

Abstract
Alzheimer disease (AD) research has mainly focused on neurodegenerative processes associated with the classic neuropathologic markers of senile plaques and neurofibrillary tangles. Additionally, cerebrovascular contributions to dementia are increasingly recognized, particularly from cerebral small vessel disease (SVD). Remarkably, in AD brains, the ApoE ε4 allele shows male excess for cerebral microbleeds (CMB), a marker of SVD, which is opposite to the female excess of plaques and tangles. Mouse transgenic models add further complexities to sex-ApoE ε4 allele interactions, with female excess of CMBs and brain amyloid. We conclude that brain aging and AD pathogenesis cannot be understood in humans without addressing major gaps in the extent of sex differences in cerebrovascular pathology.
Keywords: Small vessel disease, cerebrovasculature, cerebral amyloid angiopathy, Apolipoprotein E, sex, Cerebral Microbleeds, MRI
SEX AND APOE ALLELES
Our biological sex engenders important trade-offs: men have shorter lifespans than women, yet while women live longer, they incur more risk of Alzheimer disease (AD) throughout life. Worse yet, the ApoE ε4 allele risk factor for AD has a definitive female bias. This greater female vulnerability to AD was recognized in two benchmark post mortem studies[6,7]. Both studies showed a female excess of AD pathology (neuritic plaques and neurofibrillary tangles), which was greatest in ε4 carriers. Correspondingly, levels of cognitive deficits per unit of brain amyloid show a 5-fold excess in women[6].
In contrast to a female bias in the classical AD markers, cerebrovascular pathologies, particularly cerebral microbleeds (CMBs), showed a 3-fold male excess in three independent clinical AD cohorts - the Amsterdam Dementia Cohort [8], the Alzheimer Disease Neuroimaging Initiative (ADNI)[9], and the Karolinska Imaging Dementia Study (KIDS) [9,10].
CMBs are associated, by imaging and postmortem studies, with amyloid-β (Aβ)-containing vessels (CAA), again with ε4 bias [11], as well as hypertensive arteriopathy. Hypertension also shows a male bias (relative to premenopausal women)[12].
Moreover, we recently reported an ApoE ε4-male bias and interaction, for CMBs, in the ADNI and KIDS cohorts (Figure 1C)[9]. This is the first indication that cerebrovascular pathology may have a different sex-ApoE allele bias than the above mentioned female bias in AD mechanisms that are considered ‘neuron-based’ because of the neuronal production of Aβ. Furthermore, there may be species differences in ApoE ε4-sex interactions: By age 7 mo, EFAD transgenic mice carrying human ApoE alleles with familial AD genes (FAD) have a female excess of CMB and of CAA, opposite to the above pattern observed in humans (Figure 1A vs C). Conversely, as in humans, the cerebral cortex load of Aβ in EFAD mice had a ε4 female excess[9].
Figure 1. Cerebral microbleeds.
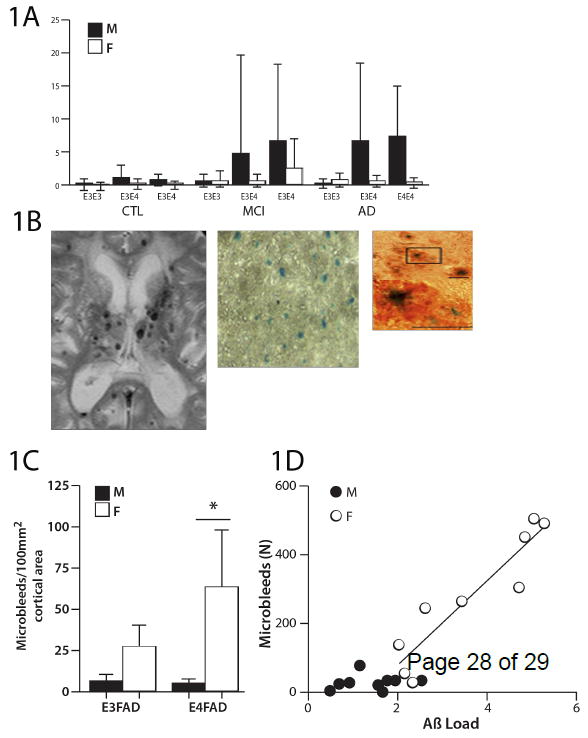
Figures are redrawn from [4] except for B panel 1. A) Human microbleed frequencies analyzed by apoE ε4 alleles in cohorts in two large memory clinics: ADNI (Alzheimer Disease Neuroimaging Initiative) and KIDS (Karolinska Imaging Dementia Study). Error bars denote the standard error of the mean. Male AD patients show a statistically significant excess of CMBs over female patients (P<0.001); the male CMB excess is further exacerbated by ε4(P<0.001), as tested by the Mann-Whitney U-test. AD, Alzheimer disease (ε3/ ε3, n= M42/F33; ε3/ ε4, n=M46/F60; ε4/ ε4, n=M25/F36); CMBs, cerebral microbleeds; F= Female; M= Male; MCI, mild cognitive impairment (ε3/ ε3, n= M144/F133; ε3/ ε4, n=M120/F88; ε4/ ε4, n=M37/F33); CTL, controls (ε3/ ε3, n= M84/F98; ε3/ ε4, n=M43/F66; ε4/ ε4, n=M5/F6).
B) CMB images: panel 1, male AD patient, with multiple CMBs on a T2* weighted MR image; panel 2, female EFAD-mouse at three magnifications showing whole field stained for hemosiderin by Prussian Blue and individual microbleeds Prussian Blue and co-immunostained for Aβ (orange). The majority (67%) of hemosiderin puncta resided within Aβ deposits.
C) EFAD mice: CMB frequencies show female excess with additive effect of ε4.
D) EFAD mice: The number of CMBs regressed against the Aβ load in the cerebral cortex of individual mice showed linear increase above a threshold level of 1.7 Aβ units; this relationship holds for females, but not males. Both apoE alleles were included.
Even without FAD genes, mice carrying human apoE ε3 and ε4 alleles have increased CMBs [4] and CAA [25]. Thus, evolution of the human apoE alleles may have introduced novel pathologies in aging that may contribute to the uniquely human severity of neurodegenerative AD processes [13]. Despite major recent advances in studying neuro-vascular interactions, there is limited data on interactions between sex and ApoE alleles, because most experimental studies of AD and human ApoE alleles have used male rodents. Notably, the reported sex differences in brain Aβ are sensitive in adults to sex steroid levels, and can be reversed by neonatal steroid manipulations[14]. These and other findings are discussed below in regards to basic mechanisms in vascular-neural mechanisms in AD.
In writing this review, we were struck by extensive gaps in the reported descriptions and analysis of sex differences (Table 1). We conclude that brain aging and AD pathogenesis cannot be understood in humans without considering cerebrovascular pathology in terms of sex and ApoE allele interactions.
Table I.
ApoE-sex interactions with brain vasculature.
| Sex | ApoE4 | ApoE x sex | EFAD mice | |
|---|---|---|---|---|
| CMB [8-10] | M+ | M+ | M++ | F++ |
| Stroke [111] | M+* | + | NR | NR |
| WMH [112-114] | F+/ - | - | NR | NR |
| CAA[115] | - | + | NR | F++ |
| CIMT [116] | M+ | M+× | NR | NR |
| Hypertension [117-119] | M+* | + | NR | - |
Compared with premenopausal women.
CAA, cerebral amyloid angiopathy; CMB, cerebral microbleed; CIMT, carotid intima-media thickness; F, female; M, male. M+, male excess; F+, female excess. NR, not reported.
= in patients with diabetes; besides [92], we did not find other reports of ApoE-sex interactions.
VASCULAR FACTORS IN AD
The axiom “A man is only as old as his arteries”, cited by Osler in 1892, reflects the long-posited contribution of cerebrovascular pathology to AD (Box 1). At the molecular level, the blood-brain barrier (BBB) becomes increasingly leaky during transitions from normal aging to mild cognitive impairment (MCI) to clinical AD [15]. These BBB changes appear distinct from cerebral small vessel disease (SVD)(Box 2), which is increasingly recognized as important to the clinical course of AD[16]. It is not well understood how SVD interacts with AD to promote cognitive decline [17]. Besides the various cerebrovascular pathologies, cognitive decline may also be associated with hippocampal sclerosis and Lewy Bodies [18]. White matter damage in association with SVD can alter attentional connectivity networks resulting in cognitive deficits [5] that may overlap with the AD clinical manifestations. The role of SVD in normal cognitive ageing is undefined for healthy elderly.
Box 1. Alzheimer’s first case also showed vascular pathology.
“Arteriosclerosis is an accompaniment of old age, and is the expression of the natural wear and tear to which tubes are subjected. Longevity is a vascular question which has been well expressed in the axiom ‘a man is only as old as his arteries’.”
From William Osler’s ‘The Principles and Practice of Medicine’[81]. This classic textbook briefly considered stroke as contributing to senility. Alzheimer’s 1907 first case report on presenile dementia in a 51 year old woman, Auguste D, with “an unusual illness of the cerebral cortex” (Uber eine eigenartige Erkrankung der Hirnrinde), also recognized atherosclerosis: “The post-mortem showed an evenly atrophic brain without macroscopic focal degeneration. The larger vascular tissues show arteriosclerotic change.” [71]. His 1911 summary of this and other cases considered, but rejected a vascular role, because these cases showed evenly distributed atrophic changes without evidence of stroke[57].
Box 2. Small vessel disease (SVD).
SVD involves microscopic changes in blood vessels below 2 mm in diameter and is increasingly implicated in dementias of aging [82,83]. SVD may develop through two etiologies, cerebral amyloid angiopathy (CAA) which typically affects cortico-subcortical structures, and hypertensive arteriopathy affecting deeper brain structures. CAA is uncommon in the basal ganglia and brain stem. CAA is associated with slowly progressive deposition of Aβ40 in the vascular wall, mainly in the tunica intima smooth muscle, which contrasts with Aβ42 predominance in senile plaques.
SVD has received much attention with the development of imaging techniques. Specifically, MRI is increasingly used in clinical setting to detect imaging markers of SVD, such as CMBs, as well as microinfarcts and cortical superficial siderosis (Figure 2). Related pathology detected by MRI includes white matter hyperintensities, lacunes, and enlarged perivascular spaces (Figure 2) [84]. These MRI markers give approaches to SVD in vivo which falls below current MRI resolution [45]. While imaging markers of SVD may develop for other reasons, SVD remains their most common etiology. All SVD markers show increased frequency in elderly with cognitive impairment [10,85-90]. Moreover, healthy elderly are also vulnerable to SVD [91-93]. Remarkably, despite the myocyte depletion in advanced SVD, the endothelial layer remains intact, with little if any of the activation characteristic of peripheral atherosclerosis [94]. Table I shows ApoE and sex interactions with brain vasculature.
CAA with the accumulation of amyloid fibrils in vessel walls is a major marker of SVD with direct links to lobar CMBs. Typically, CAA occurs at later ages in capillaries, arterioles, and arteries of small to medium size (< 2 mm diameter), particularly in the cerebral cortex and in the leptomeninges [19]. While ε4 is a major risk factor for CAA, surprisingly, ε4 does not seem to be strongly associated with hypertension. In the cerebrospinal fluid (CSF), low levels of Aβ42 are associated with CMBs, with correspondingly greater brain Aβ deposits[20-23]. Accordingly, two or more lobar CMBs, putatively of CAA origin, were associated with declining executive function in the healthy elderly [24]. Similarly, in patients with ischemic stroke and/or transient ischemic attacks, lobar CMBs are associated with executive dysfunction[25]. Lobar CMBs are also associated with lower cerebral blood flow and hypoperfusion in both healthy elderly and in a memory clinic setting [26,27], which highlights the complex interaction of SVD with healthy ageing. Again, ApoE ε4 is associated with lobar CMBs in patients with cognitive impairment[28]. White matter hyperintensities, another imaging marker of SVD common in patients with CMBs, are associated with brain Aβ42 deposits in AD[29]. White matter hyperintensities are also common in healthy ageing, and can be a normal finding even when pronounced, although the underlying etiology is most likely SVD.
In contrast to the presence of CMBs in MCI and AD patients, cognitively healthy age-matched controls from two memory clinics presented very few CMBs[9]. The higher prevalence of CMBs in elderly individuals with MCI and AD suggests a role of these processes in establishing a critical level of parenchymal Aβ accumulations (above levels observed in the healthy elderly). Critical threshold levels of Aβ for CMB and for leptomeningeal hemorrhage in AD transgenic mice are discussed below. These processes appear to differ strikingly from the earlier onset of BBB leakage in the hippocampus of normal middle-aged individuals [15], suggesting an independence of initial BBB leakage from brain Aβ accumulation.
EXPERIMENTAL STUDIES IN MICE
Aging wildtype mice are nature’s gift to the experimental analysis of brain aging because they do not develop other conditions of human aging that vary widely between individuals, particularly hypertension, CAA, or brain amyloid deposits. The absence of these human conditions is best documented in C57BL/6 male mice [30,31]. We evaluated sex by ApoE allele interactions in the EFAD mouse model transgenic for human ε3 or ε4, inserted by targeted replacement or knockin (ApoE-TR)[31], together with five distinct familial AD mutations (not targeted). EFAD mice incur very early Aβ42 deposition [32] and impaired spatial memory [33], with greater changes in ε4 vs ε3 homozygotes. Prior studies of various AD transgenic (ADtg) mice showed clear relationships between CAA and CMBs to brain levels of human Aβ42 [34]. Even without human FAD genes, ApoE-TR mice show CAA excess for the ε4 over ε3 allele [35]. We note the caveat that these mice lack distal downstream human regulatory domains that may alter ApoE expression [36].
Given that ADtg mice consistently show a female excess of Aβ42[14], we anticipated a female excess of CAA and CMBs. In the EFAD mouse model, females had excess of CAA and CMBs without overlapping distribution (Figure 1B)[9], which was opposite to the human male excess of CMBs (Figure 1A). CAA showed modest female excess in the number of Aβ-positive vessels, but without sex differences in the Aβ load per vessel.Nonetheless, ε4 increased CMBs in both mice and men. Moreover, we found a linear correlation of plaque load with both CAA load and the number of CMBs, above a defined threshold level of Aβ42 (Figure 1D). Similarly, Zipfel and colleagues [34,37] observed a threshold level of vascular Aβ42 in leptomeningeal CAA along with impaired vasodilation in a single ADtg model with endogenous murine ApoE; these authors hypothesized that microvascular Aβ42 could drive a pathological cascade from impaired dilatation to microaneurysms that in turn caused white matter lesions. The rarer ApoE ε2 allele could be relevant to these findings because it lowers AD risk, while increasing the risk for cerebral hemorrhage (Box 3).
Box 3. APOE ε2, a bloody angel.
The ApoE ε2 allele lowers AD risk, by nearly 50% per allele and is associated with a milder and later onset disease [95,96]. However, the ε2 allele takes as it gives, because it promotes earlier onset of CAA and risk of hemorrhages [97,98]. Frustratingly, as the least common of the ApoE alleles, typically 1-2%, ε2 remains the most obscure and hard to study. In the ApoE-TR and in EFAD mouse models, the APOE ε2 protein shows complex differences between ApoE ε3 and ε4 in lipid profiles of blood and brain, and in brain amyloid deposits [32,33,99]. For example, in EFAD mice, brain Aβ42 levels (total and oliogomers) were equal in the ε3 and ε2 homozygotes [33], whereas human ε2 carriers had the fewest neuritic plaques in the large NACC autopsy data set [96]. The association of CAA with ε2 was inconclusive.
Given the strong sex-ε4 interactions, we anticipate that ApoEε2 will also show interesting behavioral and biochemical sex differences. However, experimenters are challenged by the logistics of providing sufficient mice per group for adequate statistical assessment of 3 alleles in both sexes. Furthermore, it will also be desirable to include ApoE-knockouts in these experiments to assess the function of the human transgene. Thus, 4 ApoE genotypes × 2 sexes, 10 mice group = 80 mice per study (the need for large breeding colonies of ApoE-TR and 5x-FAD mice to obtain sufficient F1 EFAD mice of each allele and sex, all of the same age, encumbers additional cost). The effects on brain aging in mice without FAD genes (APOE-TR) also merit attention in view of the evidence that hippocampal atrophy during normal aging is slower in human ε2 carriers [100], suggesting slower synapse loss.
In ApoE-TR mice without FAD genes, CMBs also developed spontaneously by 7 mo, but were >90% fewer and smaller sized than in EFAD[9]. Again, the CMBs showed a female bias, but with minimal interaction with ApoE alleles. Notably, all ApoE-TR and EFAD mice had extensive CMBs, contrasting to their more limited presence in a minority of the elderly human ADNI subjects (Figure 1). In the only study of aging ApoE-TR mice, gross cerebral hemorrhages developed by 18-24 mo in a minority of both ε3 and ε4 mice, together with CAA[31]. This is remarkable, because wildtype C57BL/6 mice never develop CAA. Evidently, the human ApoE protein enhanced pro-amyloidogenic processing of mouse APP to produce fibrillizable Aβ42, despite its different sequence from the human (see below).
A specific role of the human ApoE ε3 and ε4 alleles in CAA was also shown in mice carrying the amyloid precursor protein Swedish mutation (APPSWE) on an ApoE-knockout background, which did not develop CAA or CMBs [38]. Thus, the presence of either human ApoE ε3 or ε4 causes a major increase in cerebrovascular pathology that is absent from the aging normotensive wildtype rodent. Human ApoE ε3 and ε4 expression also caused notable shifts in the location of the amyloid deposits [35]: In APPSWE mice with the endogenous murine ApoE, cerebral Aβ was accumulated as parenchymal plaques and as CAA in 100% of mice by 12 mo. In contrast, introduction of either human ApoE ε3 or ε4 caused a 20-fold reduction of parenchymal Aβ, while also decreasing and delaying the CAA. Moreover, soluble brain tissue extracts showed an increased ratio of Aβ40:42, with the inverse shift to lower Aβ40:42 in the cerebrospinal fluid; both effects were strongest for ApoE ε4. Because human ε2 carriers have greater risk of hemorrhage in CAA (Box 3), we anticipate important findings on the ε2 allele in mouse models.
Another mouse model of human apoE expression is driven by the GFAP promotor without replacement of its endogenous ApoE. These ApoE ε4 mice, but not the ε3, developed early onset BBB leakage and CMBs (hemosiderin puncta) by age 2 weeks, with ensuing cortical neuron dysfunctions by 4 months[39]. Thus, human ApoE ε4 in a mouse host that retains its endogenous ApoE can block the endogenous ApoE function to levels equivalent to the null mutant. The mechanisms involve NFkB-dependent matrix metalloproteinase 9 expression in microvascular pericytes, which is critical for BBB maintenance.
Although not widely discussed by AD researchers, the APP protein is also expressed in the peripheral vasculature, e.g. in aortic endothelia of wildtype mice [40]. ADtg2576 mice showed impaired endothelial-dependent vasodilation in response to acetylcholine in the aorta and carotid arteries, relative to C57BL/6 controls [40,41]. The impaired vasodilation was rescued by short term oral ingestion of Bosentan, an ETA/B receptor antagonist [41], and by the NADPH oxidase inhibitor VAS2870 and the PPARy ligand GW501516 [40]. Besides its presence in arterial endothelia, platelets have abundant APP, as well as the processing enzymes for Aβ which contribute the bulk of circulating plasma Aβ40 [42]. A role for APP and Aβ in peripheral atherosclerosis may be considered, with possible shared mechanisms of oxidative stress, as has been posited for CMBs in CAA.
HYPERTENSIVE ARTERIOPATHY
Humans
Hypertensive arteriopathy, often concurrent with CAA, increases the risk of intracerebral hemorrhage[43]. Hypertensive arteriopathy is attributed to impaired auto-regulation, with consequent dilatation of arterioles and capillaries (‘resistance vessels’) [44]. CMBs in deep brain regions show associations with hypertension [10,44,45], but no association with CSF amyloid markers,[28] or brain amyloid in PET-imaging[46]. As expected from risk factors for hypertension, male sex and advanced age are common in patients with deep CMBs[10]. Hypertension may increase the risk of ICH, despite the lack of strong association with CAA [19]; most patients with hypertension in fact are very well medicated. However, the interaction of hypertension and the ε4 allele has shown to increase amyloid burden in the brain, in healthy middle-aged individuals, and even more so in individuals with unmedicated hypertension [47].
Mice
Associations of CMBs with hypertension in elderly humans were modeled in wildtype C57BL/6 male mice with drug-induced hypertension, with an age comparison of 24 months (equivalent to human age 60) vs 4 months (equivalent to human age 20) [30]. As expected, aging did not alter blood pressure in controls. In contrast, hypertensive older mice had 2-fold more CMBs than the young. Moreover, the first CMBs appeared 4 days earlier in the older mice, with correspondingly greater arterial redox stress (3-nitrotyrosine and Nox4). Gait dysfunction was also utilized as a novel neurological measure of CMBs; specifically, irregularities in paw placement preceded the onset of CMBs. This motor dysfunction is reminiscent of the increased risk of falls in AD and in mild cognitive impairment (MCI) [48,49]. We anticipate future studies of sex interactions in cerebrovascular responses to hypertension using ApoE-TR mice, which show major sex-ApoE interactions in inflammatory responses[50].
REACTIVE OXYGEN SPECIES AND SMALL VESSEL DISEASE
Experimental studies have shown a role of reactive oxygen species (ROS) in CMBs. In ADtg mice (Tg2576), orally ingested apocyanin (NADPH oxidase inhibitor) and tempol (non-specific ROS scavenger, also known as mitoTEMPO) diminished CMBs by 30-50%, together with lowering Nox isoforms and other makers of redox stress in cerebral arteries[51]. Notably, apocyanin also decreased cerebral cortex ApoE protein levels by 50%, consistent with findings that the ADtg in ApoE-knockout background had >50% fewer CMBs[38]. Similarly, in the induced hypertension model of wildtype mice discussed above, apocyanin and tempol diminished both CMBs and redox stress[30]. Further in vitro analysis, with segments of middle cerebral artery subjected to acute hypertension, showed a marked age-dependent increase in ROS and redox-sensitive MMP; again, apocyanin and mitoTEMP attenuated the exacerbating effect of age.
Of potential interest to therapeutics development is the response to resveratrol, [30] a putative anti-aging drug which attenuates ROS in many models and systems[52]. Ingestion of resveratrol for ten days before inducing hypertension sharply attenuated the elevation of CMBs, oxidative stress, and gait disturbances, without altering the degree of hypertension[30]. Thus, resveratrol may have therapeutic benefit to drug-resistant hypertension and its associated CMBs. However, resveratrol also lowered hippocampal ApoE by >50% in C57BL/6 mice [53]. This might be counter-productive, due to ApoE’s importance as a lipid carrier in neuronal remodeling and repair[3].
For humans, data are limited on the relationships of ROS to CMBs and SVD. Certain gene variants in endothelial nitric acid synthase (eNOS) were associated with lacunar infarcts[54,55]; however, these studies, did not directly assess endothelial functions regulated by NO. Systemic associations of CMBs were shown in the Framingham Heart Study, where a stroke-free subgroup with CMBs had elevated plasma myeloperoxidase (MPO), an oxidative enzyme associate with inflammation[56].
SMALL VESSEL DISEASE and AD pathogenesis
Could CMBs be directly involved in AD pathogenesis? A possible vascular role in the neuropathology of this index case of pre-senile dementia was considered, and rejected, by Alzheimer himself (Box 3). In a valuable review, Stone traced the subsequent history of hypotheses linking plaque induction to local microhemorrhages[57]. Similarly, the vasocentric nature of plaques was described in detail by Kumar-Singh and colleagues [58]. In two ADtg models (tg2576 and PSAPP), all plaques were associated with vessels and that CMBs occurred near to vessels with CAA and ‘around dense plaques’. These findings extended the prior analysis of vasocentric plaques observed in the Flemish variant of heritable AD with early onset cerebral hemorrhage[59]. In our studies of EFAD mice, most Aβ deposits included a CMB (Fig. 3). Moreover, in AD brains, haeme and hemoglobin co-localize with fibrillary Aβ42 [60,61]. A histological study of the cerebral cortex in 20 patients with AD and Down’s syndrome with AD also co-localized CMBs with amyloid plaques around arterioles, capillaries and venules [60]. Lastly, we note that in vivo imaging localized amyloid density near lobar CMBs [46,62].
A direct role for microvascular leakage in plaque formation was suggested by the rapid induction of Aβ42 deposits in FAD mice by a needle stab into the hippocampus that caused a fine trace of blood; injections of hemoglobin stimulated larger amounts of Aβ around the needle track [63]. In vitro, low levels of iron (<1 μmole) promote the aggregation of Aβ peptides, a catalytic effect blocked by chelation [64]. Thus, much evidence now supports the microvascular leakage of hemoglobin in Aβ plaque genesis. The ApoE ε4 protein could contribute to pre-clinical Aβ deposition by its greater membrane interactions (Box 4), which putatively could exacerbate blood-brain barrier leakiness in normal human aging. SVD shows a progression in relation to tau pathology in which early Braak stages are associated with artery wall remodeling with decreased smooth muscle actin in small-medium size leptomeningeal vessels, while leptomeningeal arteriole remodeling at late Braak stages was associated with presence of CAA[65]. The mechanisms behind CAA-independent remodeling are unknown.
Box 4. Evolution of human APOE alleles.
| 61 | 112 | 158 | global average frequency[101] | |
| human ε4 | R | R | R | 0.14 + 0.036 |
| ε3 | R | C | R | 0.78 + 0.042 |
| ε2 | R | C | C | 0.08 + 0.026 |
| mouse | T | R | R | 1.0 |
| chimpanzee | T | R | R | 1.0 |
C, cysteine; R, arginine; T, threonine
Humans are the only species shown to have ApoE allele variants. The ApoE ε4 allele was present in earlier Homo at least 600,000 years ago, but differs from the chimpanzee, as discussed below[3,101]. The ε3 allele emerged concurrently with anatomically modern H. sapiens about 250,000 years ago, while ε2 descended from ε3, about 100,000 years ago[3]. Allele frequencies in a global sample showed ε3 is the major allele in all populations, generally followed by ε4 and ε2. Populations differ widely in allele frequencies along regional gradients, e.g. ε4 ranges >5-fold between northern and southern Europe, while ε2 may be absent from some northern Asian-derived groups[3,102]. These gradients do not obviously correspond to differences in AD or cerebrovascular diseases. Besides the well-known 3 alleles, there are 30 coding variants at other sites. Four sites in ApoE lipid binding domains show positive selection[103,104].
The human ε4 protein has a unique structure among apoE isoforms, the molten globule, a partly unfolded 4-helix bundle with increased β-structure, and different lipid binding affinity [105-106]. The molten globule structure has not been described in the ApoE of other species. Also relevant to AD is the greater lipophilic disruption by ε4 in vitro [105], also manifested as greater Aβ-induced lysosomal leakage in vitro[1]. Structural analysis of the mouse and chimpanzee apoE protein suggests that their lipid binding properties and peptide chain organization is closest to human ε3, despite the R112 and R158 shared with human ε4[104,106]. The critical residue for these activities is the R61 of humans, which if introduced to mice, renders their apoE more ε4-like in lipid binding[106].
The persistence of ε4 in modern populations, despite its late life costs to brain aging, may represent adaptive benefits to younger ages in some environments [107]. For example ε4 carrying children in a Brazilian slum had fewer episodes of diarrhea and better cognitive development[108]. Similarly, in ApoE transgenic mice, ε4 increased resistance to intestinal cryptosporidial infections[109]. The role of infections in AD pathogenesis is increasingly discussed[107].
THE BLOOD BRAIN BARRIER, SMALL VESSEL DISEASE, and AGING
SVD patients show increased permeability of the BBB [66]. Independent imaging markers of small vessel disease and associations with increased BBB permeability include: CMBs [22], white matter hyperintensities and lacunes [66,67]. In AD, accelerated degeneration of pericytes is higher in ApoE ε4 vs ε3 carriers and is associated with greater BBB disruption[68].
Cerebrovascular aging without classical atherosclerotic pathology may also contribute to increased BBB permeability. As elegantly shown for healthy humans with a new gadolinium MRI marker, BBB leakage increased progressively after age 30 in a linear fashion beyond age 70 [15]. Aging mice showed the same trends: C57BL/6 males showed a 75% increase of IgG leakage in the hippocampus in 7 vs 24 mo old mice[69], while ApoE-TR mice showed thinning of brain capillary basement membranes by 12 mo that was greater in ε4 vs ε3 carriers [70]. Sex differences were not considered in these studies.
SPECIES DIFFERENCES
Mice and humans have functionally important differences in coding sequences of Aβ42 and ApoE peptides. Departing from the large-sweep identity of the Aβ peptide throughout vertebrate phyla, the mouse Aβ42 differs from the human at three sites (R5G, H13R, Y19P) that attenuate its aggregation and cytoxicity [71,72]. Nonetheless, as noted above, in the presence of human ApoE ε3 or ε4, the endogenous murine Aβ formed fibrils and CAA during aging, which are unknown in wildtype mice [31]. Among the numerous coding differences between mouse and human ApoE (72% homology), some may influence APP processing, which involves isoform-specific binding [73]. One likely site is T61R (Box 4). We do not know which of the many sequence differences between mice and men sites mediate to the ApoE effects underlying the uniquely human sex- apoE interactions.
SEX DIFFERENCES AND SMALL VESSEL DISEASE
Could developmental variations in sex steroid levels alter the penetrance of ApoE ε4 in AD risk? In FADtg mice, blood levels of sex steroids influence neurodegenerative changes [14,74]. Besides such ‘activating’ effects of sex steroids, sex steroids have profound ‘organizational’ effects on the developing brain[75]. In humans, exposure to excess androgens during development in congenital adrenal hyperplasia (CAH) modifies sex-linked behaviors during both childhood and adulthood [76]. Moreover, masculinization of female ADtg mice by neonatal testosterone decreased adult brain Aβ; conversely, demasculinization of ADTG males with neonatal flutamide increased adult Aβ[14,74].
Could cerebral arteries be subject to such organizational effects of sex steroids? One suggestive example is aneurysms, which are male biased in humans and mice, but can be induced in female mice by neonatal androgenization [77]. Given that the sex differences in mouse brain Aβ42 and in aortic aneurysms can be switched by neonatal sex steroid treatment, we hypothesize that cerebrovascular amyloid is also subject to developmental influences of sex steroids. Even neonatal mouse neurons and cardiomyocytes in primary culture show greater male vulnerability to ischemia [78].
The cerebral arteries have different embryonic cell origins than neurons and astrocytes: In day 7 embryo mice, the first cerebral vessels originate from migrating blood islands of a different anlagen than the neural crest [79]. Nonetheless, their development shares some of the same homeobox transcription factors. Sex differences in cerebral vascular development are unexplored. These sex differences may be understood as resulting from coevolutionary responses of autosomal genes to intralocus sexual conflict, such as detected for systolic blood pressure and cholesterol in the Framingham Heart Study [80].
CONCLUDING REMARKS AND FUTURE PERSPECTIVES
Biological differences in the cerebrovascular and neuronal aspects of aging promise to increase our understanding of these complex interactions during normal aging and in the uniquely human extreme degeneration that arises during AD. The role of small vessel disease in cognitive age changes is rapidly gaining in importance. Brain aging and AD pathogenesis cannot be understood in humans without considering cerebrovascular pathology in terms of sex and ApoE allele interactions. We also hope to stimulate further inquiry relevant to precision drug therapy by ApoE genotype and sex.
Figure 2.
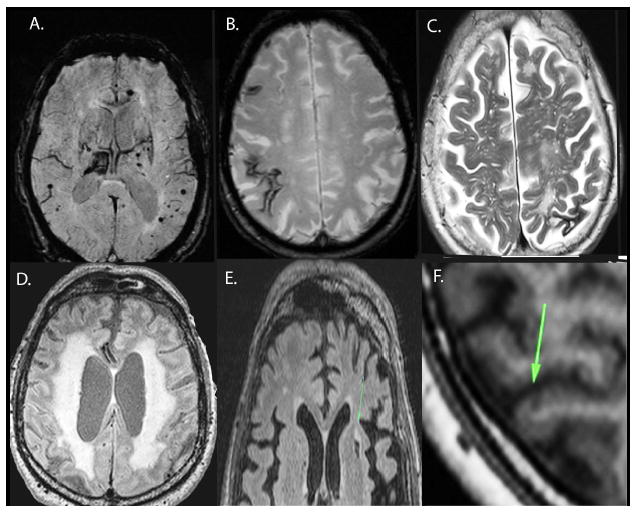
MR imaging markers of small vessel disease: A) Cerebral microbleeds (black arrows) are seen as dark round hypointensities on the gradient echo T2* MR image. Note the larger bleed in the right thalamus. The size of microbleeds on the image slightly exaggerates their size in vivo.
B) Cortical superficial siderosis (black arrows) are seen on MR as characteristically dark, linear, and gyriform, and are considered a sensitive marker of CAA [110].
C) Enlarged perivascular spaces in the centrum semiovale on the T2-weighted MRI sequence. Perivascular spaces in the centrum semiovale and basal ganglia are thought to represent CAA, and hypertensive arteriopathy respectively. D) White matter hyperintensities are seen best on FLAIR MRI. White matter hyperintensities are thought to be multifactorial, and a marker of SVD. E) Lacune (arrow). Seen as CSF-signal on T1 and T2, sometimes with a surrounding hyperintense rim. F) Cortical microinfarct (arrow), seen as <5 mm, hypointensities, perpendicular to the cortex. Images are from the KIDS cohort[10].
Trend box.
-
▪
Older men have a higher risk of cerebral microbleeds (CMBs), augmented by the ApoE4 allele. In contrast, EFAD-mice show the opposite sex effect, with a 3-10-fold female excess of CMBs which dwarfed possible effects of ApoE alleles.
-
▪
Cerebral small vessel disease is increasingly implicated in cognitive impairment of aging, particularly in conjunction with AD.
-
▪
Reactive oxygen species (ROS) are mechanistically associated with CMBs in several mouse models. In humans, the limited data also support mechanistic links of ROS to CMBs.
-
▪
Most amyloid plaques in EFAD-mice include a cerebral microbleed; similar vasocentric plaques are seen in humans with the Flemish AD variant.
Outstanding questions box.
-
▪
Do cerebral microbleeds trigger amyloid plaques in the brain?
-
▪
Is the combination of hypertensive arteriopathy and cerebral amyloid angiopathy the reason males have more microbleeds than females?
-
▪
Do sexes differ in the causes and mechanisms of cerebral small vessel disease?
-
▪
How does introduction of the human ApoE transgene and protein to mice cause cerebral amyloid angiopathy, a condition that is absent from wildtype rodents?
Acknowledgments
CE Finch is grateful for grant support from the Cure Alzheimer’s Fund and the National Institute on Aging: R01 AG051521; R21-AG040683; P01-AG026572 (RD Brinton PI), Project 2 (CE Finch and TE Morgan). SS is grateful for grant support from the Swedish Dementia Association, the Swedish Stroke Foundation and Stohnes research foundation. Because the reference format limits the number of authors shown, we wish to list all of our colleagues who contributed to these extensive studies. Mafalda Cacciottolo (lead author) with Amy Christensen, Alexandra Moser, Jiahui Liu, Christian J Pike, Todd E Morgan, Egor Dolzhenko, Andreas Charidimou, Lars-Olaf Wahlund, Maria Kristofferson Wiberg, Gloria Chia-Yi Chiang, and the Alzheimer’s Disease Neuroimaging Initiative (ADNI).
Glossary box
- Apolipoprotein E (ApoE)
ApoE was first recognized in blood cholesterol transport where it is secreted by the liver[1]. Of the three apoE alleles, ε4 was associated with elevated blood cholesterol levels [2], with a widely varying frequency between human populations (Box 4). In the brain, ApoE is secreted by astrocytes as a transporter of lipids to neurons [3]. Because the major lipoproteins ApoA1 and ApoB are at minimal levels in brain, ApoE in conjunction with a ApoJ, has prime role in synaptic membrane remodeling[4].
- Cerebral amyloid angiopathy or congophilic amyloid angiopathy (CAA)
Small cerebral vessels with amyloid deposits that stain with Congo red dye. The amyloid causes vascular fragility and often results in microbleeds in the cerebral lobes.
- Cerebral small vessel disease (SVD)
Pathology of brain microscopic vessels smaller than 2 mm diameter, including arterioles, capillaries and venules. Because these vessels are below the resolution of current brain imaging, surrogate markers of the disease (Box 1) are used. The two most common etiologies of small vessel disease are hypertensive arteriopathy and cerebral amyloid angiopathy. SVD prevalence increases strongly at later ages and may be 5-10-fold more prevalent than large vessel stroke[5]. At advanced ages, multiple cerebrovascularpathologies are increasingly common.
- EFAD-mice
Transgenic mice with familial Alzheimer disease (FAD) mutations crossed with mice carrying human ApoE alleles by targeted gene replacement (gene knockin).
- Hypertensive arteriopathy
Hypertensive damage to vessels causing arteriosclerotic changes and vascular fragility and microbleeds, particularly in the basal ganglia.
Footnotes
Disclosures: None for all authors
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mahley RW, et al. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J Lipid Res. 2009;50(Suppl):S183–188. doi: 10.1194/jlr.R800069-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sing CF, Davignon J. Role of the apolipoprotein E polymorphism in determining normal plasma lipid and lipoprotein variation. Am J Hum Genet. 1985;37:268–285. [PMC free article] [PubMed] [Google Scholar]
- 3.Fullerton SM, et al. Apolipoprotein E Variation at the Sequence Haplotype Level: Implications for the Origin and Maintenance of a Major Human Polymorphism. Am J Hum Genet. 2000;67:881–900. doi: 10.1086/303070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leduc V, et al. APOE and cholesterol homeostasis in Alzheimer’s disease. Trends Mol Med. 2010;16:469–477. doi: 10.1016/j.molmed.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 5.Dey AK, et al. Pathoconnectomics of cognitive impairment in small vessel disease: A systematic review. Alzheimers Dement J Alzheimers Assoc. 2016 doi: 10.1016/j.jalz.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Barnes LL, et al. Sex differences in the clinical manifestations of Alzheimer disease pathology. Arch Gen Psychiatry. 2005;62:685–691. doi: 10.1001/archpsyc.62.6.685. [DOI] [PubMed] [Google Scholar]
- 7.Corder EH, et al. The biphasic relationship between regional brain senile plaque and neurofibrillary tangle distributions: modification by age, sex, and APOE polymorphism. Ann N Y Acad Sci. 2004;1019:24–28. doi: 10.1196/annals.1297.005. [DOI] [PubMed] [Google Scholar]
- 8.Benedictus MR, et al. Microbleeds, Mortality, and Stroke in Alzheimer Disease: The MISTRAL Study. JAMA Neurol. 2015 doi: 10.1001/jamaneurol.2015.14. [DOI] [PubMed] [Google Scholar]
- 9.Cacciottolo M, et al. The APOE4 allele shows opposite sex bias in microbleeds and Alzheimer’s disease of humans and mice. Neurobiol Aging. 2016;37:47–57. doi: 10.1016/j.neurobiolaging.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shams S, et al. Cerebral microbleeds: different prevalence, topography, and risk factors depending on dementia diagnosis—the Karolinska Imaging Dementia Study. AJNR Am J Neuroradiol. 2015;36:661–666. doi: 10.3174/ajnr.A4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rannikmäe K, et al. Genetics of cerebral amyloid angiopathy: systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2013;84:901–908. doi: 10.1136/jnnp-2012-303898. [DOI] [PubMed] [Google Scholar]
- 12.Reckelhoff JF. Gender Differences in the Regulation of Blood Pressure. Hypertension. 2001;37:1199–1208. doi: 10.1161/01.hyp.37.5.1199. [DOI] [PubMed] [Google Scholar]
- 13.Finch CE, Austad SN. Commentary: is Alzheimer’s disease uniquely human? Neurobiol Aging. 2015;36:553–555. doi: 10.1016/j.neurobiolaging.2014.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vest RS, Pike CJ. Gender, sex steroid hormones, and Alzheimer’s disease. Horm Behav. 2013;63:301–307. doi: 10.1016/j.yhbeh.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montagne A, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296–302. doi: 10.1016/j.neuron.2014.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cordonnier C, van der Flier WM. Brain microbleeds and Alzheimer’s disease: innocent observation or key player? Brain. 2011 doi: 10.1093/brain/awq321. [DOI] [PubMed] [Google Scholar]
- 17.Marnane M, Hsiung GYR. Could Better Phenotyping Small Vessel Disease Provide New Insights into Alzheimer Disease and Improve Clinical Trial Outcomes? Curr Alzheimer Res. 2016;13:750–763. doi: 10.2174/1567205013666160222112634. [DOI] [PubMed] [Google Scholar]
- 18.Neltner JH, et al. Brain pathologies in extreme old age. Neurobiol Aging. 2016;37:1–11. doi: 10.1016/j.neurobiolaging.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Charidimou A, et al. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry. 2012;83:124–137. doi: 10.1136/jnnp-2011-301308. [DOI] [PubMed] [Google Scholar]
- 20.Goos JDC, et al. Microbleeds relate to altered amyloid-beta metabolism in Alzheimer’s disease. Neurobiol Aging. 2012;33:1011.e1–9. doi: 10.1016/j.neurobiolaging.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 21.Goos JDC, et al. Patients with Alzheimer disease with multiple microbleeds: relation with cerebrospinal fluid biomarkers and cognition. Stroke J Cereb Circ. 2009;40:3455–3460. doi: 10.1161/STROKEAHA.109.558197. [DOI] [PubMed] [Google Scholar]
- 22.Shams S, et al. Cerebrospinal fluid profiles with increasing number of cerebral microbleeds in a continuum of cognitive impairment. J Cereb Blood Flow Metab. 2015 doi: 10.1177/0271678X15606141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kester MI, et al. Associations between cerebral small-vessel disease and Alzheimer disease pathology as measured by cerebrospinal fluid biomarkers. JAMA Neurol. 2014;71:855–862. doi: 10.1001/jamaneurol.2014.754. [DOI] [PubMed] [Google Scholar]
- 24.Meier IB, et al. Lobar microbleeds are associated with a decline in executive functioning in older adults. Cerebrovasc Dis Basel Switz. 2014;38:377–383. doi: 10.1159/000368998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gregoire SM, et al. Strictly lobar microbleeds are associated with executive impairment in patients with ischemic stroke or transient ischemic attack. Stroke J Cereb Circ. 2013;44:1267–1272. doi: 10.1161/STROKEAHA.111.000245. [DOI] [PubMed] [Google Scholar]
- 26.Gregg NM, et al. Incidental Cerebral Microbleeds and Cerebral Blood Flow in Elderly Individuals. JAMA Neurol. 2015;72:1021–1028. doi: 10.1001/jamaneurol.2015.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doi H, et al. Analysis of Cerebral Lobar Microbleeds and a Decreased Cerebral Blood Flow in a Memory Clinic Setting. Intern Med. 2015;54:1027–1033. doi: 10.2169/internalmedicine.54.3747. [DOI] [PubMed] [Google Scholar]
- 28.Chiang GC, et al. Cerebral Microbleeds, CSF p-Tau, and Cognitive Decline: Significance of Anatomic Distribution. AJNR Am J Neuroradiol. 2015 doi: 10.3174/ajnr.A4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Westen D, et al. Cerebral white matter lesions - associations with Aβ isoforms and amyloid PET. Sci Rep. 2016;6:20709. doi: 10.1038/srep20709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toth P, et al. Aging exacerbates hypertension-induced cerebral microhemorrhages in mice: role of resveratrol treatment in vasoprotection. Aging Cell. 2015;14:400–408. doi: 10.1111/acel.12315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sullivan PM, et al. Human apolipoprotein E4 targeted replacement mice show increased prevalence of intracerebral hemorrhage associated with vascular amyloid deposition. J Stroke Cerebrovasc Dis Off J Natl Stroke Assoc. 2008;17:303–311. doi: 10.1016/j.jstrokecerebrovasdis.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 32.Youmans KL, et al. APOE4-specific changes in Aβ accumulation in a new transgenic mouse model of Alzheimer disease. J Biol Chem. 2012;287:41774–41786. doi: 10.1074/jbc.M112.407957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu D, et al. APOE4 enhances age-dependent decline in cognitive function by down-regulating an NMDA receptor pathway in EFAD-Tg mice. Mol Neurodegener. 2015;10:7. doi: 10.1186/s13024-015-0002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han BH, et al. Cerebrovascular Dysfunction in Amyloid Precursor Protein Transgenic Mice: Contribution of Soluble and Insoluble Amyloid-β Peptide, Partial Restoration via γ-Secretase Inhibition. J Neurosci. 2008;28:13542–13550. doi: 10.1523/JNEUROSCI.4686-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fryer JD, et al. Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci Off J Soc Neurosci. 2005;25:2803–2810. doi: 10.1523/JNEUROSCI.5170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grehan S, et al. Two distal downstream enhancers direct expression of the human apolipoprotein E gene to astrocytes in the brain. J Neurosci Off J Soc Neurosci. 2001;21:812–822. doi: 10.1523/JNEUROSCI.21-03-00812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zipfel GJ, et al. Cerebral amyloid angiopathy: progressive disruption of the neurovascular unit. Stroke J Cereb Circ. 2009;40:S16–19. doi: 10.1161/STROKEAHA.108.533174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fryer JD, et al. Apolipoprotein E markedly facilitates age-dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. J Neurosci Off J Soc Neurosci. 2003;23:7889–7896. doi: 10.1523/JNEUROSCI.23-21-07889.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bell RD, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.d’Uscio LV, et al. Activation of PPARδ prevents endothelial dysfunction induced by overexpression of amyloid-β precursor protein. Cardiovasc Res. 2012;96:504–512. doi: 10.1093/cvr/cvs266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elesber AA, et al. Bosentan preserves endothelial function in mice overexpressing APP. Neurobiol Aging. 2006;27:446–450. doi: 10.1016/j.neurobiolaging.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 42.EHRLICH D, et al. Effects of oxidative stress on amyloid precursor protein processing in rat and human platelets. Platelets. 2013;24:26–36. doi: 10.3109/09537104.2012.661104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Charidimou A, et al. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry. 2012;83:124–137. doi: 10.1136/jnnp-2011-301308. [DOI] [PubMed] [Google Scholar]
- 44.Werring DJ, editor. Cerebral Microbleeds: Pathophysiology to Clinical Practice. 1. Cambridge University Press; 2011. [Google Scholar]
- 45.Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9:689–701. doi: 10.1016/S1474-4422(10)70104-6. [DOI] [PubMed] [Google Scholar]
- 46.Park JH, et al. Pathogenesis of cerebral microbleeds: In vivo imaging of amyloid and subcortical ischemic small vessel disease in 226 individuals with cognitive impairment. Ann Neurol. 2013;73:584–593. doi: 10.1002/ana.23845. [DOI] [PubMed] [Google Scholar]
- 47.Rodrigue KM, et al. Risk factors for β-amyloid deposition in healthy aging: Vascular and genetic effects. JAMA Neurol. 2013;70:600–606. doi: 10.1001/jamaneurol.2013.1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mignardot JB, et al. Postural sway, falls, and cognitive status: a cross-sectional study among older adults. J Alzheimers Dis JAD. 2014;41:431–439. doi: 10.3233/JAD-132657. [DOI] [PubMed] [Google Scholar]
- 49.Delbaere K, et al. Mild cognitive impairment as a predictor of falls in community-dwelling older people. Am J Geriatr Psychiatry Off J Am Assoc Geriatr Psychiatry. 2012;20:845–853. doi: 10.1097/JGP.0b013e31824afbc4. [DOI] [PubMed] [Google Scholar]
- 50.Colton CA, et al. Sex steroids, APOE genotype and the innate immune system. Neurobiol Aging. 2005;26:363–372. doi: 10.1016/j.neurobiolaging.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 51.Han BH, et al. Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc Natl Acad Sci U S A. 2015;112:E881–890. doi: 10.1073/pnas.1414930112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Novelle MG, et al. Resveratrol supplementation: Where are we now and where should we go? Ageing Res Rev. 2015;21:1–15. doi: 10.1016/j.arr.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thomas J, et al. Dietary supplementation with resveratrol and/or docosahexaenoic acid alters hippocampal gene expression in adult C57Bl/6 mice. J Nutr Biochem. 2013;24:1735–1740. doi: 10.1016/j.jnutbio.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 54.Hassan A, et al. Endothelial nitric oxide gene haplotypes and risk of cerebral small-vessel disease. Stroke J Cereb Circ. 2004;35:654–659. doi: 10.1161/01.STR.0000117238.75736.53. [DOI] [PubMed] [Google Scholar]
- 55.Yemisci M, et al. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med. 2009;15:1031–1037. doi: 10.1038/nm.2022. [DOI] [PubMed] [Google Scholar]
- 56.Shoamanesh A, et al. Inflammatory biomarkers, cerebral microbleeds, and small vessel disease: Framingham Heart Study. Neurology. 2015;84:825–832. doi: 10.1212/WNL.0000000000001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stone J. What initiates the formation of senile plaques? The origin of Alzheimer-like dementias in capillary haemorrhages. Med Hypotheses. 2008;71:347–359. doi: 10.1016/j.mehy.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 58.Kumar-Singh S, et al. Dense-core plaques in Tg2576 and PSAPP mouse models of Alzheimer’s disease are centered on vessel walls. Am J Pathol. 2005;167:527–543. doi: 10.1016/S0002-9440(10)62995-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kumar-Singh S, et al. Dense-core senile plaques in the Flemish variant of Alzheimer’s disease are vasocentric. Am J Pathol. 2002;161:507–520. doi: 10.1016/S0002-9440(10)64207-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cullen KM, et al. Microvascular pathology in the aging human brain: evidence that senile plaques are sites of microhaemorrhages. Neurobiol Aging. 2006;27:1786–1796. doi: 10.1016/j.neurobiolaging.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 61.Wu CW, et al. Hemoglobin promotes Abeta oligomer formation and localizes in neurons and amyloid deposits. Neurobiol Dis. 2004;17:367–377. doi: 10.1016/j.nbd.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 62.Dierksen GA, et al. Spatial relation between microbleeds and amyloid deposits in amyloid angiopathy. Ann Neurol. 2010;68:545–548. doi: 10.1002/ana.22099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chuang J-Y, et al. Interactions between amyloid-β and hemoglobin: implications for amyloid plaque formation in Alzheimer’s disease. PloS One. 2012;7:e33120. doi: 10.1371/journal.pone.0033120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang X, et al. Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer’s Abeta peptides. J Biol Inorg Chem JBIC Publ Soc Biol Inorg Chem. 2004;9:954–960. doi: 10.1007/s00775-004-0602-8. [DOI] [PubMed] [Google Scholar]
- 65.Merlini M, et al. Tau pathology-dependent remodelling of cerebral arteries precedes Alzheimer’s disease-related microvascular cerebral amyloid angiopathy. Acta Neuropathol (Berl) 2016;131:737–752. doi: 10.1007/s00401-016-1560-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Topakian R, et al. Blood-brain barrier permeability is increased in normal-appearing white matter in patients with lacunar stroke and leucoaraiosis. J Neurol Neurosurg Psychiatry. 2010;81:192–197. doi: 10.1136/jnnp.2009.172072. [DOI] [PubMed] [Google Scholar]
- 67.Taheri S, et al. Blood-brain barrier permeability abnormalities in vascular cognitive impairment. Stroke J Cereb Circ. 2011;42:2158–2163. doi: 10.1161/STROKEAHA.110.611731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Halliday MR, et al. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2015 doi: 10.1038/jcbfm.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tucsek Z, et al. Aging exacerbates obesity-induced cerebromicrovascular rarefaction, neurovascular uncoupling, and cognitive decline in mice. J Gerontol A Biol Sci Med Sci. 2014;69:1339–1352. doi: 10.1093/gerona/glu080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alata W, et al. Human apolipoprotein E ε4 expression impairs cerebral vascularization and blood-brain barrier function in mice. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab. 2015;35:86–94. doi: 10.1038/jcbfm.2014.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.De Strooper B, et al. Production of intracellular amyloid-containing fragments in hippocampal neurons expressing human amyloid precursor protein and protection against amyloidogenesis by subtle amino acid substitutions in the rodent sequence. EMBO J. 1995;14:4932–4938. doi: 10.1002/j.1460-2075.1995.tb00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boyd-Kimball D, et al. Rodent Abeta(1-42) exhibits oxidative stress properties similar to those of human Abeta(1-42): Implications for proposed mechanisms of toxicity. J Alzheimers Dis JAD. 2004;6:515–525. doi: 10.3233/jad-2004-6509. [DOI] [PubMed] [Google Scholar]
- 73.Theendakara V, et al. Neuroprotective Sirtuin ratio reversed by ApoE4. Proc Natl Acad Sci U S A. 2013;110:18303–18308. doi: 10.1073/pnas.1314145110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carroll JC, et al. Sex differences in β-amyloid accumulation in 3xTg-AD mice: role of neonatal sex steroid hormone exposure. Brain Res. 2010;1366:233–245. doi: 10.1016/j.brainres.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arnold AP. Conceptual frameworks and mouse models for studying sex differences in physiology and disease: why compensation changes the game. Exp Neurol. 2014;259:2–9. doi: 10.1016/j.expneurol.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hines M, et al. Early androgen exposure and human gender development. Biol Sex Differ. 2015;6:3. doi: 10.1186/s13293-015-0022-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang X, et al. Transient exposure of neonatal female mice to testosterone abrogates the sexual dimorphism of abdominal aortic aneurysms. Circ Res. 2012;110:e73–85. doi: 10.1161/CIRCRESAHA.111.253880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fairbanks SL, et al. Sex stratified neuronal cultures to study ischemic cell death pathways. J Vis Exp JoVE. 2013 doi: 10.3791/50758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vasudevan A, Bhide PG. Angiogenesis in the embryonic CNS: a new twist on an old tale. Cell Adhes Migr. 2008;2:167–169. doi: 10.4161/cam.2.3.6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stearns SC, et al. Constraints on the coevolution of contemporary human males and females. Proc Biol Sci. 2012;279:4836–4844. doi: 10.1098/rspb.2012.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Osler W. The principles and practice of medicine, designed for the use of practitioners and students of medicine. Edinburgh ; London: Young J Putland; 1892. p. c1892. [Google Scholar]
- 82.Cordonnier C, van der Flier WM. Brain microbleeds and Alzheimer’s disease: innocent observation or key player? Brain J Neurol. 2011;134:335–344. doi: 10.1093/brain/awq321. [DOI] [PubMed] [Google Scholar]
- 83.Prins ND, Scheltens P. White matter hyperintensities, cognitive impairment and dementia: an update. Nat Rev Neurol. 2015;11:157–165. doi: 10.1038/nrneurol.2015.10. [DOI] [PubMed] [Google Scholar]
- 84.Wardlaw JM, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013;12:822–838. doi: 10.1016/S1474-4422(13)70124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cordonnier C, et al. Prevalence and severity of microbleeds in a memory clinic setting. Neurology. 2006;66:1356–1360. doi: 10.1212/01.wnl.0000210535.20297.ae. [DOI] [PubMed] [Google Scholar]
- 86.Pettersen JA, et al. Microbleed topography, leukoaraiosis, and cognition in probable Alzheimer disease from the Sunnybrook dementia study. Arch Neurol. 2008;65:790–795. doi: 10.1001/archneur.65.6.790. [DOI] [PubMed] [Google Scholar]
- 87.Wollenweber FA, et al. Prevalence of cortical superficial siderosis in patients with cognitive impairment. J Neurol. 2014;261:277–282. doi: 10.1007/s00415-013-7181-y. [DOI] [PubMed] [Google Scholar]
- 88.Martinez-Ramirez S, et al. Topography of dilated perivascular spaces in subjects from a memory clinic cohort. Neurology. 2013;80:1551–1556. doi: 10.1212/WNL.0b013e31828f1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van Veluw SJ, et al. Cortical microinfarcts on 3T MRI: Clinical correlates in memory-clinic patients. Alzheimers Dement J Alzheimers Assoc. 2015 doi: 10.1016/j.jalz.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 90.Na HK, et al. Cortical superficial siderosis: a marker of vascular amyloid in patients with cognitive impairment. Neurology. 2015;84:849–855. doi: 10.1212/WNL.0000000000001288. [DOI] [PubMed] [Google Scholar]
- 91.Poels MMF, et al. Cerebral microbleeds are associated with worse cognitive function: the Rotterdam Scan Study. Neurology. 2012;78:326–333. doi: 10.1212/WNL.0b013e3182452928. [DOI] [PubMed] [Google Scholar]
- 92.Vernooij MW, et al. Superficial siderosis in the general population. Neurology. 2009;73:202–205. doi: 10.1212/WNL.0b013e3181ae7c5e. [DOI] [PubMed] [Google Scholar]
- 93.Jeerakathil T, et al. Cerebral microbleeds: prevalence and associations with cardiovascular risk factors in the Framingham Study. Stroke J Cereb Circ. 2004;35:1831–1835. doi: 10.1161/01.STR.0000131809.35202.1b. [DOI] [PubMed] [Google Scholar]
- 94.Hainsworth AH, et al. Endothelial cells and human cerebral small vessel disease. Brain Pathol Zurich Switz. 2015;25:44–50. doi: 10.1111/bpa.12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Roses AD. Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu Rev Med. 1996;47:387–400. doi: 10.1146/annurev.med.47.1.387. [DOI] [PubMed] [Google Scholar]
- 96.Serrano-Pozo A, et al. APOEε2 is associated with milder clinical and pathological Alzheimer disease. Ann Neurol. 2015;77:917–929. doi: 10.1002/ana.24369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Charidimou A, et al. Cerebral amyloid angiopathy with and without hemorrhage: evidence for different disease phenotypes. Neurology. 2015;84:1206–1212. doi: 10.1212/WNL.0000000000001398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nicoll JA, et al. High frequency of apolipoprotein E epsilon 2 in patients with cerebral hemorrhage due to cerebral amyloid angiopathy. Ann Neurol. 1996;39:682–683. doi: 10.1002/ana.410390521. [DOI] [PubMed] [Google Scholar]
- 99.Pankiewicz JE, et al. Blocking the apoE/Aβ interaction ameliorates Aβ-related pathology in APOE ε2 and ε4 targeted replacement Alzheimer model mice. Acta Neuropathol Commun. 2014;2:75. doi: 10.1186/s40478-014-0075-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chiang GC, et al. Hippocampal atrophy rates and CSF biomarkers in elderly APOE2 normal subjects. Neurology. 2010;75:1976–1981. doi: 10.1212/WNL.0b013e3181ffe4d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McIntosh AM, et al. The apolipoprotein E (APOE) gene appears functionally monomorphic in chimpanzees (Pan troglodytes) PloS One. 2012;7:e47760. doi: 10.1371/journal.pone.0047760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Singh PP, et al. APOE distribution in world populations with new data from India and the UK. Ann Hum Biol. 2006;33:279–308. doi: 10.1080/03014460600594513. [DOI] [PubMed] [Google Scholar]
- 103.Vamathevan JJ, et al. The role of positive selection in determining the molecular cause of species differences in disease. BMC Evol Biol. 2008;8:273. doi: 10.1186/1471-2148-8-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Finch CE. Evolution in health and medicine Sackler colloquium: Evolution of the human lifespan and diseases of aging: roles of infection, inflammation, and nutrition. Proc Natl Acad Sci U S A. 2010;107(Suppl 1):1718–1724. doi: 10.1073/pnas.0909606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Legleiter J, et al. The modulating effect of mechanical changes in lipid bilayers caused by apoE-containing lipoproteins on Aβ induced membrane disruption. ACS Chem Neurosci. 2011;2:588–599. doi: 10.1021/cn2000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Raffai RL, et al. Introduction of human apolipoprotein E4 “domain interaction” into mouse apolipoprotein E. Proc Natl Acad Sci U S A. 2001;98:11587–11591. doi: 10.1073/pnas.201279298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Finch C, Martin G. Dementias of the Alzheimer type: views through the lens of evolutionary biology suggest amyloid-driven brain aging is a trade-off with host defense. Evolutionary thinking in medicine: from research to policy and practice. 2016 In Press. [Google Scholar]
- 108.Mitter SS, et al. Apolipoprotein E4 influences growth and cognitive responses to micronutrient supplementation in shantytown children from northeast Brazil. Clin São Paulo Braz. 2012;67:11–18. doi: 10.6061/clinics/2012(01)03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Azevedo OGR, et al. Apolipoprotein E plays a key role against cryptosporidial infection in transgenic undernourished mice. PloS One. 2014;9:e89562. doi: 10.1371/journal.pone.0089562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Feldman HH, et al. Superficial siderosis: a potential diagnostic marker of cerebral amyloid angiopathy in Alzheimer disease. Stroke J Cereb Circ. 2008;39:2894–2897. doi: 10.1161/STROKEAHA.107.510826. [DOI] [PubMed] [Google Scholar]
- 111.Chen C, Hu Z. ApoE Polymorphisms and the Risk of Different Subtypes of Stroke in the Chinese Population: A Comprehensive Meta-Analysis. Cerebrovasc Dis Basel Switz. 2016;41:119–138. doi: 10.1159/000442678. [DOI] [PubMed] [Google Scholar]
- 112.Amar K, et al. Are genetic factors important in the aetiology of leukoaraiosis? Results from a memory clinic population. Int J Geriatr Psychiatry. 1998;13:585–590. doi: 10.1002/(sici)1099-1166(199809)13:9<585::aid-gps825>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 113.Hirono N, et al. Effect of the apolipoprotein E epsilon4 allele on white matter hyperintensities in dementia. Stroke J Cereb Circ. 2000;31:1263–1268. doi: 10.1161/01.str.31.6.1263. [DOI] [PubMed] [Google Scholar]
- 114.Sawada H, et al. Cerebral white matter lesions are not associated with apoE genotype but with age and female sex in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2000;68:653–656. doi: 10.1136/jnnp.68.5.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.McCarron MO, Nicoll JA. Apolipoprotein E genotype and cerebral amyloid angiopathy-related hemorrhage. Ann N Y Acad Sci. 2000;903:176–179. doi: 10.1111/j.1749-6632.2000.tb06366.x. [DOI] [PubMed] [Google Scholar]
- 116.Elosua R, et al. Association of APOE genotype with carotid atherosclerosis in men and women: the Framingham Heart Study. J Lipid Res. 2004;45:1868–1875. doi: 10.1194/jlr.M400114-JLR200. [DOI] [PubMed] [Google Scholar]
- 117.Doumas M, et al. Gender differences in hypertension: myths and reality. Curr Hypertens Rep. 2013;15:321–330. doi: 10.1007/s11906-013-0359-y. [DOI] [PubMed] [Google Scholar]
- 118.Fuzikawa AK, et al. Association of ApoE polymorphisms with prevalent hypertension in 1406 older adults: the Bambuí Health Aging Study (BHAS) Braz J Med Biol Res Rev Bras Pesqui Médicas E Biológicas Soc Bras Biofísica Al. 2008;41:89–94. doi: 10.1590/s0100-879x2008000200002. [DOI] [PubMed] [Google Scholar]
- 119.Niu W, et al. Association of an apolipoprotein E polymorphism with circulating cholesterols and hypertension: a meta-based Mendelian randomization analysis. Hypertens Res Off J Jpn Soc Hypertens. 2012;35:434–440. doi: 10.1038/hr.2011.202. [DOI] [PubMed] [Google Scholar]