

Abstract
During DNA replication, forks often stall and require restart. One mechanism for restart requires that the fork be moved in a direction opposite to that of replication. This reaction is known as fork regression. For this reaction to occur, the enzyme must couple unwinding of the nascent heteroduplex fork arms to the rewinding of nascent strands ahead of itself and to the parental duplex in its wake. As the arms of the fork are complementary, this reaction is isoenergetic making it challenging to study. To overcome this, a novel adaptation of magnetic tweezers was developed by the Croquette group. Here, a 1,200 bp hairpin was attached at opposite ends to a flow cell surface and a magnetic bead. By manipulating the bead with the magnets, force can be applied to unwind the hairpin or alternatively, released to allow the hairpin to rewind. This adaptation was used to study fork regression by RecG. The results show that this is an efficient regression enzyme, able to work against a large opposing force. Critically, it couples DNA unwinding to duplex rewinding and in the process, can displace bound proteins from fork arms.
Keywords: magnetic tweezers, hairpin substrate, DNA helicase, RecG, SSB, replication fork, fork regression, Holliday junction
1. Introduction
Successful genome duplication relies on the close interplay between the genetic recombination and DNA repair machinery (1–3). The need for this interplay arises due to the replication machinery frequently encountering roadblocks that stall or collapse replication forks (4–7). In bacteria, stalled replication forks can directly restarted or regressed (8–11). That is, moved in the direction opposite to that of replisome movement (Figure 1). Although replication fork regression can in principle be spontaneous as demonstrated previously (12), it can also, in theory, be catalyzed by a number of proteins (4, 13–15). Over the past several years it has become increasingly clear that the branched-DNA specific molecular motor RecG, is responsible for the regression of stalled replication forks (16–18).
Figure 1. Fork regression requires coupling of DNA unwinding to duplex rewinding.

(A). A stalled replication fork is shown impeded and the nascent leading (blue) and lagging (orange) strands indicated. Fork regression results in rightward movement of the fork, away from the site of the replication road-block, concomitant with the extrusion of a duplex region resulting from the annealing of the nascent leading and lagging strands. The resulting DNA structure has been termed the “chicken foot” and is structurally equivalent to a Holliday junction. Fork readvancement occurs after regression and repair has taken place and results in leftward movement of the fork. Once complete, replisome reloading ensues. (B). Efficient regression requires the combined actions of nascent heteroduplex arm unwinding with DNA rewinding that occurs ahead of the advancing enzyme as well as in its wake. This results in reforming of the parental duplex and extrusion of a heteroduplex toe. The resulting three “toed” structure is known as the chicken foot intermediate.
To do this, RecG must possess several key activities that operate concurrently. First, it must operate as an atypical DNA helicase. That is, it must be capable of both unwinding nascent duplex regions while simultaneously rewinding DNA both ahead of the advancing enzyme, as well as in its wake (Figure 1). Second, during the process of translocation and duplex DNA remodeling, it must generate sufficient force so as to displace proteins which may be bound to either single- or double-stranded DNA domains of the fork. Finally, it should be able to catalyze fork regression leading to the formation of a Holliday junction, a central intermediate in most fork rescue pathways (19).
Here, a novel adaptation of the replication fork processing assay developed by the Croquette group is outlined. This adaptation takes advantage of magnetic tweezers to manipulate a 1,200bp hairpin substrate or fork mimic. In these manipulations, reactions essential to fork processing are studied: single-strand DNA annealing; nascent duplex unwinding; protein displacement and extrusion of a Holliday junction from a fork.
2. Materials and methods
2.1 Proteins
RecG protein was purified to near homogeneity as described previously (16, 17, 20). Purification involved the use of four, sequential chromatographic steps: Q-Sepharose, heparin FF, and hydroxylapatite and MonoS. The protein concentration was determined spectrophotometrically using an extinction coefficient of 49,500 M−1 cm−1 (35). The activity of the protein was determined using a coupled spectrophotometric ATPase assay using M13 ssDNA as cofactor (16, 17, 20). This purification procedure yielded a 4-fold increase in specific activity relative to that used previously (21).
The single stranded DNA-binding protein (SSB) was purified from strain K12ΔH1Δtrp as described (22). The concentration of purified SSB protein was determined at 280 nM using ε = 30, 000 M−1.cm−1. The site size of SSB protein was determined to be 10 nucleotides per monomer by monitoring the quenching of the intrinsic fluorescence of SSB that occurs on binding to ssDNA, as described (23).
2.2 DNA substrate
The DNA substrate consists of a 1,239 bp hairpin with a 4 nt loop, a 76 nt 5′-biotinylated ssDNA tail at one end, and a 146 bp 3′-digoxigenin labeled dsDNA tail at the opposite end (Figure 2). The original description can be found in references (24–26) with similar types of hairpins described in (27, 28) for the study of the NS3 and XPD proteins. The hairpin substrate used for RecG was constructed from a 1.1kB insert of the pGEM plasmid pNo-GTT that was released by sequential digestion with ApaI and NotI (24). The oligonucleotides described below were ligated to modify ends to facilitate attachment to beads (avidin-biotin) or to the flow cell surface (antibody-digoxygenin).
Figure 2. DNA hairpin substrate construction.

A schematic of the substrate is shown with each of the oligonucleotides shown. Sequences of each are provided in section 2.2. The purified insert from plasmid pNo-GTT is coloured black (center). The hairpin oligonucleotide (purple) is ligated to the 3′-overhang created by ApaI restriction enzyme cleavage. The annealed complex of flap 1 (green) and template 1 (red) is ligated to the 5′-overhang created by NotI restriction enzyme cleavage. Next, the primer (blue) is annealed to the 3′-end of template 1. Thereafter, T4 DNA polymerase and dNTPS including digoxgenin-labeled dUTP are added, extending the primer (zig-zag line) adding 6 DIG bases (orange) at the extreme 3′end of template 1.
Oligonucleotides used to construct the hairpin substrates were purchased from Integrated DNA Technologies, Coralville, IA. Unmodified oligonucleotides were purified using either 8 or 12% Urea-PAGE depending on the length of the DNA molecule, followed by desalting. Biotinylated oligonucleotides were purified by binding to Immobilized Monomeric Avidin agarose resin (Pierce) followed by elution with 2 mM biotin. Prior to ligation, the hairpin and template oligonucleotides of both substrates were phosphorylated using T4 Polynucleotide Kinase. Thereafter, they were annealed to the 1.1 kb insert at a ten-fold molar excess and ligated using T4 DNA ligase at 4 °C for a minimum of 24 hours. A schematic of the substrate is shown in Figure 2.
The sequences of the oligonucleotides used in the construction of the 1,200bp hairpin are: hairpin 1 (5′-GTCAGATGCCTTTTGGCATCTGACGGCC-3′), flap 1 (5′-Biotin-AATTGCATGTATTACTTGGTAGGATCCGTCATAGCTTTAGCGATTTGGGACACTTCA TCAAGACTTCCAGAGCAGCCGGAGACATATAGCTACAGG-3′), and template 1 (5′-GGCCCCTGTAGCTATATGTCTCCGCCCCCCCCCCTGTGTGTGTGTGTGGTTGTGT GGTGTGTGGTTGTGTGTTGGTGGTTGCATACTTCCGGGAACGCAG-3′). The hairpin 1 oligonucleotide forms a 4nt hairpin, a 10bp duplex DNA region and a 4 base, 3′-overhang (5′-GGCC-3′) suitable for ligation to ApaI. The flap 1 and template 1 oligonucleotides partially anneal to one another, creating a 20bp duplex region (the underlined regions in each sequence) and a 4 base, 5′overhang (5′-GGCC-3′) suitable for ligation to NotI. The remaining base pairs are not complementary creating a 76 nucleotide fork region with an available 5′-biotin moiety for binding to streptavidin-coated beads on one arm with an available sequence on the opposite arm for primer annealing and extension as described in the next paragraph.
Ligated products from the above-mentioned reactions were purified from 1% agarose gels using gel extraction kits (Qiagen). A primer of sequence 5′-AAAAAAGTGTTGTGTGGTGTTGTTTGGGTGTTGTTTGTGTGTTGTTTGGTGTTGTTT GGGTGTTGTTTGTGTGTTGTTTGCTGCGTTCCCGGAAGTATGC-3′, was annealed to the 3′-end of the template 1 oligonucleotide, forming a 20bp duplex DNA region (bold sequence) followed by a tail 80 nucleotides in length, that can be extended by a DNA polymerase. Following annealing, T4 DNA polymerase (exo-) was used to fill in the overhang and incorporate six digoxygenin-labeled dUTP nucleotides at the extreme 3′-end. Thereafter, completed hairpin substrates were purified by agarose gel electrophoresis.
The final substrate, a 1,200 bp hairpin that forms the core of the experiments described below is shown in Figures 2 and 3. It contains a central 4nt hairpin, followed by a GC-clamp and two arms of complementary sequence. Under minimal applied force, the arms reanneal forming a hairpin. The design of this molecule mimics that of a nascent fork: the duplex region corresponds to unreplicated, parental DNA, with the two single stranded arms corresponding to the nascent leading and lagging strands. In the schematic in Figure 3, both arms are single stranded in character corresponding to a fork with gaps in both strands.
Figure 3. Magnetic tweezers manipulate a novel fork substrate.

A schematic of the 1,200 bp hairpin substrate under the careful control of magnetic tweezers is shown. The DNA molecule is held in place by site specific attachment to two surfaces: antibody-digoxygenin at the 3′-end (flow cell surface) and biotin-streptavidin at the 5′-end (bead surface). Tension in the DNA molecule and consequently its height (Z-extension) is carefully controlled by magnets of the tweezers, with opposing force directed away from the surface as indicated by the yellow arrow. Simultaneously, the bead is illuminated by an LED light source and the image captured by a CCD camera. Software is then used to calculate bead position (in nm) and to convert position data in DNA length (bp).
Consequently, and due to sequence complementarity, this molecule can be easily unwound (arms pulled apart) or rewound (arms reannealed). These opposing reactions, critical to the study of fork rescue, can be implemented by the application of force or by enzyme action. When force is increased, the duplex region is unwound as the substrate is pulled apart (Figure 4, top right). Similarly, if an enzyme were unwinding this DNA molecule (indicated by the red arrow), the length of the duplex region would decrease, and that of the ssDNA arms would increase. This is observed as an increase in Z as a function of time, as shown in the graph.
Figure 4. Key fork rescue reactions can be studied using a hairpin substrate.

The hairpin substrate is held under tension by the magnetic tweezers (left panel). Introduction of enzyme and an energy source (ATP) can produce one of two reactions. If the enzyme catalyzes fork readvancement, it will preferentially unwind the duplex region of the substrate and the length of the DNA will increase as shown in the graph (top right). Thus the change in Z that occurs as a function of time can be attributed to the translocating enzyme. In contrast, if the enzyme catalyzes fork regression, motion will occur in the opposite direction (red arrow), and the two single stranded arms will be rewound (bottom right). Consequently, the net length of the DNA molecule will decrease as shown in the graph. In these reactions, duplex rewinding occurs and is terminated when the enzyme stalls permanently on the DNA or dissociates. This results in a virtually instantaneous increase in Z-height resulting from the opposing force applied by the magnetic tweezers (indicated by the red lines). The slope of the lines (ΔZ/Δt) is used to determine reaction rate and the length of the unwinding (or rewinding period) corresponds to processivity of the enzyme.
In contrast, when the force pulling the substrate apart is decreased, the complementary arms can reanneal. In like fashion, if the enzyme were rewinding the two ssDNA arms, the length of duplex region being extruded increases (enzyme movement is indicated by the red arrow). In both instances, this is observed as a net decrease in Z (Figure 4, bottom right). In the graph shown, the reaction is initiated with the substrate almost fully unwound. When rewinding occurs, Z height decreases, until the enzyme dissociates or force is applied. This causes the Z height to increase to its maximal value (graph, Figure 4, bottom right).
The hairpin substrate mimics a fork with parental duplex and two single stranded arms (Figure 3). During DNA replication and fork stalling, it is conceivable that only one of the arms will have a ssDNA gap. Therefore, to construct gapped substrates, a simple modification is used. Assembly begins under low force conditions with the DNA hairpin fully formed (Figure 5A, panel 1). Then, force is applied by the magnetic tweezers to completely unzipper the substrate (panel 2). This is followed by the introduction of oligonucleotides 90 bases in length and complementary to either the upper or lower regions of the substrate. These oligonucleotides are introduced in separate reactions and allowed to anneal. Once the force is lowered, the hairpin is extruded leaving a fork with one duplex arm (formed by the annealed oligonucleotide) and a ssDNA gap in either the nascent leading or lagging strands (panel 3).
Figure 5. Stalled replication fork substrates can be constructed in situ.
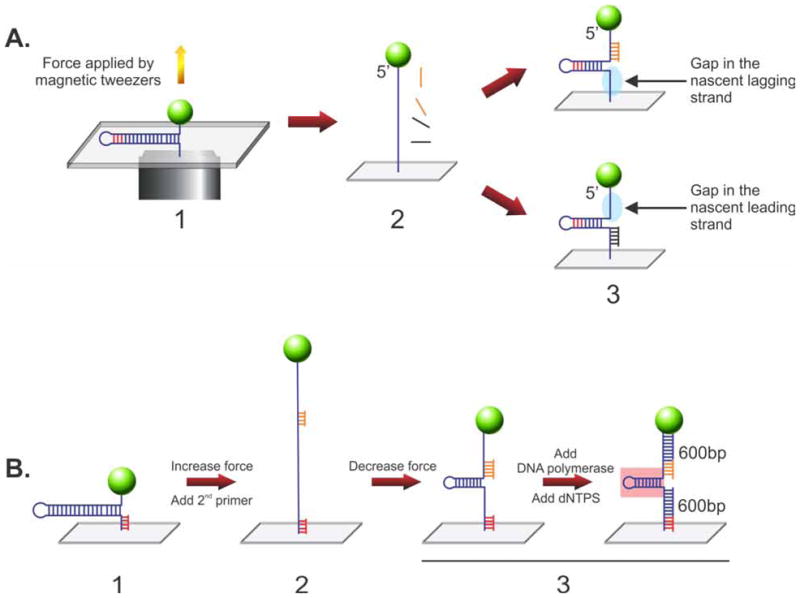
Two types of DNA substrates relevant to fork rescue can be constructed within the confines of the flow cell. (A) Construction of forks with gaps in either the leading or lagging strands. Here, the 1,200 bp hairpin is fully stretched by the application of force form the magnetic tweezers. Then, oligonucleotides complementary to the 5′- or 3′-proximal regions are introduced in separate reactions and allowed to bind. Once the opposing force is decreased, a partial hairpin is extruded and as the oligonucleotide remains annealed to reveal a fork with a gap on the opposite side, either the lagging (top) or leading strand (bottom). (B) Construction of a fork with duplex arms. As before the starting point is the 1,200 bp hairpin except now, a short DNA primer is annealed to 3′end of the substrate. Then, force is applied to unwind the hairpin and a second primer is introduced and allowed to bind. When the force is reduced, the DNA length decreases, a partial hairpin is extruded (~600bp in length) with primers bound to the opposing arms as indicated. When DNA polymerase is added, the tailed duplex regions are extended from each primer, producing a fork with 600 bp duplex arms. Pink box, parental duplex DNA region of the fork. The graph indicates the corresponding changes in Z-height as the hairpin is sequentially pulled apart and allow to reform as substrate construction proceeds.
The final fork substrate was originally used in a study for fork processing by UvsW (26). Here it is used to test whether RecG can regress a fork into a Holliday junction. It is constructed in situ in the flow cell in sequential steps. First, a primer (red) is annealed to the 1,200 bp hairpin (Figure 5B, panel 1). Then, force is applied to unzipper the substrate, a second primer (orange) is added and allowed to anneal (panel 2). The force is then decreased, allowing the hairpin to partially reform, producing a fork with primers bound to each arm (panel 3, left). Next, T4 DNA polymerase and dNTPs are added and synthesis ensues using the red and orange primers. Finally, enzyme and unincorporated dNTPs are washed out, revealing a fork with a 600bp parental duplex region (pink box) and two nascent heteroduplex arms, each 600bp in length (panel 3, right).
The progression of substrate assembly reactions can be followed in real-time as shown in the example in Figure 6B. At t<118s, the hairpin is fully formed so Z-height is at a minimum. Once force is applied, the molecule is fully stretched and Z-height reaches its maximum (t=118–122s). Finally, following oligonucleotide annealing, the force is lowered to an intermediate value and the nascent substrate is revealed (t=122–130s).
Figure 6. RecG catalyzes the requisite reactions needed to regress a stalled replication fork.
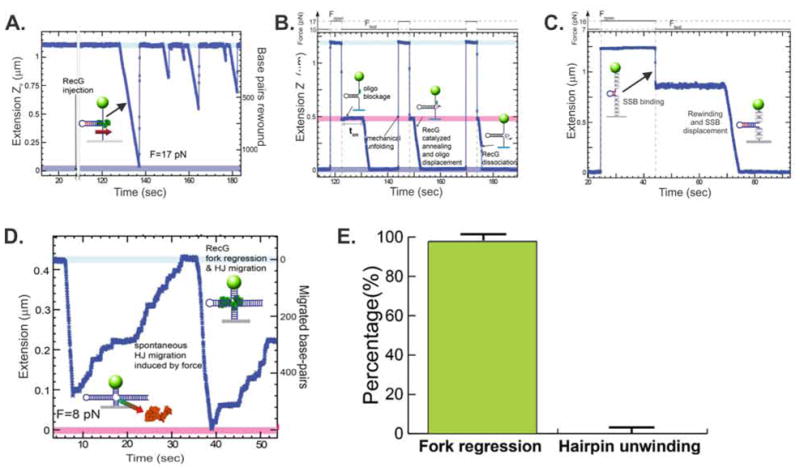
The data presented were published in (18) and are reused here in modified form.
(A). RecG efficiently rewinds ssDNA fork arms. The DNA substrate is the core 1,200bp hairpin (Fig. 2 3). Reactions are done while the substrate is held under a constant tension of 17pN. Following the introduction of RecG multiple, separate events are observed where the extension of the DNA molecule decreases as a function of time. These events correspond to single, rewinding events where RecG reanneals the complementary arms. While the reactions rates are similar, the processivity of each event varies.
(B). RecG couples DNA unwinding to duplex rewinding. In these reactions, the hairpin substrate is pulled apart by the application of force between t=118 and 122sec. Then an oligonucleotide is introduced and allowed to bind. Once the force is lowered, a partial hairpin is extruded to reveal a fork with a gap in the nascent lagging strand (Z-height indicated by the pink region). Once RecG is introduced, binding occurs, with the length of the on rate dictated by the nature of the fork (ton). The reaction then ensues and RecG unwinds the oligonucleotide resulting in its displacement, concomitant with rewinding of the ssDNA arms, resulting in extrusion of the hairpin. The hairpin can be repeatedly mechanically unfolded by the magnetic tweezers, allowing the displaced oligonucleotide to rebind and the reaction repeated. The length of ton was used to demonstrate substrate discrimination by RecG. For substrates with gaps in the leading strand ton= 15±1sec, whereas for forks with gaps in the lagging strand, ton= 1.8±0.1sec.
(C). During rewinding, RecG displaces proteins bound to fork arms. Here, the hairpin was mechanically unfolded to the GC clamp and SSB (grey ovals) allowed to bind. As the protein wraps the ssDNA the net length of the molecule decreases. Once RecG is added, the extension decreased rapidly indicating fork arm rewinding, concomitant with SSB displacement.
(D). RecG regresses a stalled fork resulting in formation of a Holliday junction. In these assays, the fork substrate was constructed using the scheme shown in Figure 4B. The resulting substrate is held in place by the magnetic tweezers using an opposing force of 8pN. Once RecG is introduced, extension rapidly decreases terminating at 0.08μm as RecG dissociates. As the now four arms of the resulting Holliday junction are equivalent, spontaneous junction migration occurs. When RecG rebinds, the reaction is repeated, this time at a slightly reduced rate
(E). RecG catalyzes fork regression and not fork readvancement. A critical feature of the action of RecG at a fork is the direction in which it catalyzes fork movement. This was tested using the gapped substrates constructed in Figure 4A.
2.3 Magnetic Tweezers assay
A PicoTwist magnetic tweezers instrument (www.picotwist.com) was used to manipulate individual, asymmetrically-modified, DNA hairpin molecules attached at one end to the lower surface of a glass flow cell and at the opposite end, to a magnetic bead (25). The DNA substrates were modified with digoxigenin on the 3′-end and biotin on the 5′-end. Next, the glass surface was treated with anti-digoxigenin antibody (Roche) and then passivated with BSA to minimize non-specific binding sites (25). Separately, DNA was incubated with 1μm, streptavidin-coated Dynal magnetic beads (Invitrogen). Thereafter, DNA-bead complexes were injected into the flow cell and allowed to bind to the surface. The DNA hairpins are manipulated by capturing the bead in the magnetic trap generated by a pair of permanent magnets (Figure 3).
To determine whether the DNA (i.e., the bead) is attached to the surface and whether tethering is via one or two DNA molecules, testing is done using the characterized mechanical properties of the hairpin. For this DNA substrate, extension remains almost constant below ~15 pN and abruptly increases when the hairpin is unzipped by the application of force above ~15 pN. In contrast, if a bead is tethered by two DNA hairpins, the force needed to unzip them is doubled (~30 pN). Thus, to determine how a bead is attached to the surface, the position of the magnets is changed so the applied force is varied from 1–20pN. The difference in extension of the DNA between these low and high forces is then calculated (Δz). If this value is consistent with the length of the completely unzipped and extended hairpin (1nm/bp, therefore 1.2 μm), then that bead is selected for the subsequent experiment. In contrast, if the change in extension is zero, then the bead is either bound to the surface non-specifically or may be tethered by more than one DNA substrate.
To track the position of the magnetic bead in three dimensions with nanometer resolution, video-microscopy was used at 30 Hz. From the resulting bead positions, the extension of the DNA molecule and the strength of the stretching force were deduced (29). A calibration curve of the applied force versus the vertical position of the magnets is then used to exert forces within 10% error on the DNA molecules. Experiments are typically done in a single stream flow cell, with the inlet port connected to a syringe pump and the outlet port to waste. Details of the flow cell assembly are presented in (25). Once bead attachment is confirmed, reaction buffer (20mM Tris-HCl, pH 8.0, 1mM DTT and either 3 or 10mM MgCl2) containing RecG and ATP (1mM) is introduced to initiate reactions.
2.4 Data analysis
The raw data, corresponding to the real-time changes of the DNA extension in nm, were converted into the number of base pairs rewound or base pairs migrated by RecG using a calibration factor determined from the elastic properties of ssDNA and dsDNA (26). Reaction rates were obtained from a linear fit to the traces filtered with a third-order Savitzky-Golay filter over a time window of 0.1s. The histogram of the instantaneous rates was fit to Gaussian functions. The error bars shown in the histograms are proportional to the inverse of the square root of the number of points for each individual bin.
Reaction progress was deduced from changes in the extension of the DNA molecule, Ze. A controlled force was applied to the ends of the hairpin using the magnets, with Ze(t) obtained by tracking the position of the magnetic bead (29). Consequently, the unwinding of the hairpin corresponds to an increase in Ze (Figure 4, top right). This follows because the DNA molecule has unwound and the constant opposing force pulls the DNA molecule to an ever increasing length, with the final position dictated by the GC-clamp. In contrast, DNA rewinding is observed as a decrease in Ze (Figure 4, bottom right). This makes sense because the complementary arms have been reannealed, extruding an increasing longer hairpin, whose final length is dictated by the length of the complementary ssDNA arms.
3. RecG catalyzes multiple reactions critical to efficient fork regression
a. Efficient DNA strand rewinding (Figure 6A)
To determine whether RecG could catalyze duplex rewinding the 1,200 bp hairpin was partially denatured leaving the GC rich region intact. The addition of RecG and ATP resulted in rapid rewinding of the ssDNA arms of the fork. Once RecG dissociation occurs, spontaneous force induced strand separation occurred, resulting in rapid extension of the molecule back to its original length. The changes in molecular extension were converted to the number of base pairs rewound by measuring the elastic response of ssDNA (18, 25, 26). The average rewinding rate was calculated to be 123 ± 2 bp/s.
b. Coupling of DNA unwinding to duplex rewinding (Figure 6B)
During the course of a fork regression reaction (Fig. 1), two simultaneously operating enzymatic activities should be in play. These are DNA unwinding where the nascent heteroduplex arms of the fork are separated and subsequently rewound as parental duplex in the enzyme wake and a nascent toe extruded ahead of the advancing enzyme. To test whether RecG could catalyze these reactions simultaneously, the hairpin was pulsed apart by the application of force and in separate reactions two different 90mer oligonucleotides introduced (Figure 5A). The resulting substrates have gaps in either the leading or lagging strands. Then, DNA forks were maintained at a moderate force between 5 and 13 pN, and following introduction of RecG, a rapid decrease in extension was observed. This corresponds to unwinding of the oligonucleotide:hairpin duplex, concomitant with reannealing of the forms arms, leading to extrusion of the parental duplex region in the wake of the enzyme. As RecG binds to fork substrates with very high affinity relative to short oligonucleotides (20), reactions can be repeated in cyclical fashion. Following complete rewinding and helicase dissociation. The opposing force unzippers the hairpin allowing the 90mers to rebind. Once RecG rebinds to the fork, the reaction is repeated.
This strategy (i.e., the cyclic fork regression assay) was used to determine the substrate preference for RecG. Previous work has shown that the enzyme preferentially acts on forks with a gap in the nascent leading strand (16, 17, 20, 30). To determine substrate preference here, the on rate or time taken for RecG to bind each of the fork substrates was determined, as indicated by ton in Figure 6B. At t=148 and 174sec, two additional measurements of ton are visible, each with increasingly shorter duration. For RecG it is possible to analyze multiple events in cyclical fashion as the enzyme is not inhibited by the 90mer oligonucleotide. Collectively, the results show that the on rate for RecG binding to a fork with a gap in the nascent leading strand occurred 8.3-fold faster than to forks with gaps in the nascent lagging strand. In addition, these measurements permit estimations of kon for binding forks with nascent lagging (kon1) or leading (kon2) strand gaps, where kon=〈ton〉−1[RecG]−1 resulting in values of kon1~ 6 × 107 M−1 s−1 and kon2~7 × 106 M−1 s−1 being obtained.
c. SSB is displaced by RecG during duplex rewinding (Figure 6C)
As the local concentration of multiple proteins in the vicinity of the fork is likely to be high, their association with the fork might prove deleterious to RecG. To test this, the single strand binding protein (SSB) was used as it is likely to be present at higher concentrations and it binds to ssDNA with high affinity, requiring 2M salt for dissociation (23, 31). The results show that during duplex rewinding, RecG displaces SSB protein at a rate comparable to that observed on free DNA (Fig. 5A). These data show that protein obstacles do not represent a barrier to RecG. This is consistent with data showing that the enzyme can rewind the duplex while working against an opposing force as large as 35pN (18).
d. RecG regresses stalled forks resulting in Holliday junction extrusion (Figure 6D)
A key intermediate in most fork rescue models is the formation of a Holliday junction (19). This is structurally equivalent to a regressed fork, or chicken foot intermediate (Fig. 1). Formation of this intermediate is important as it is the preferred substrate for RuvAB. This enzyme is known to play a key role in the late stages of homologous recombination and there is a strong genetic dependence on ruvAB in fork rescue (4, 32).
To test whether RecG catalyzes fork unwinding/rewinding leading to Holliday junction extrusion, a substrate containing 3 duplex arms was constructed in situ using the strategy shown in Figure 5B. The results show that following the introduction of RecG and ATP, the length of the DNA molecule decreased, consistent with helicase mediated regression resulting in extrusion of a Holliday junction. As the application of the force by the magnetic tweezers favors the fork readvancement over regression, the original 3-armed substrate was restored as soon as RecG dissociated.
e. RecG catalyzes fork regression, not fork readvancement (Figure 6E)
A key aspect in the rescue of stalled replication forks is the movement of the fork away from the site of DNA damage (Fig. 1). As the fork arms are equivalent, and if each is duplex in nature, then the forward (regression) and backward (readvancement) reactions are isoenergetic. Consequently, an enzyme such as RecG should be able to drive both. To determine whether this occurs, gapped DNA substrates were constructed as before and the directional preference of the enzyme tested. Surprisingly, RecG catalyzes reactions in the regression direction almost exclusively (99% vs 1%).
4. Concluding remarks
The regression of stalled replication forks requires an atypical DNA helicase – RecG. This enzyme must be able to unwind nascent duplex arms of a fork, and couple this to ssDNA rewinding resulting in extrusion of a Holliday junction. Reactions should be exclusively in the regression direction and in the process, the helicase should be able to work against large opposing forces that can be used to displace bound proteins from the DNA.
By using a novel fork DNA, magnetic tweezers were used to modify and manipulate the substrate to provide insight into the molecule mechanism of fork rescue. The results clearly show that RecG is indeed a specialized DNA helicase. Furthermore, the results are consistent with bulk-phase biochemical studies demonstrating that RecG acts at a fork first, producing substrates for RuvAB.
This beautiful assay system developed by the Croquette group and described herein, is easily adapted to a variety of enzymes that have been proposed to act at stalled forks in E.coli, including Rep, UvrD, and DinG (33). By modifying the substrate appropriately, DNA replication can also be studied and the effects of discontinuities such as nicks, repetitive sequences, mismatches and DNA damage (e.g., pyrimidine dimers) can be assessed. Finally, this system is ideally suited for studies of recently identified fork rescue enzymes of eukaryotes including SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A-like1 (SMARCAL1) and the Helicase-like transcription factor (HTLF) (34, 35)
Highlights.
A description of a magnetic tweezer assay developed by the Croquette laboratory is presented
The assay uses a novel 1,200bp hairpin substrate that is readily modified to mimic a variety of structures that may form at a stalled DNA replication fork
The E. coli RecG was studied to reveal it is a novel DNA helicase
RecG catalyzes an efficient fork regression reaction that couples DNA unwinding to duplex rewinding
The enzyme works against large opposing forces (i.e., >35pN), can displace bound proteins from the fork (e.g., SSB)
Acknowledgments
Work in the Bianco laboratory is supported by NIH grant GM100156 to PRB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kogoma T. Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiology & Molecular Biology Reviews. 1997;61(2):212–38. doi: 10.1128/mmbr.61.2.212-238.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuzminov A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev. 1999;63(4):751–813. doi: 10.1128/mmbr.63.4.751-813.1999. table of content. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kowalczykowski SC. Initiation of genetic recombination and recombination-dependent replication. Trends in Biochemical Sciences. 2000;25(4):156–65. doi: 10.1016/s0968-0004(00)01569-3. [DOI] [PubMed] [Google Scholar]
- 4.Seigneur M, Bidnenko V, Ehrlich S, Michel B. RuvAB acts at arrested replication forks. Cell. 1998;95(3):419–30. doi: 10.1016/s0092-8674(00)81772-9. [DOI] [PubMed] [Google Scholar]
- 5.Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. The importance of repairing stalled replication forks. Nature. 2000;404(6773):37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- 6.Cox MM. Historical overview: searching for replication help in all of the rec places. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(15):8173–80. doi: 10.1073/pnas.131004998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox MM. Recombinational DNA repair of damaged replication forks in Escherichia coli: questions. Annu Rev Genet. 2001;35:53–82. doi: 10.1146/annurev.genet.35.102401.090016. [DOI] [PubMed] [Google Scholar]
- 8.Marians KJ. Replication and recombination intersect. Current Opinion in Genetics & Development. 2000;10(2):151–6. doi: 10.1016/s0959-437x(00)00059-9. [DOI] [PubMed] [Google Scholar]
- 9.Lusetti SL, Cox MM. The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annual Review of Biochemistry. 2002;71:71–100. doi: 10.1146/annurev.biochem.71.083101.133940. [DOI] [PubMed] [Google Scholar]
- 10.Marians KJ. Mechanisms of replication fork restart in Escherichia coli. Philos Trans R Soc Lond B Biol Sci. 2004;359(1441):71–7. doi: 10.1098/rstb.2003.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michel B, Grompone G, Flores MJ, Bidnenko V. Multiple pathways process stalled replication forks. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(35):12783–8. doi: 10.1073/pnas.0401586101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Postow L, Ullsperger C, Keller RW, Bustamante C, Vologodskii AV, Cozzarelli NR. Positive torsional strain causes the formation of a four-way junction at replication forks. Journal of Biological Chemistry. 2001;276(4):2790–6. doi: 10.1074/jbc.M006736200. [DOI] [PubMed] [Google Scholar]
- 13.McGlynn P, Lloyd R. Action of RuvAB at replication fork structures. J Biol Chem. 2001;276(45):41938–44. doi: 10.1074/jbc.M107945200. [DOI] [PubMed] [Google Scholar]
- 14.Robu ME, Inman RB, Cox MM. Situational repair of replication forks: roles of RecG and RecA proteins. Journal of Biological Chemistry. 2004;279(12):10973–81. doi: 10.1074/jbc.M312184200. [DOI] [PubMed] [Google Scholar]
- 15.Robu ME, Inman RB, Cox MM. RecA protein promotes the regression of stalled replication forks in vitro. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(15):8211–8. doi: 10.1073/pnas.131022698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abd Wahab S, Choi M, Bianco PR. Characterization of the ATPase activity of RecG and RuvAB proteins on model fork structures reveals insight into stalled DNA replication fork repair. J Biol Chem. 2013;288(37):26397–409. doi: 10.1074/jbc.M113.500223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buss J, Kimura Y, Bianco P. RecG interacts directly with SSB: implications for stalled replication fork regression. Nucleic Acids Res. 2008;36(22):7029–42. doi: 10.1093/nar/gkn795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manosas M, Perumal SK, Bianco PR, Ritort F, Benkovic SJ, Croquette V. RecG and UvsW catalyse robust DNA rewinding critical for stalled DNA replication fork rescue. Nat Commun. 2013;4:2368. doi: 10.1038/ncomms3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atkinson J, McGlynn P. Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 2009;37(11):3475–92. doi: 10.1093/nar/gkp244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slocum SL, Buss JA, Kimura Y, Bianco PR. Characterization of the ATPase activity of the Escherichia coli RecG protein reveals that the preferred cofactor is negatively supercoiled DNA. J Mol Biol. 2007;367(3):647–64. doi: 10.1016/j.jmb.2007.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whitby MC, Vincent SD, Lloyd RG. Branch migration of Holliday junctions: identification of RecG protein as a junction specific DNA helicase. EMBO J. 1994;13(21):5220–8. doi: 10.1002/j.1460-2075.1994.tb06853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lohman TM, Green JM, Beyer RS. Large-scale overproduction and rapid purification of the Escherichia coli ssb gene product. Expression of the ssb gene under lambda PL control. Biochemistry. 1986;25(1):21–5. doi: 10.1021/bi00349a004. [DOI] [PubMed] [Google Scholar]
- 23.Lohman TM, Overman LB. Two binding modes in Escherichia coli single strand binding protein-single stranded DNA complexes. Modulation by NaCl concentration. J Biol Chem. 1985;260(6):3594–603. [PubMed] [Google Scholar]
- 24.Manosas M, Spiering MM, Zhuang Z, Benkovic SJ, Croquette V. Coupling DNA unwinding activity with primer synthesis in the bacteriophage T4 primosome. Nat Chem Biol. 2009;5(12):904–12. doi: 10.1038/nchembio.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manosas M, Meglio A, Spiering MM, Ding F, Benkovic SJ, Barre FX, et al. Magnetic tweezers for the study of DNA tracking motors. Methods Enzymol. 2010;475:297–320. doi: 10.1016/S0076-6879(10)75013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manosas M, Perumal SK, Croquette V, Benkovic SJ. Direct observation of stalled fork restart via fork regression in the T4 replication system. Science. 2012;338(6111):1217–20. doi: 10.1126/science.1225437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng W. Mechanisms of HCV NS3 helicase monitored by optical tweezers. Methods Mol Biol. 2015;1259:229–55. doi: 10.1007/978-1-4939-2214-7_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spies M. Two steps forward, one step back: determining XPD helicase mechanism by single-molecule fluorescence and high-resolution optical tweezers. DNA Repair (Amst) 2014;20:58–70. doi: 10.1016/j.dnarep.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gosse C, Croquette V. Magnetic tweezers: micromanipulation and force measurement at the molecular level. Biophys J. 2002;82(6):3314–29. doi: 10.1016/S0006-3495(02)75672-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singleton MR, Scaife S, Wigley DB. Structural analysis of DNA replication fork reversal by RecG. Cell. 2001;107(1):79–89. doi: 10.1016/s0092-8674(01)00501-3. [DOI] [PubMed] [Google Scholar]
- 31.Liu J, Choi M, Stanenas AG, Byrd AK, Raney KD, Cohan C, et al. Novel, fluorescent, SSB protein chimeras with broad utility. Protein Sci. 2011;20(6):1005–20. doi: 10.1002/pro.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.West SC. Processing of recombination intermediates by the RuvABC proteins. Annual Review of Genetics. 1997;31:213–44. doi: 10.1146/annurev.genet.31.1.213. [DOI] [PubMed] [Google Scholar]
- 33.Boubakri H, de Septenville AL, Viguera E, Michel B. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J. 2010;29(1):145–57. doi: 10.1038/emboj.2009.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Betous R, Mason AC, Rambo RP, Bansbach CE, Badu-Nkansah A, Sirbu BM, et al. SMARCAL1 catalyzes fork regression and Holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012;26(2):151–62. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kile AC, Chavez DA, Bacal J, Eldirany S, Korzhnev DM, Bezsonova I, et al. HLTF’s Ancient HIRAN Domain Binds 3′ DNA Ends to Drive Replication Fork Reversal. Mol Cell. 2015;58(6):1090–100. doi: 10.1016/j.molcel.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]