

Abstract
Hemophilia A and B are coagulation disorders resulting from the loss of functional coagulation factor VIII (FVIII) or factor IX proteins, respectively. Gene therapy for hemophilia with adeno-associated virus vectors has shown efficacy in hemophilia B patients. Although hemophilia A patients are more prevalent, the development of therapeutic adeno-associated virus vectors has been impeded by the size of the F8 cDNA and impaired secretion of FVIII protein. Further, it has been reported that over-expression of the FVIII protein induces endoplasmic reticulum stress and activates the unfolded protein response pathway both in vitro and in hepatocytes in vivo, presumably due to retention of misfolded FVIII protein within the endoplasmic reticulum. Engineering of the F8 transgene, including removal of the B domain (BDD-FVIII) and codon optimization, now allows for the generation of adeno-associated virus vectors capable of expressing therapeutic levels of FVIII. Here we sought to determine if the risks of inducing the unfolded protein response in murine hepatocytes extend to adeno-associated virus gene transfer. Although our data show a mild activation of unfolded protein response markers following F8 gene delivery at a certain vector dose in C57BL/6 mice, it was not augmented upon further elevated dosing, did not induce liver pathology or apoptosis, and did not impact FVIII immunogenicity.
Introduction
Hemophilia A and B are coagulations disorders resulting from the loss of functional coagulation factor VIII (FVIII) or factor IX (FIX) proteins, respectively.1 Untreated patients may spontaneously or through trauma develop internal bleeds leading to progressive joint damage and potentially fatal bleeds into closed spaces such as the cranium. The recommend treatment for hemophilia is the prophylactic infusion of recombinant or plasma derived coagulation factors. While effective, these exogenously administered coagulation factors have short biological half-lives and thus have to be recurrently administered, which requires frequent intravenous access. Although longer-acting factors are currently in clinical development, half-life of FVIII in circulation is only slightly prolonged.2 In contrast, gene therapy holds the promise of multiyear and perhaps even life-long correction of the disease from a single drug administration. This concept is supported by demonstration of lasting therapy in studies in large animals and recent clinical trial results, which are based on liver-directed gene transfer with adeno-associated virus (AAV) vectors.3–6
Stable therapeutic levels of the vitamin K-dependent serine protease FIX or its cofactor, FVIII, have been reached using multiple viral based delivery systems and target tissues in both small and large hemophilia animal models.4,7 AAV gene transfer to hepatocytes has had translational success in early phase clinical trials in hemophilia B patients, resulting in FIX expression levels that have significantly reduced or completely eliminated the need for exogenous FIX protein therapy.3,8 While hemophilia A is more common (1 in 5,000 male births) than hemophilia B (1 in 25,000 male births), progress on developing an AAV-F8 vector has been complicated by multiple factors. Prominent among these factors is the low efficiency of human FVIII protein expression and secretion (due to retention in the endoplasmic reticulum, ER),9,10 and the size of the F8 coding sequence.11,12 To overcome these limitations, B domain deleted (BDD) FVIII has been chosen as a sufficiently small sequence to fit within the packaging limit of an AAV vector. Expression levels have been substantially improved by use of codon-optimized sequences,13,14 and secretion and biological activity have been improved through protein engineering.14–16
Nonetheless, several concerns remain. One is the high immunogenicity of FVIII, with 25–30% of hemophilia A patients forming antidrug antibodies (termed “inhibitors”). Although FVIII normally circulates at low levels (200 ng/ml), thus requiring only low protein doses to correct the bleeding disorder, potent antibody responses against FVIII occur even at these low antigen doses. Supporting a gene therapy approach, AAV-mediated gene transfer to hepatocytes often induces immune tolerance to the transgene product, which is in part mediated by induction of CD4+CD25+FoxP3+ regulatory T cells.17 However, expression levels and changes in the FVIII sequence may impact the immune response. Furthermore, the potential for inhibitor formation upon AAV liver gene transfer has varied between species in various preclinical studies.4,14 Encouragingly, hepatic AAV gene transfer has been able to reverse pre-existing inhibitors in a canine model of hemophilia A.18
A second concern is the potential for overexpression of FVIII in hepatocytes to cause a cellular stress response, as reported by others for plasmid gene transfer.19 The unfolded protein response (UPR) pathway is a conserved cellular stress response and quality control mechanism that is activated upon accumulation of misfolded proteins in the ER, which may result from increased translation.20–24 The UPR sentinel chaperone protein BiP (“binding immunoglobulin protein”, also known as “glucose-regulated protein 78” or GRP78) selectively binds misfolded proteins. When a threshold level of BiP is engaged with misfolded proteins, the UPR is initiated. The resulting signaling cascade directs a halt in cellular translation, upregulation of chaperone proteins specialized in protein folding, and—if unresolved—leads to apoptotic cell death. In vitro heterologous overexpression of the human FVIII protein was reported to lead to the accumulation of misfolded FVIII and UPR chaperone protein aggregate in the ER and induce programmed cell death.25 Similarly, Malhotra et al. demonstrated that in vivo hydrodynamic delivery of plasmids encoding full-length or BDD-FVIII proteins resulted in activation of the UPR in murine hepatocytes and apoptosis.10 However, such hydrodynamic injections are done under high pressure, and transfection with plasmid DNA is a different process from receptor-mediated viral infection.
The relevance of these findings for hepatic FVIII gene transfer with the clinically used AAV vector system has not yet been demonstrated. Therefore, we investigated the potential for induction of UPR in mouse livers following administration of high doses of AAV serotype 8 vector expressing codon-optimized BDD-FVIII. Although we found clear evidence for an initial cellular stress response at vector doses ≥ 1011 vg in C57BL/6 mice, corresponding to expression of ≥ 0.04 IU/ml circulating FVIII, there was no indication of hepatotoxicity or heightened immune response to FVIII.
Results
High-dose gene transfer of codon-optimized AAV8-F8 does not elevate liver enzyme levels
Wild-type C57BL/6 mice were used to investigate the potential of BDD-FVIII expressed from a codon-optimized AAV8-F8 (AAV8-coF8) vector to induce the UPR in transduced livers using vector doses of 1 × 1010, 1 × 1011, and 1 × 1012 vg/mouse. C57BL/6 mice were selected for these experiments, because (i) this strain shows the highest transduction of hepatocytes with most AAV serotypes, and (ii) this strain was used by the original study on UPR to FVIII in vivo expression in hepatocytes by Malhotra et al. An AAV8-null vector, containing a promoterless transgene, and an AAV8-F9 vector at a dose of 1 × 1012 vg were included as controls to rule out potential contributions of capsid protein to induce ER stress and hepatotoxicity or effects that are not specific to FVIII.26,27 Livers were collected 4 weeks following gene transfer, where we typically observe maximal BDD-FVIII expression. Plasma was collected from mice at 2 and 4 weeks to measure circulating human BDD-FVIII protein and alanine transaminase (ALT) levels, a sensitive plasma marker for hepatotoxicity. We observed a vector dose-dependent increase in circulating BDD-FVIII antigen levels (Figure 1a). None of the AAV8-coF8 transduced mice formed antibodies against FVIII (Figure 1b). Therefore, the cellular stress response in C57BL/6 mice did not increase immunogenicity of FVIII. There was also no elevation of ALT levels (Figure 1c). For unknown reasons, the AAV8-null vector transduced mice had ~twofold increase in ALT compared with naive mice, although this was still within normal range for this strain. This cannot be attributed to a response to capsid, as the same dose of 1 × 1012 vg of AAV8-coF8 or AAV8-F9, containing the same capsid load, did not have this effect.
Figure 1.

Hepatic AAV8 gene transfer of codon-optimized BDD-FVIII (coFVIII) to C57BL/6 mice. (a) Systemic FVIII antigen levels as a function of time after gene transfer of 1 × 1010 vg (n = 5), 1 × 1011 vg (n = 4–8), or 1 × 1012 vg (n = 4–8) to C57BL/6 mice or to hemophilia A mice (129/BL6-F8−/Y, labeled with “HA” in caption). (b) Anti-FVIII IgG1 levels at 4 weeks post gene transfer in C57BL/6 mice treated with the indicated vectors. A previously identified anti-FVIII IgG1 positive plasma sample from a FVIII immunized 129/BL6-F8−/Y mouse was included as a control. (c) Liver enzyme levels (ALT, alanine transaminase) in plasma 4 weeks after hepatic gene transfer to C57BL/6 mice. For comparison, data from AAV-F9 transduced and naive mice are shown. Data are mean ± SD. AAV, adeno-associated virus; BDD, B domain deleted; FVIII, functional factor VIII; SD, standard deviation.
Evidence for induction of cellular stress by BDD-FVIII expression in the liver at doses ≥1011 vg
To further address if overexpression of BDD-FVIII lead to activation of the UPR in mouse livers, we assayed for changes in the expression of the sentinel UPR chaperone protein BiP and the downstream signaling, proapoptotic protein CCAAT/enhancer-binding protein homologous protein (CHOP) 4 weeks after gene transfer (n = 3–7 per vector and dose). Western blot of liver lysates clearly showed upregulation in BiP expression in mouse livers treated at the higher vector doses of 1 × 1011 and 1 × 1012 vg of AAV8-coF8 (Figure 2a). These results were reproducible when Western blots were repeated (data not shown). In contrast, no elevation of BiP expression was seen in livers transduced with the lowest dose of AAV8-coF8 (1 × 1010 vg) or at 1012 vg with AAV8-null or AAV8-F9 vectors (Figure 2a and data not shown; a total of 3 livers were analyzed from AAV8-F9 transduced mice, which expressed on average 26–30 μg FIX/ml plasma, i.e., 5.2–6x higher than normal, during the first month after gene transfer). Livers collected from mice 48 hours after treatment with the ER stress inducing agent tunicamycin were used as a positive control.28 Interestingly, the levels of BiP expression did not further increase from the mid to the high dose of AAV8-coF8. The expression pattern observed for CHOP protein levels matched that seen for BiP (Figure 2b).
Figure 2.
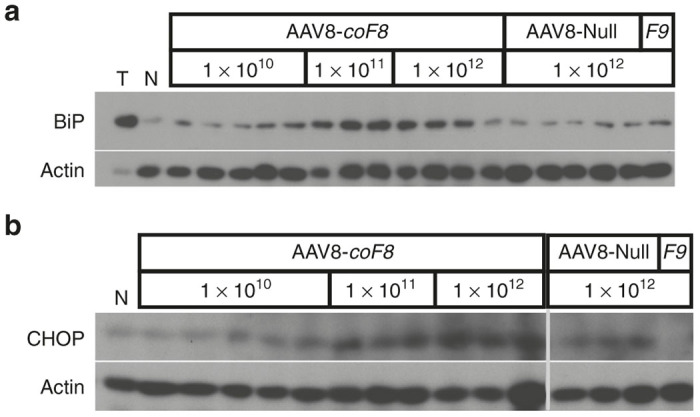
Analysis of the expression status of UPR chaperones as a consequence of hepatic gene transfer of coF8 in C57BL/6 mice 4 weeks post treatment. (a) Western Blotting of liver tissue lysates for detection of BiP and (b) of CHOP in animals receiving 1 × 1010 vg (n = 5), 1 × 1011 vg (n = 3), or 1 × 1012 vg (n = 4) of AAV8-coF8 or 1 × 1012 vg of AAV8 null vector (n = 5). For additional comparison, protein extract from a mouse treated with tunicamycin (T), that was untreated (N, “naive”), or that received 1 × 1012 vg of AAV8-F9 vector (F9). AAV, adeno-associated virus; BiP, binding immunoglobulin protein; CHOP, CCAAT/enhancer-binding protein homologous protein; UPR, unfolded protein response.
ER stress response in C57BL/6 mice declines over time
To rule out the possibility of a delayed ER stress response or potential differences in background strains we conducted two additional studies in wild-type C57BL/6 and hemophilia A 129/BL6-F8−/Y mice receiving a dose of 1 × 1011 vg of the AAV8-coF8 vector, where we had previously observed induction of the ER stress response (Figure 2a). Circulating BDD-FVIII levels were measured at 2, 4, 8, and 12 weeks gene transfer. BDD-FVIII protein reached peak levels at 4 weeks and remained stable for the duration of the study (Figure 3a). Endpoint analysis of plasma at 4 weeks (129/BL6-F8−/Y mice) and 12 weeks (C57BL/6 mice) showed that vector treated mice remained free of anti-FVIII IgG1 antibodies and ALT levels were within normal limits (Figure 3b,c), suggesting that ER stress was not severe enough to cause substantial death of murine hepatocytes. Western blots of liver lysates showed no evidence for BiP upregulation in 4/5 C57BL/6 mice at 12 weeks, while CHOP was still somewhat upregulated. Thus, the ER stress response appeared to subside rather than escalate over time.
Figure 3.

ER stress response following hepatic AAV8-coF8 gene transfer declines over time in C57BL/6 mice. (a) Systemic levels of FVIII antigen as a function of time in C57BL/6 mice (n = 5) injected with 1 × 1011 vg of an AAV8-coF8 vector. (b) Anti-FVIII IgG1 levels in plasma at 12 weeks. (c) Liver enzyme levels (ALT, alanine transaminase) in plasma 12 weeks after hepatic gene transfer. Dotted lines represent mean ± 1 SD of ALT measured in naive C57BL/6 mice. (d) Analysis of the expression status of UPR chaperones as a consequence of hepatic gene transfer of coF8 in C57BL/6 mice 12 weeks post-treatment. Western Blotting of liver tissue lysates for detection of BiP, CHOP, and actin in C57BL/6 mice (n = 5) receiving 1 × 1011 vg. For additional comparison, protein extract from a C57BL/6 mouse treated with tunicamycin (T) or that was untreated (N, “naive”). AAV, adeno-associated virus; BiP, binding immunoglobulin protein; CHOP, CCAAT/enhancer-binding protein homologous protein; ER, endoplasmic reticulum; FVIII, functional factor VIII; UPR, unfolded protein response.
AAV8-coF8 gene transfer does not cause liver pathology or apoptosis of hepatocytes
Finally, mouse liver tissue was stained with hematoxylin and eosin to assess pathology and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) fluorescent staining was used to assay for apoptosis. Images of TUNEL stained sections were captured on a fluorescent microscope. As expected, tunicamycin treated positive control mice had the highest overall level of TUNEL+ hepatocytes. In contrast, we did not observe an elevation in TUNEL+ hepatocytes in liver sections from AAV8-coF8 vector treated mice at either 4 weeks or 12 weeks as compared with age-matched controls (that did not receive gene transfer). Three of five mice injected with 1 × 1012 vg of the AAV8-null vector had some defined areas of TUNEL+ hepatocytes (Figure 4a), in line with the elevation of plasma ALT. Hematoxylin and eosin stained sections were blindly scored for pathology. No significant difference in pathological scores was observed between control (no gene transfer) and vector treated mice (data not shown). None of the experimental treatments resulted in inflammation or other histopathology (Figure 4b and data not shown).
Figure 4.
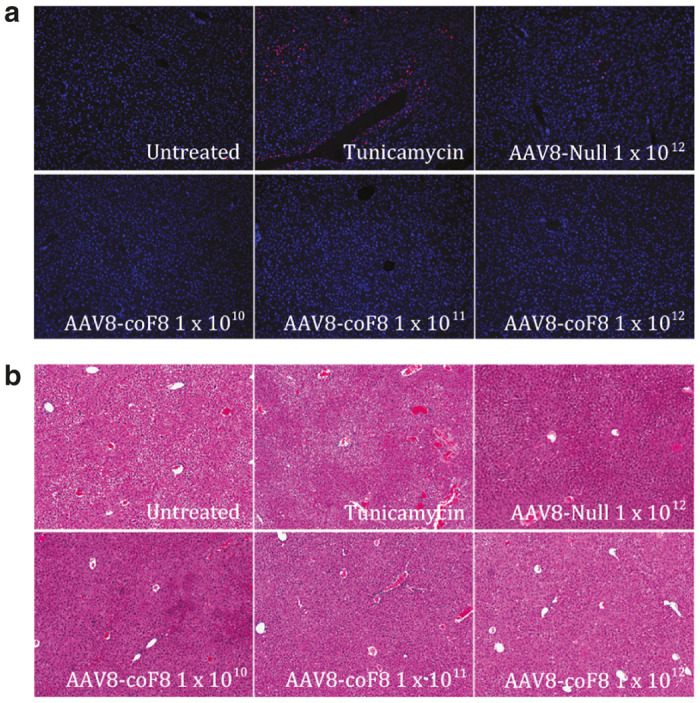
Histological analysis of C57BL/6 mouse liver tissue 4 weeks after AAV8-coF8 gene transfer. Paraffin embedded liver sections stained with (a) Terminal deoxynucleotidyl transferase dUTP nick end labeling kit (TUNEL) for presence of apoptotic cells, (blue –DAPI (4’, 6-Diamidino-2-Phenylindole, Dihydrochloride), red –fluorescein-dUTP, pseudocolored) and (b) Hematoxylin and Eosin (H&E) for general tissue pathology. Representative images for each group are shown. AAV, adeno-associated virus.
High-dose gene transfer of codon-optimized AAV8-FVIII does not increase the risk of inhibitor formation in hemophilia A mice
In previous experiments, we found that liver-directed gene transfer with AAV8 vectors expressing human BDD-FVIII from wild-type or codon-optimized cDNA under a hepatocyte-specific promoter, unlike repeated intravenous (i.v.) injections of BDD-FVIII protein, did not cause inhibitor formation in hemophilia A mice at a vector dose of 1 × 1011 vg/mouse.13 However, use of the codon-optimized sequence improved systemic expression by up to 10-fold.13 Moreover, only the codon-optimized vector tolerized the animals, so that they remained hypo-responsive to BDD-FVIII after subsequent i.v. challenge with protein.13 Here, we injected 1 × 1011 vg (n = 3) or 1 × 1012 vg (n = 4) of the codon-optimized AAV8-F8 vector into the tail vein of hemophilia A mice with a gene deletion in exon-16 coding for murine F8 on 129/BL6 genetic background (129/BL6-F8−/Y). By 1 month after gene transfer, mice expressed ~10% of normal FVIII activity at 1 × 1011 vg and ~100% of normal at 1 × 1012 vg, which slightly declined by 2 months (Figure 5a). Circulating BDD-FVIII levels as measured by enzyme-linked immunosorbent assay (ELISA) were comparable with those in C57BL/6 mice for the same vector doses (Figures 1a and 5b). FVIII levels measured by ELISA were lower compared with activity measurements, likely reflecting an underestimation because of masking of FVIII by von Willebrand factor in the ELISA.
Figure 5.

Correction of murine hemophilia A mice and antibody formation against FVIII by hepatic AAV8 gene transfer of codon-optimized BDD-F8 (coF8). (a) Systemic FVIII activity levels as a function of time after gene transfer of 1 × 1011 vg (n = 3) or 1 × 1012 vg (n = 4) to 129/BL6-F8−/Y mice (HA). (b) Systemic FVIII antigen levels as determined by ELISA. (c) Anti-FVIII IgG1 levels and (d) Bethesda titers, measured 1 week after repeated i.v. injections of BDD-FVIII protein (1 IU, weekly for 4 weeks) in mice that received gene transfer and in control mice. Data are mean ± SD. Panels c and d additionally shows values for individual mice. (e) Analysis of the expression status of UPR chaperones as a consequence of hepatic gene transfer of coF8 in C57BL/6 mice 12 weeks and 129/BL6-F8−/Y mice 4 weeks post-treatment. Western Blotting of liver tissue lysates for detection of BiP, CHOP, and actin in 129/BL6-F8−/Y mice receiving 1 × 1011 vg (n = 4) or that were untreated (naive, n = 3). For positive control, protein extract from a C57BL/6 mouse treated with tunicamycin (T) is shown (same control as in Figure 3d, as these samples were run on the same blot). AAV, adeno-associated virus; BiP, binding immunoglobulin protein; CHOP, CCAAT/enhancer-binding protein homologous protein; BDD, B domain deleted; ELISA, enzyme-linked immunosorbent assay; FVIII, functional factor VIII; UPR, unfolded protein response; SD, standard deviation.
During the 4-week time course of the experiment, there was no evidence for antibody formation against FVIII by ELISA or Bethesda assay (data not shown). All mice were subsequently challenged with 4 weekly i.v. doses of BDD-FVIII protein. Of the seven animals, only one in the high-dose group formed an antibody against FVIII (17BU), while control mice that had not received gene transfer all formed inhibitors in response to the i.v. treatment with protein (Figure 5c,d). Moreover, 6/7 control mice developed very high-titer inhibitors of > 50BU.
Another cohort of hemophilia A mice received 1 × 1011 vg of AAV8-coF8 (n = 4), and livers were collected 4 weeks after gene transfer. In contrast to the data shown for C57BL/6 mice in Figure 2, 129/BL6-F8−/Y mice had no evidence for increase in BiP or CHOP expression at 4 weeks after gene transfer (Figure 5e). These results indicate a mouse-strain dependence of ER stress in hepatocytes in response to FVIII expression. TUNEL staining of liver sections also had no increase over naive 129/BL6-F8−/Y colony background, and no elevation of ALT levels was observed (data not shown).
Discussion
While AAV gene therapy for hemophilia B has been successful and is tested in multiple clinical trials, there are ~4 times more patients with hemophilia A. Preclinical development of an optimal protocol to express high levels of human FVIII has been more challenging, but have seen recent success with the use of BDD-FVIII and codon optimization. Our data presented here raise the concern that high expression of FVIII protein may prompt an UPR in hepatocytes. UPR was not seen following administration of a null vector or a FIX vector, indicating that elevated FVIII levels were the cause of the response. However, this cellular stress response was limited, without abrogating tolerance induction by transgene expression or causing toxicity.
Boosting hepatocyte-derived FVIII expression—risks versus rewards
Recent studies have documented that FVIII is normally synthesized in the liver by endothelial cells rather than by hepatocytes therefore endogenous murine FVIII protein is unlikely to contribute to the hepatocyte ER stress response from AAV8-coF8 gene transfer.29–32 The wild-type FVIII sequence, also referred to as full-length FVIII, includes a heavily glycosylated B domain that is involved in intracellular processing of FVIII but is dispensable for coagulation activity in the blood.33 Because of limited packaging size of the genome, capsid tropism to hepatocytes, and relative abundance of hepatocytes over liver endothelial cells, the currently prevailing strategy is to use AAV vectors to transfer BDD-FVIII into hepatocytes for heterologous expression from a hepatocyte-specific promoter. Currently, the prevailing approach for hemophilia gene therapy is gene transfer to hepatocytes. Use of a hepatocyte-specific promoter combined with a serotype with high hepatic tropism allowed us to study the effect of ectopic FVIII overexpression in hepatocytes (while endothelial cells are naturally the predominant site of FVIII synthesis). Further, given the increased potential of immune responses directed against the BDD-FVIII protein, our previous studies have shown that AAV mediated tolerance is less effective when using an ubiquitous promoter.34 In mice, the most efficient serotype for this task is AAV8, while a debate is ongoing about the optimal serotype for human hepatocytes.35–37 Regardless, additional modifications are needed to achieve therapeutic expression with this approach, such as the >5% of normal that have now been achieved in hemophilia B patients with the more robustly expressed FIX.3,8
One broadly adapted strategy has been codon optimization of therapeutic transgenes as a means of enhancing transgene expression. Yet there is evidence that low frequency codons and stable secondary structures may play a role in protein folding and assembly by inducing a stall of protein synthesis.38–40 Thus, codon optimization may impact protein folding and function, particularly in larger proteins such as BDD-FVIII. Nonetheless, substantial increases in AAV-encoded, hepatocyte-derived BDD-FVIII of up to 1 log have been achieved using codon-optimized sequences.13,14 Hence, there is a clear therapeutic benefit. Furthermore, the use of the codon-optimized sequence facilitated tolerance induction to FVIII in hemophilia A mice, which correlated with increased regulatory T cells induction upon hepatic gene transfer.13 However, the predisposition of human BDD-FVIII protein to accumulate in the ER and engage with UPR chaperones may cause a cellular stress response at elevated levels of expression, which is what we found here.19,25
A major concern in designing this study was the selection of an optimal window of time to analyze plasma and livers for UPR and liver damage, as the kinetics of AAV expression differ from hydrodynamic plasma delivery.19 Our experience with liver directed AAV8-coF8 gene transfer in mice is that maximal expression in the circulation is reached between 2 and 4 weeks following gene delivery, which is supported by this study. Therefore, we selected 4 and 12 weeks as relevant time points to address the impact of maximal BDD-FVIII protein expression in hepatocyte in the short-term and over an extended time frame. While we cannot rule out that ER stress already occurred at earlier time points, the objective of this study was to assess the effect of long-term AAV-derived FVIII expression in the liver. Induction of a cellular stress response by hepatic gene transfer to C57BL/6 mice was seen at the mid and high dose tested here, with the mid dose achieving levels of transgene expression that are a therapeutic target for current and future clinical trials. The response was also mouse strain dependent and not seen in 129/BL6-F8−/Y at a dose and time point where C57BL/6 mice had clearly up-regulated BiP and CHOP. While we did not investigate UPR for the highest vector dose used in 129/BL6-F8−/Y mice, Lange et al., as reported in a companion article (Mol Ther Methods Clin Dev 3, 16064, 2016), achieved similar high levels of FVIII activity in this strain at their highest vector dose and found only a delayed and transient cellular stress response in the liver.
No evidence that stress response to FVIII causes toxicity or increased immunogenicity
Overall, our data show that expression of a codon-optimized human BDD-FVIII transgene following AAV liver-targeted gene transfer is not sufficient to induce UPR mediated apoptosis in murine hepatocytes or pathology. In fact, induction of stress response markers occurred at or above a certain vector dose (1 × 1011 vg/mouse or 4 × 1012 vg/kg for a 25-g animal), equivalent to ~0.05 IU/ml BDD-FVIII protein in circulation, but was not further increased at a 10-fold higher dose. One unanticipated result in our study was the observation of liver damage in control mice receiving 1 × 1012 vg of the AAV8-Null vector, confirmed by both a rise in plasma ALT and TUNEL+ stained hepatocytes. As there is no evidence supporting toxicity with viral capsids in mice and this particular vector has no transgene expression, the most likely explanation for this mild increase in ALT and positive TUNEL staining is the copurification of some toxic agent in this particular viral preparation (which was produced as research grade but not clinical grade material and had to be concentrated to achieve vector doses). This conclusion is supported by an absence of elevated ALT and TUNEL+ hepatocytes in mice receiving similar doses of coF8 and F9 vectors.
A previous study by Malhotra et al. reported that BDD-FVIII expression in C57BL/6 mice by hydrodynamic plasmid delivery induced UPR responses in mouse liver that caused apoptotic cell death in hepatocytes.19 However, hydrodynamic gene delivery induces varying degrees of changes in hepatocyte morphology and cytosol content and thus may sensitize hepatocytes to cellular stress induction following overexpression of BDD-FVIII protein and/or heightened the consequences of the stress response.41,42 These conclusions are further supported by the study by Lange et al. (Mol Ther Methods Clin Dev 3, 16064, 2016), in which hepatocyte apoptosis was only observed in plasmid hydrodynamic delivery of canine BDD-FVIII (cF8) but was absent following i.v. AAV8-cF8 gene delivery. In contrast, a recent study using hydrodynamic delivery of a piggy-Bac transposon expressing a codon optimized B domain deleted FVIII failed to detect activation of the ER stress response compared with sham treated mice, albeit these authors only examined late time points.43
Importantly, we did not observe any increase in apoptotic hepatocytes over naive strain background with our AAV8-coF8 vector at doses up to 1 × 1012 vg, corresponding to circulating levels of BDD-FVIII protein in the range of 0.12–0.3 IU/ml in both hemophilia A and C57BL/6 mice by ELISA and 0.46–1.8 IU/ml (i.e., 46–180% of normal) in hemophilia A mice using a functional coagulation assay. It is likely that the ELISA technique underestimates the BDD-FVIII levels, which is often observed because of shielding by von Willebrand factor (vWF) protein bound to FVIII.44,45 Finally, the stress response we observed in C57BL/6 mice did not cause antibody formation against the FVIII transgene product.
Implications for human gene therapy
With the initiation of the first clinical trial of AAV-F8 gene therapy in hemophilia A patients (NCT02576795) it is important to model and understand potential barriers to successful therapy.
While we focused on studying our codon-optimized construct, which substantially improved therapy and tolerance induction in our previous experiments, Lange and colleagues (see companion article, Mol Ther Methods Clin Dev 3, 16064, 2016) found that high levels of canine BDD-FVIII protein expressed from an AAV8 vector (without codon optimization) induced a delayed and transient cellular stress response at 12 weeks after gene transfer. It is likely that these different outcomes are in part caused by mouse strain differences, as we also failed to detect upregulation of BiP in 129/BL/6-F8−/Y mice 4 weeks following gene delivery (1 × 1011 vg AAV8-coF8) compared with identically dosed wild-type C57BL/6 mice. While our studies observed differences in immune responses against human and canine FVIII proteins, in aggregate the data strongly suggest that the risk for antibody formation is not linked to an ER stress response.
Overall, data from both of our studies support that the magnitude and/or duration of the stress response induced by AAV-F8 mediated gene transfer is insufficient to drive apoptosis in murine hepatocytes up to 12 weeks following gene transfer. Nonetheless, our studies show that there is at least a potential for cellular stress response upon overexpression of FVIII protein in hepatocytes, which investigators should be aware of when taking vectors forward to clinical trial. Even though we found no evidence for liver toxicity and that the stress response subsided over time, we cannot entirely rule out longer-term effects. Finally, these results support further development of strategies aimed at improving secretion of FVIII, which may limit ER stress. Examples include incorporation of or mimicking sequences from other species, such as porcine or canine, that have more efficiently secreted and/or more active FVIII molecules (albeit one needs to also consider that such modified molecules may introduce novel B and T cell epitopes).15,46–48
Materials and Methods
AAV vectors
Recombinant AAV8 vectors were produced by triple transfection method of HEK-293 cells and purified by iodixanol gradient centrifugation as previously described.49 Codon-optimized human BDD-F8 gene (GeneArt, Regensburg, Germany) and F9 genes are expressed from the hepatocyte specific ApoE/human α-antitrypsin (hAAT) enhancer/promoter as published.13,34 The AAV-null vector contains the β-gal and neo genes both with a mutation in the initiating methionine (ATG to CTG) and lack both promoters and polyadenylation signals.27 Virus titers were determined by dot blot assay. Vectors were administered at a dose of 1 × 1010, 1 × 1011, or 1 × 1012 vg per mouse via tail vein injection.
Animal studies
Male C57BL/6 mice (6–8 weeks of age) were purchased from Jackson Laboratories (Bar Harbor, ME). Hemophilia A mice with a targeted deletion of exon 16 of the murine F8 gene have been maintained on a mixed C57BL/6–129/sv background as previously published and were obtained from our in-house colony.13 Viral vectors were administered by i.v. injection in a 200 μl volume at the indicated doses. In some studies hemophilia A mice received four weekly i.v. injections of 1 IU BDD-FVIII (Xyntha, Pfizer, New York, NY) following vector administration. Blood for plasma analysis was obtained by tail bleed into citrate buffer (0.38%) for hemophilia A mice or into heparinized capillary tubes for studies with C57BL/6 mice. As a positive control for ER stress induction, C57BL/6 mice received an intraperitoneal injection of 25 μg tunicamycin dissolved in 20% dimethylsulfoxide (DMSO)/phosphate-buffered saline (PBS)/150 mmol/l dextrose, and livers were collected 48 hours later.
FVIII activity, antigen levels, and antibody assays
FVIII activity in hemophilia A mouse plasma was determined using an activated partial thromboplastin time assay. Activity was calculated using a standard curve generated from normal human plasma over a range of 0–40% normal FVIII levels. Levels of circulating human Factor VIII protein in mouse plasma samples were determined using the VisuLize FVIII Antigen Kit (Affinity Biologicals, Ancaster, Canada) per manufacturer’s instructions. BDD-FVIII levels were normalized against naive and control vector treated mice to correct for cross reactivity of the ELISA with murine FVIII protein. Inhibitory antibody titers against FVIII were determined using the Bethesda inhibitor assay. One Bethesda unit is defined as the amount of inhibitory antibody that can neutralize 50% FVIII activity following a 2-hour incubation at 37°C. An immunocapture ELISA was used to determine titers of FVIII-specific IgG1 using purified mouse IgG1 as a standard.
Analysis of ER stress response in murine livers
Liver tissue lysates were generated using lysing matrix beads (MP Biomedicals, Santa Ana, CA) in radioimmunoprecipitation assay (RIPA) buffer and total protein levels were adjusted using OD280 adsorption. For Western blotting, 100 ng of total protein was run on 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE), transferred to polyvinylidene difluoride membrane (PVDF) membrane and probed overnight with one of the following antibodies: BiP (1:1,000), CHOP (1:1,000), pan-actin (1:1,000), or Ph/PERK (1:1,000), all from Cell Signaling (Danvers, MA), and the corresponding secondary antibodies. Specific protein visualized with Pierce ECL2 Western Blotting Kit (Thermo Fisher Scientific, Waltham, MA).
Toxicity studies
Liver function test was run on mouse plasma using a colorimetric Alanine Transaminase (ALT) Activity Assay kit (ab105134, Abcam, Cambridge, UK) following manufacturer’s instructions. For histological analysis, mouse liver tissue was rinsed with PBS and fixed in 10% buffered Formalin Solution overnight and embedded in paraffin blocks. Sections (4 μm) were stained with hematoxylin and eosin or processed for TUNEL staining with In Situ Cell Death Detection Kit, Roche (Basel, Switzerland).
Author Contributions
I.Z., D.M.M., B.P., B.E.H., and M.A.S. performed experiments; I.Z., D.M.M., B.E.H., and R.W.H. analyzed results; I.Z., D.M.M., M.A.S., and R.W.H. wrote the manuscript; D.M.M. and R.W.H. supervised the study.
Acknowledgments
This work was funded by NIH grants R01 AI51390 and R01 HL097088 to RWH. DMM was supported by an ASPIRE award from the Pfizer Hemophilia Program.
R.W.H. received royalty payments for AAV gene transfer technology from Spark Therapeutics.
The first two authors contributed equally to this work.
References
- Furie, B and Furie, BC (1988). The molecular basis of blood coagulation. Cell 53: 505–518. [DOI] [PubMed] [Google Scholar]
- Tiede, A (2015). Half-life extended factor VIII for the treatment of hemophilia A. J Thromb Haemost 13 Suppl 1: S176–S179. [DOI] [PubMed] [Google Scholar]
- Nathwani, AC, Reiss, UM, Tuddenham, EG, Rosales, C, Chowdary, P, McIntosh, J et al. (2014). Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med 371: 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers, GL and Herzog, RW (2015). Gene therapy for hemophilia. Front Biosci (Landmark Ed) 20: 556–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatino, DE, Lange, AM, Altynova, ES, Sarkar, R, Zhou, S, Merricks, EP et al. (2011). Efficacy and safety of long-term prophylaxis in severe hemophilia A dogs following liver gene therapy using AAV vectors. Mol Ther 19: 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, H, Lillicrap, D, Patarroyo-White, S, Liu, T, Qian, X, Scallan, CD et al. (2006). Multiyear therapeutic benefit of AAV serotypes 2, 6, and 8 delivering factor VIII to hemophilia A mice and dogs. Blood 108: 107–115. [DOI] [PubMed] [Google Scholar]
- Markusic, DM and Herzog, RW (2012). Liver-directed adeno-associated viral gene therapy for hemophilia. J Genet Syndr Gene Ther 1: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani, AC, Tuddenham, EG, Rangarajan, S, Rosales, C, McIntosh, J, Linch, DC et al. (2011). Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 365: 2357–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquette, KA, Pittman, DD and Kaufman, RJ (1995). A 110-amino acid region within the A1-domain of coagulation factor VIII inhibits secretion from mammalian cells. J Biol Chem 270: 10297–10303. [DOI] [PubMed] [Google Scholar]
- Tagliavacca, L, Wang, Q and Kaufman, RJ (2000). ATP-dependent dissociation of non-disulfide-linked aggregates of coagulation factor VIII is a rate-limiting step for secretion. Biochemistry 39: 1973–1981. [DOI] [PubMed] [Google Scholar]
- Toole, JJ, Pittman, DD, Orr, EC, Murtha, P, Wasley, LC and Kaufman, RJ (1986). A large region (approximately equal to 95 kDa) of human factor VIII is dispensable for in vitro procoagulant activity. Proc Natl Acad Sci USA 83: 5939–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman, DD, Alderman, EM, Tomkinson, KN, Wang, JH, Giles, AR and Kaufman, RJ (1993). Biochemical, immunological, and in vivo functional characterization of B-domain-deleted factor VIII. Blood 81: 2925–2935. [PubMed] [Google Scholar]
- Sack, BK, Merchant, S, Markusic, DM, Nathwani, AC, Davidoff, AM, Byrne, BJ et al. (2012). Transient B cell depletion or improved transgene expression by codon optimization promote tolerance to factor VIII in gene therapy. PLoS One 7: e37671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh, J, Lenting, PJ, Rosales, C, Lee, D, Rabbanian, S, Raj, D et al. (2013). Therapeutic levels of FVIII following a single peripheral vein administration of rAAV vector encoding a novel human factor VIII variant. Blood 121: 3335–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siner, JI, Iacobelli, NP, Sabatino, DE, Ivanciu, L, Zhou, S, Poncz, M et al. (2013). Minimal modification in the factor VIII B-domain sequence ameliorates the murine hemophilia A phenotype. Blood 121: 4396–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, HC, Gangadharan, B and Doering, CB (2011). Enhanced biosynthesis of coagulation factor VIII through diminished engagement of the unfolded protein response. J Biol Chem 286: 24451–24457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack, BK, Herzog, RW, Terhorst, C and Markusic, DM (2014). Development of gene transfer for induction of antigen-specific tolerance. Mol Ther Methods Clin Dev 1: 14013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn, JD, Ozelo, MC, Sabatino, DE, Franck, HW, Merricks, EP, Crudele, JM et al. (2010). Eradication of neutralizing antibodies to factor VIII in canine hemophilia A after liver gene therapy. Blood 116: 5842–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra, JD, Miao, H, Zhang, K, Wolfson, A, Pennathur, S, Pipe, SW et al. (2008). Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci USA 105: 18525–18530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, HP, Zhang, Y, Zeng, H, Novoa, I, Lu, PD, Calfon, M et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633. [DOI] [PubMed] [Google Scholar]
- Tu, BP and Weissman, JS (2004). Oxidative protein folding in eukaryotes: mechanisms and consequences. J Cell Biol 164: 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder, M and Kaufman, RJ (2005). The mammalian unfolded protein response. Annu Rev Biochem 74: 739–789. [DOI] [PubMed] [Google Scholar]
- Malhotra, JD and Kaufman, RJ (2007). Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal 9: 2277–2293. [DOI] [PubMed] [Google Scholar]
- Ron, D and Walter, P (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8: 519–529. [DOI] [PubMed] [Google Scholar]
- Dorner, AJ, Bole, DG and Kaufman, RJ (1987). The relationship of N-linked glycosylation and heavy chain-binding protein association with the secretion of glycoproteins. J Cell Biol 105(6 Pt 1): 2665–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, M, Nayak, S, Hoffman, BE, Terhorst, C, Cao, O and Herzog, RW (2009). Improved induction of immune tolerance to factor IX by hepatic AAV-8 gene transfer. Hum Gene Ther 20: 767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arruda, VR, Fields, PA, Milner, R, Wainwright, L, De Miguel, MP, Donovan, PJ et al. (2001). Lack of germline transmission of vector sequences following systemic administration of recombinant AAV-2 vector in males. Mol Ther 4: 586–592. [DOI] [PubMed] [Google Scholar]
- Pratt, RM, Yamada, KM, Olden, K, Ohanian, SH and Hascall, VC (1979). Tunicamycin-induced alterations in the synthesis of sulfated proteoglycans and cell surface morphology in the chick embryo fibroblast. Exp Cell Res 118: 245–252. [DOI] [PubMed] [Google Scholar]
- Everett, LA, Cleuren, AC, Khoriaty, RN and Ginsburg, D (2014). Murine coagulation factor VIII is synthesized in endothelial cells. Blood 123: 3697–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahani, T, Covens, K, Lavend’homme, R, Jazouli, N, Sokal, E, Peerlinck, K et al. (2014). Human liver sinusoidal endothelial cells but not hepatocytes contain factor VIII. J Thromb Haemost 12: 36–42. [DOI] [PubMed] [Google Scholar]
- Fahs, SA, Hille, MT, Shi, Q, Weiler, H and Montgomery, RR (2014). A conditional knockout mouse model reveals endothelial cells as the principal and possibly exclusive source of plasma factor VIII. Blood 123: 3706–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanolini, D, Merlin, S, Feola, M, Ranaldo, G, Amoruso, A, Gaidano, G et al. (2015). Extrahepatic sources of factor VIII potentially contribute to the coagulation cascade correcting the bleeding phenotype of mice with hemophilia A. Haematologica 100: 881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipe, SW (2009). Functional roles of the factor VIII B domain. Haemophilia 15: 1187–1196. [DOI] [PubMed] [Google Scholar]
- Mingozzi, F, Liu, YL, Dobrzynski, E, Kaufhold, A, Liu, JH, Wang, Y et al. (2003). Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest 111: 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisowski, L, Dane, AP, Chu, K, Zhang, Y, Cunningham, SC, Wilson, EM et al. (2014). Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature 506: 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S, Ling, C, Zhong, L, Li, M, Su, Q, He, R et al. (2015). Efficient and targeted transduction of nonhuman primate liver with systemically delivered optimized AAV3B vectors. Mol Ther 23: 1867–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L, Bell, P, Somanathan, S, Wang, Q, He, Z, Yu, H et al. (2015). Comparative study of liver gene transfer with AAV vectors based on natural and engineered AAV capsids. Mol Ther 23: 1877–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro, VP and Chappell, SA (2014). A critical analysis of codon optimization in human therapeutics. Trends Mol Med 20: 604–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum, G, Chen, C, Kaur, J, Cui, X, Zhang, H, Asahara, H et al. (2013). Quantifying elongation rhythm during full-length protein synthesis. J Am Chem Soc 135: 11322–11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komar, AA (2009). A pause for thought along the co-translational folding pathway. Trends Biochem Sci 34: 16–24. [DOI] [PubMed] [Google Scholar]
- Suda, T, Gao, X, Stolz, DB and Liu, D (2007). Structural impact of hydrodynamic injection on mouse liver. Gene Ther 14: 129–137. [DOI] [PubMed] [Google Scholar]
- Suda, T and Liu, D (2007). Hydrodynamic gene delivery: its principles and applications. Mol Ther 15: 2063–2069. [DOI] [PubMed] [Google Scholar]
- Staber, JM, Pollpeter, MJ, Arensdorf, A, Sinn, PL, Rutkowski, DT and McCray, PB Jr (2014). piggyBac-mediated phenotypic correction of factor VIII deficiency. Mol Ther Methods Clin Dev 1: 14042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlot, AJ, Koppelman, SJ, van den Berg, MH, Bouma, BN and Sixma, JJ (1995). The affinity and stoichiometry of binding of human factor VIII to von Willebrand factor. Blood 85: 3150–3157. [PubMed] [Google Scholar]
- Terraube, V, O’Donnell, JS and Jenkins, PV (2010). Factor VIII and von Willebrand factor interaction: biological, clinical and therapeutic importance. Haemophilia 16: 3–13. [DOI] [PubMed] [Google Scholar]
- Lytle, AM, Brown, HC, Paik, NY, Knight, KA, Wright, JF, Spencer, HT et al. (2016). Effects of FVIII immunity on hepatocyte and hematopoietic stem cell-directed gene therapy of murine hemophilia A. Mol Ther Methods Clin Dev 3: 15056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering, CB, Denning, G, Dooriss, K, Gangadharan, B, Johnston, JM, Kerstann, KW et al. (2009). Directed engineering of a high-expression chimeric transgene as a strategy for gene therapy of hemophilia A. Mol Ther 17: 1145–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooriss, KL, Denning, G, Gangadharan, B, Javazon, EH, McCarty, DA, Spencer, HT et al. (2009). Comparison of factor VIII transgenes bioengineered for improved expression in gene therapy of hemophilia A. Hum Gene Ther 20: 465–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolotukhin, S, Byrne, BJ, Mason, E, Zolotukhin, I, Potter, M, Chesnut, K et al. (1999). Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther 6: 973–985. [DOI] [PubMed] [Google Scholar]