Ca2+ signaling storage, while binding to misfolded proteins to prevent their export from SER. Calnexin is an integral protein that assists in protein folding and quality control and is upregulated as a primary intracellular defense mechanism against cellular stress such as ischemia. Aging drastically downregulated both protein levels (calreticulin, young axons, mean ± SEM, 100 ± 8%, n = 16; aging axons, 49 ± 7%, n = 8, p = 0.0003, two-tailed unpaired Student's t test; calnexin, young axons, mean ± SEM, 100 ± 6%, n = 16; aging axons, 64 ± 6%, n = 8; p = 0.0006, two-tailed unpaired Student's t test), suggesting impaired Ca2+ regulation and SER stress responses in aging axons (Fig. 6I,J).
Discussion
We characterized and showed that aging caused prominent ultrastructural modifications in CNS WM axons, axonal mitochondria, and axonal SER networks, leading to impaired mitochondrial function. Key findings that correlated with these modifications included a prominent decrease in axon numbers, increased myelination, and increased nodal and paranodal distances. Moreover, aging led to fusion of mitochondria and impaired mitochondria–SER interactions with reduced ATP production and increased oxidative stress. Our findings serve as an important baseline for understanding how aging affects axon–mitochondria–SER interactions while maintaining baseline axon function, inevitably rendering tissue more vulnerable to increased oxidative stress due to impaired mitochondrial morphology and function. We suggest that the abrupt increase in the vulnerability of WM function to injury with aging is mediated by these ultrastructural changes.
Consistent with previous reports, aging caused a prominent decrease in the number of optic nerve axons (Peters, 1999; Bowley et al., 2010); however, optic nerve diameter remained constant due to a proportional increase in axonal diameter and volume. The increase in axon diameter led to an increase in the thickness of the surrounding myelin sheaths with aging, and, moreover, the increase in myelin thickness surpassed the increase in axon diameter such that the g ratio of axons decreased with age. Although loss of myelin in humans (Kemper, 1994) and myelin changes characterized by splitting, ballooning, and exuberant formation (Feldman and Peters, 1998; Peters et al., 2000; Sandell and Peters, 2002) in brain and optic nerves with aging have been reported, overproduction of myelin allows sheaths to enlarge and accommodate the subsequent increases in the diameters of enclosed axons. Moreover, during aging, the activity of genes involved in myelin synthesis is upregulated (Blalock et al., 2003), so more myelin is added to an axon and the myelin sheath gets thicker and begins to occupy an increasing proportion of the surrounding volume (Lasiene et al., 2009). These results suggest that oligodendrocytes remain active during aging and continue to add lamella to the myelin sheaths of intact nerve fibers so that the myelin becomes thicker (Peters, 2009; Bowley et al., 2010). Whether myelin becomes less compact with aging is currently under investigation.
We also observed increased nodal and internodal lengths with aging, despite previous reports of shorter internodal distances in aging spinal cord (Lasiene et al., 2009) and primate CNS (Peters, 2009). A majority of aging axon profiles exhibited diffuse Nav1.6 immunoreactivity with prominent elongation of Caspr labeling, as described previously (Hinman et al., 2006; Lasiene et al., 2009; Baltan, 2014). Displacement or aberrant localization of nodal, paranodal, and juxtaparanodal structures suggests that axonal function at the nodes of Ranvier may be compromised with age. In contrast, these modifications to axons and myelin indices seem to be age-related adaptations acquired to preserve axon conduction properties and spatiotemporal summation of axon function as depicted by an increase in CAP area (Baltan et al., 2010). Although our focus in this current study was on investigating the architectural changes that underlie the increased vulnerability of axons to ischemia at 12 months of age, it will be interesting to characterize these parameters in older axons in future studies.
A central finding of this study is that aging axons have fewer mitochondria than young axons, but that they are thicker and longer. Our previous observation of longer and thicker CFP(+) mitochondria in aging axons could not differentiate whether several mitochondria clustered to appear as longer mitochondria or whether individual mitochondria became elongated (Baltan, 2014; Parpura et al., 2016). Images obtained by 3-D EM for this study unequivocally showed that individual mitochondria became elongated and thicker with expanded volume in aging axons. Age-dependent expansion of mitochondrial morphology correlated with a mismatch among mitochondrial shaping proteins such that increases in levels of mfn-1 and mfn-2 and decreases in Drp-1 levels favored mitochondrial fusion in aging axons. The processes of fusion and fission contribute to mitochondrial quality control and the response of mammalian cells to stress such that fusion is stimulated by energy demand and stress, while fission generates new organelles and facilitates quality control (Frank et al., 2001; Skulachev, 2001; Tondera et al., 2009). Because fusion allows mitochondria to compensate for defects by sharing components and thereby helping maintain energy output during stress (Westermann, 2010), it is possible that this conformation of mitochondria is an adaptation to the lower energy status that occurs with aging. On the other hand, a reduction in mitochondrial numbers together with an increase in axon volume resulted in large portions of aging axons without mitochondrial coverage. The number of mitochondria directly correlates with the level of cellular metabolic activity and with an interruption in mitochondrial dynamics due to a mismatch in mitochondrial shaping proteins, leading to reduced ATP production in aging axons. Further evidence supporting that alterations in mitochondrial shape and numbers impaired mitochondrial function stems from increases in the levels of oxidative stress markers (4-HNE, 3-NT, and NO). Morphological alterations compromise mitochondrial function and the resultant reduced energy production and increased oxidative stress underlie the increased vulnerability of WM to an ischemic attack.
Another significant finding of our study is the demonstration of impaired mitochondria–SER interactions, specifically with reduced points of contact. Mitochondrial division occurs at positions where SER tubules contact mitochondria and mediate constriction before Drp-1 recruitment (Friedman et al., 2011). Drp-1, which is recruited from the cytosol, forms spirals around mitochondria and then constricts to sever both inner and outer membranes. Therefore, SER may play an active role in defining the position of mitochondrial division sites (Friedman et al., 2011). Fusion between mitochondrial outer membranes is mediated by membrane-anchored mfn-1 and mfn-2, whereas fusion between mitochondrial inner membranes is mediated by Opa-1 (Olichon et al., 2003; Cipolat et al., 2004; Chen et al., 2005). Therefore, the age-dependent mismatch between mitochondrial shaping proteins and altered mitochondria–SER interactions all favor longer, thicker axonal mitochondria with increased volume.
Furthermore, age-dependent reductions in mitochondria–SER interactions may lead to a disruption in Ca2+ signaling and lipid import between SER and mitochondria because SER and mitochondria form close contacts, termed the mitochondria-associated membrane (MAM). On the MAM, calnexin and calreticulin regulate Ca2+ flux via IP3 receptors and SERCA (sarcoplasmic/endoplasmic reticulum Ca2+ ATPase; Camacho et al., 2003). As a result of Ca2+ signaling between these two organelles, mitochondria take up these local high amounts of Ca2+ in a quasi-synaptic manner (Kirichok et al., 2004; Graier et al., 2007), and increased intramitochondrial Ca2+ levels promote activity of ATP synthase (Jouaville et al., 1999; Griffiths and Rutter, 2009). Consistent with this, the reduction in calreticulin and calnexin levels in aging axons is associated with lower ATP production, suggesting an impaired Ca2+ signaling between SER and mitochondria in axons. Furthermore, mfn-2, one of the three MAM-regulating proteins that regulate SER morphology and Ca2+ homeostasis to mediate tethering of the SER to mitochondria (de Brito and Scorrano, 2008), is drastically increased in aging axons. In addition to promoting mitochondrial fusion, overexpression of mfn-2 has been shown to inhibit apoptosis (Neuspiel et al., 2005; Jahani-Asl et al., 2007), which is perhaps another adaptive mechanism to sustain aging axon function.
Calreticulin and calnexin show a high degree of structural and functional similarities, and both of these SER chaperone proteins exhibited a significant age-dependent decrease in WM, suggesting that both chaperone proteins are closely related with the aging process. It is likely that aging, as a result of lower calreticulin and calnexin levels, impairs SER Ca2+ homeostasis (Coe and Michalak, 2009) and impedes aging axon function recovery following OGD (Baltan et al., 2008; Baltan, 2015). This is an appealing suggestion particularly because removal or attenuation of Ca2+ influx confers complete protection to axon function in young MONs after OGD (60 min) becomes ineffective or injurious in MONs from 12-month-old mice, even after short durations of OGD (30–45 min; Baltan et al., 2008). The implications and role of calreticulin/calnexin on SER and mitochondrial function require further investigation in young and aging WM.
The importance of how WM ages under normal conditions and in clinical manifestations of neurodegenerative diseases has been greatly underestimated, and failure to consider WM remains an impediment to successful translation of preclinical data. Our results have direct implications for the pathophysiology of age-related WM diseases, including glaucoma and stroke. Age is an established major risk factor for glaucoma, and recent studies suggest that age-related changes inherent to optic nerve significantly contribute to the onset and prognosis of this leading cause of blindness (Baltan et al., 2010; Guedes et al., 2011). Similarly, stroke occurs in the elderly and WM is injured in most strokes, contributing to the disability associated with clinical deficits. Our results suggest that aging interrupts mitochondrial function, leading to ATP depletion and increased oxidative stress in WM. Consequently, interventions that attenuate oxidative injury, preserve mitochondrial structure and function, and restore ATP levels effectively promote axon function recovery, even when applied after ischemia (Baltan et al., 2011; Baltan, 2012, 2014). Therefore, consideration of age-dependent changes in WM structure in disease models of glaucoma, multiple sclerosis, traumatic brain injury, and Alzheimer's disease may lead to more successful translation of preclinical findings to clinical trials.
Notes
Supplemental material for this article is available at https://www.youtube.com/playlist?list=PLMG0zKOgNqNk0Io4JTYiuXMndkHMGX41-. The video shows young and aging axons and their mitochondria. This material has not been peer reviewed.
Footnotes
This work was supported by NIA Grant AG033720 to S.B. We thank Dr. Chris Nelson for his help in editing this paper.
G.J.K. is a consultant for Renovo Neural Inc. and scientific director for 3-D EM at Renovo Neural Inc., which is a company that provides serial block-face imaging on a fee-for-service basis.
References
- Baltan S. Ischemic injury to white matter: an age-dependent process. Neuroscientist. 2009;15:126–133. doi: 10.1177/1073858408324788. [DOI] [PubMed] [Google Scholar]
- Baltan S. Histone deacetylase inhibitors preserve function in aging axons. J Neurochem. 2012;123:108–115. doi: 10.1111/j.1471-4159.2012.07949.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S. Age-dependent mechanisms of white matter injury after stroke. In: Baltan S, Carmichael ST, Matute C, Xi G, Zhang JH, editors. White matter injury in stroke and CNS disease. New York: Springer; 2014. pp. 373–403. [Google Scholar]
- Baltan S. Age-specific localization of NMDA receptors on oligodendrocytes dictates axon function recovery after ischemia. Neuropharmacology. 2015 doi: 10.1016/j.neuropharm.2015.09.015. Advance online publication. Retrieved February 25, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S, Besancon EF, Mbow B, Ye Z, Hamner MA, Ransom BR. White matter vulnerability to ischemic injury increases with age because of enhanced excitotoxicity. J Neurosci. 2008;28:1479–1489. doi: 10.1523/JNEUROSCI.5137-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S, Inman DM, Danilov CA, Morrison RS, Calkins DJ, Horner PJ. Metabolic vulnerability disposes retinal ganglion cell axons to dysfunction in a model of glaucomatous degeneration. J Neurosci. 2010;30:5644–5652. doi: 10.1523/JNEUROSCI.5956-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S, Murphy SP, Danilov CA, Bachleda A, Morrison RS. Histone deacetylase inhibitors preserve white matter structure and function during ischemia by conserving ATP and reducing excitotoxicity. J Neurosci. 2011;31:3990–3999. doi: 10.1523/JNEUROSCI.5379-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bay V, Butt AM. Relationship between glial potassium regulation and axon excitability: a role for glial Kir4.1 channels. Glia. 2012;60:651–660. doi: 10.1002/glia.22299. [DOI] [PubMed] [Google Scholar]
- Black JA, Renganathan M, Waxman SG. Sodium channel Nav1.6 is expressed along nonmyelinated axons and it contributes to conduction. Mol Brain Res. 2002;105:19–28. doi: 10.1016/S0169-328X(02)00385-6. [DOI] [PubMed] [Google Scholar]
- Blalock EM, Chen KC, Sharrow K, Herman JP, Porter NM, Foster TC, Landfield PW. Gene microarrays in hippocampal aging: statistical profiling identifies novel processes correlated with cognitive impairment. J Neurosci. 2003;23:3807–3819. doi: 10.1523/JNEUROSCI.23-09-03807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS, Matthews G. Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron. 2001;30:91–104. doi: 10.1016/S0896-6273(01)00265-3. [DOI] [PubMed] [Google Scholar]
- Bowley MP, Cabral H, Rosene DL, Peters A. Age changes in myelinated nerve fibers of the cingulate bundle and corpus callosum in the rhesus monkey. J Comp Neurol. 2010;518:3046–3064. doi: 10.1002/cne.22379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho P, John L, Li Y, Paredes RM, Roderick HL. Calnexin and calreticulin, ER associated modulators of calcium transport in the ER. In: Eggleton P, Michalak M, editors. Calreticulin. Boston: Springer; 2003. pp. 126–132. [Google Scholar]
- Cavallotti C, Pacella E, Pescosolido N, Tranquilli-Leali FM, Feher J. Age-related changes in the human optic nerve. Can J Ophthalmol. 2002;37:389–394. doi: 10.1016/S0008-4182(02)80040-0. [DOI] [PubMed] [Google Scholar]
- Cavallotti C, Cavallotti D, Pescosolido N, Pacella E. Age-related changes in rat optic nerve: morphological studies. Anat Histol Embryol. 2003;32:12–16. doi: 10.1046/j.1439-0264.2003.00431.x. [DOI] [PubMed] [Google Scholar]
- Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe H, Michalak M. Calcium binding chaperones of the endoplasmic reticulum. Gen Physiol Biophys. 2009;28:96–103. [PubMed] [Google Scholar]
- Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci U S A. 2004;101:8168–8173. doi: 10.1073/pnas.0402765101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins KL, Perkel DH, Dorfman LJ. Nerve fiber conduction-velocity distributions. I. Estimation based on the single-fiber and compound action potentials. Electroencephalogr Clin Neurophysiol. 1979;46:634–646. doi: 10.1016/0013-4694(79)90101-9. [DOI] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- Feldman ML, Peters A. Ballooning of myelin sheaths in normally aged macaques. J Neurocytol. 1998;27:605–614. doi: 10.1023/A:1006926428699. [DOI] [PubMed] [Google Scholar]
- Fiala JC. Reconstruct: a free editor for serial section microscopy. J Microsc. 2005;218:52–61. doi: 10.1111/j.1365-2818.2005.01466.x. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Harris KM. Extending unbiased stereology of brain ultrastructure to three-dimensional volumes. J Am Med Informatics Assoc. 2001;8:1–16. doi: 10.1136/jamia.2001.0080001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster RE, Connors BW, Waxman SG. Rat optic nerve: electrophysiological, pharmacological and anatomical studies during development. Dev Brain Res. 1982;3:371–386. doi: 10.1016/0165-3806(82)90005-0. [DOI] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell. 2001;1:515–525. doi: 10.1016/S1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graier WF, Frieden M, Malli R. Mitochondria and Ca2+ signaling: old guests, new functions. Pflügers Arch. 2007;455:375–396. doi: 10.1007/s00424-007-0296-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths EJ, Rutter GA. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim Biophys Acta. 2009;1787:1324–1333. doi: 10.1016/j.bbabio.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Guedes G, Tsai JC, Loewen NA. Glaucoma and aging. Curr Aging Sci. 2011;4:110–117. doi: 10.2174/1874609811104020110. [DOI] [PubMed] [Google Scholar]
- Heflin M, Lum H. Overview of aging. In: Garner A, Klintworth GK, editors. Pathobiology of ocular disease. New York: CRC; 2008. pp. 361–370. [Google Scholar]
- Hinman JD, Peters A, Cabral H, Rosene DL, Hollander W, Rasband MN, Abraham CR. Age-related molecular reorganization at the node of Ranvier. J Comp Neurol. 2006;495:351–362. doi: 10.1002/cne.20886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Katz B. The effect of sodium ions on the electrical activity of the giant axon of the squid. J Physiol. 1949;108:37–77. doi: 10.1113/jphysiol.1949.sp004310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahani-Asl A, Cheung EC, Neuspiel M, MacLaurin JG, Fortin A, Park DS, McBride HM, Slack RS. Mitofusin 2 protects cerebellar granule neurons against injury-induced cell death. J Biol Chem. 2007;282:23788–23798. doi: 10.1074/jbc.M703812200. [DOI] [PubMed] [Google Scholar]
- Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci U S A. 1999;96:13807–13812. doi: 10.1073/pnas.96.24.13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan MR, Cho MH, Ullian EM, Isom LL, Levinson SR, Barres BA. Differential control of clustering of the sodium channels Nav1.2 and Nav1.6 at developing CNS nodes of Ranvier. Neuron. 2001;30:105–119. doi: 10.1016/S0896-6273(01)00266-5. [DOI] [PubMed] [Google Scholar]
- Kemper TL. Neuroanatomical and neuropathological changes during aging and dementia. In: Albert ML, Knoefel JE, editors. Clinical neurology of aging. New York: Oxford UP; 1994. pp. 3–67. [Google Scholar]
- Khan HA. Bioluminometric assay of ATP in mouse brain: Determinant factors for enhanced test sensitivity. J Biosci. 2003;28:379–382. doi: 10.1007/BF02705114. [DOI] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- Lasiene J, Matsui A, Sawa Y, Wong F, Horner PJ. Age-related myelin dynamics revealed by increased oligodendrogenesis and short internodes. Aging Cell. 2009;8:201–213. doi: 10.1111/j.1474-9726.2009.00462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, Lichtman JW. Imaging axonal transport of mitochondria in vivo. Nat Methods. 2007;4:559–561. doi: 10.1038/nmeth1055. [DOI] [PubMed] [Google Scholar]
- Murphy SP, Lee RJ, McClean ME, Pemberton HE, Uo T, Morrison RS, Bastian C, Baltan S. MS-275, a Class I histone deacetylase inhibitor, protects the p53-deficient mouse against ischemic injury. J Neurochem. 2014;129:509–515. doi: 10.1111/jnc.12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuspiel M, Zunino R, Gangaraju S, Rippstein P, McBride H. Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J Biol Chem. 2005;280:25060–25070. doi: 10.1074/jbc.M501599200. [DOI] [PubMed] [Google Scholar]
- Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278:7743–7746. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- Parpura V, Fisher ES, Lechleiter JD, Schousboe A, Waagepetersen HS, Brunet S, Baltan S, Verkhratsky A. Glutamate and ATP at the interface between signaling and metabolism in astroglia: examples from pathology. Neurochem Res 1–16. 2016 doi: 10.1007/s11064-016-1848-6. Advance online publication. Retrieved February 25, 2016. [DOI] [PubMed] [Google Scholar]
- Peters A. Normal aging in the cerebral cortex of primates. In: Peters A, Morrison JH, editors. Cerebral cortex. Boston: Springer; 1999. pp. 49–80. [Google Scholar]
- Peters A. The effects of normal aging on myelinated nerve fibers in monkey central nervous system. Front Neuroanat. 2009;3:1. doi: 10.3389/neuro.05.011.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Moss MB, Sethares C. Effects of aging on myelinated nerve fibers in monkey primary visual cortex. J Comp Neurol. 2000;419:364–376. doi: 10.1002/(SICI)1096-9861(20000410)419:3<364::AID-CNE8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Ross MH, Pawlina W. Nerve tissue. In: Ross M. H., Pawlina W, editors. Histology: a text and atlas with correlated cell and molecular biology. Philadelphia: Wolters Kluwer Health; 2016. pp. 356–403. [Google Scholar]
- Sandell JH, Peters A. Effects of age on the glial cells in the rhesus monkey optic nerve. J Comp Neurol. 2002;445:13–28. doi: 10.1002/cne.10162. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Matsuki N, Ikegaya Y. Action-potential modulation during axonal conduction. Science. 2011;331:599–601. doi: 10.1126/science.1197598. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skulachev VP. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem Sci. 2001;26:23–29. doi: 10.1016/S0968-0004(00)01735-7. [DOI] [PubMed] [Google Scholar]
- Stys PK, Waxman SG, Ransom BR. Na(+)-Ca2+ exchanger mediates Ca2+ influx during anoxia in mammalian central nervous system white matter. Ann Neurol. 1991;30:375–380. doi: 10.1002/ana.410300309. [DOI] [PubMed] [Google Scholar]
- Szabadkai G, Duchen MR. Mitochondria: the hub of cellular Ca2+ signaling. Physiology (Bethesda) 2008;23:84–94. doi: 10.1152/physiol.00046.2007. [DOI] [PubMed] [Google Scholar]
- Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, Da Cruz S, Clerc P, Raschke I, Merkwirth C, Ehses S, Krause F, Chan DC, Alexander C, Bauer C, Youle R, Langer T, Martinou JC. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009;28:1589–1600. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uo T, Veenstra TD, Morrison RS. Histone deacetylase inhibitors prevent p53-dependent and p53-independent Bax-mediated neuronal apoptosis through two distinct mechanisms. J Neurosci. 2009;29:2824–2832. doi: 10.1523/JNEUROSCI.6186-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]