

Abstract
Enhanced glioma-stem-cell (GSC) motility and therapy resistance are considered to play key roles in tumor cell dissemination and recurrence. As such, a better understanding of the mechanisms by which these cells disseminate and withstand therapy could lead to more efficacious treatments. Here, we introduce a novel micro-/nanotechnology-enabled chip platform for performing live-cell interrogation of patient-derived GSCs with single-clone resolution. On-chip analysis revealed marked intertumoral differences (>10-fold) in single-clone motility profiles between two populations of GSCs, which correlated well with results from tumor-xenograft experiments and gene-expression analyses. Further chip-based examination of the more-aggressive GSC population revealed pronounced interclonal variations in motility capabilities (up to ∼4-fold) as well as gene-expression profiles at the single-cell level. Chip-supported therapy resistance studies with a chemotherapeutic agent (i.e., temozolomide) and an oligo RNA (anti-miR363) revealed a subpopulation of CD44-high GSCs with strong antiapoptotic behavior as well as enhanced motility capabilities. The living-cell-interrogation chip platform described herein enables thorough and large-scale live monitoring of heterogeneous cancer-cell populations with single-cell resolution, which is not achievable by any other existing technology and thus has the potential to provide new insights into the cellular and molecular mechanisms modulating glioma-stem-cell dissemination and therapy resistance.
Keywords: Living single-cell analysis, cell motility, nanochannel electroporation, glioblastoma, cancer stem cell, anti-microRNA
Graphical abstract
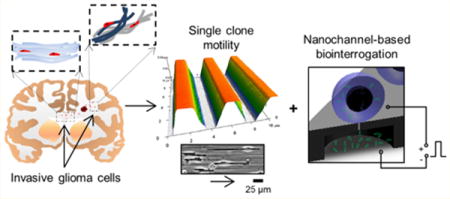
Glioblastoma (GBM) is a lethal primary brain cancer with a highly invasive nature, which inevitably results in post-therapeutic recurrence beyond the resection margin.1 This aggressive tumor exhibits distinct intracranial spreading patterns, disseminating preferentially as single cells along prealigned anatomical microstructures within the brain parenchyma, including white-matter tracts, the basal lamina of blood vessels, and the subpial space, among others (Figure 1a).1–3 Indeed, a growing amount of evidence suggests that the invasive phenotype of GBM cells is highly dependent on cell motility.4 Moreover, recurrence seems to be primarily driven by a subpopulation of highly invasive tumor-initiating cells, known as glioma stem cells (GSCs), which are reportedly more motile and resistant to therapy-induced apoptosis and thus presumably contribute to tumor recurrence.5,6 As GSCs continue to draw significant interest from the scientific and medical communities, new tools capable of probing behavioral singularities at the single-cell level are needed to better understand and counteract the mechanisms by which these cells spread to develop new foci of recurrent tumors in the brain.
Figure 1.
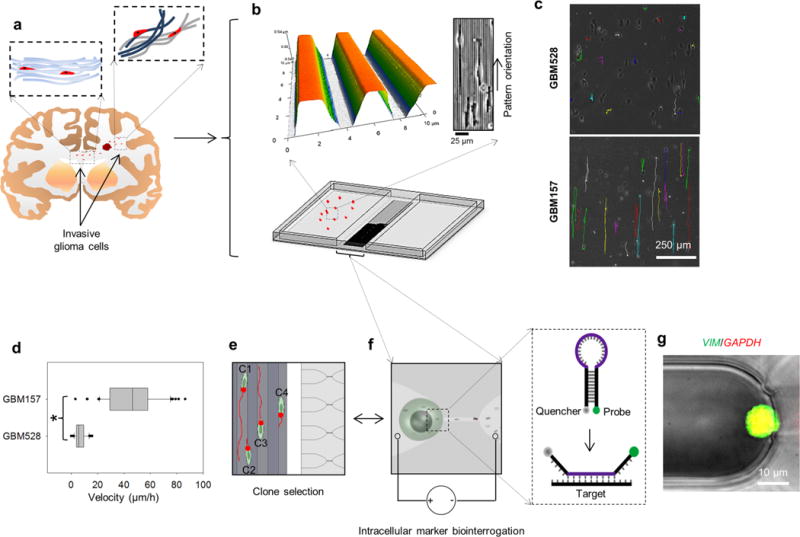
Patient-derived GSC exhibition of significant inter- and intrapopulation differences in disseminative capabilities. (a) Schematic diagram illustrating the concept of single-cell dissemination in invasive gliomas. GSCs migrate along highly oriented fiberlike structures (e.g., white-matter tracts) and invade the surrounding brain parenchyma. (b) Micro-/nanofabricated chip for GSC motility biointerrogation at the single-cell level. Top images show an atomic-force-microscopy micrograph (left) of the topography mimicking the fiberlike structures within the tissue. GSCs exhibit guided alignment and motility in response to the topography (right). (c) Motility patterns followed by single GSCs. Patient-derived GSC populations exhibit different motility capabilities. GBM528 remain immobile on the biomimetic surface, while GBM157 show enhanced and guided single-cell motility along the textured surface. (d) Single-cell quantification of motility for GBM528 and GBM157. Although GBM157 show overall improved motility capabilities compared to GBM528, there are significant interclonal variations in motility within the GBM157 population. (e,f) On-chip nanochannel-based electroporation (NEP) was then used to identify interclonal differences in the expression patterns of migration-related genes by in situ hybridization. (g) NEP-delivered molecular beacons for vimentin mRNA showed significant interclonal differences in the expression pattern of its gene, which correlated with the motility capabilities of the cells. Asterisks indicate p < 0.05 (Dunn’s method).
Although a better understanding of the single-cell motility of GSCs could possibly help unlock more-effective treatments for this disease, the research on the mechanisms underlying this phenomenon has been comparatively limited. This could be due, in part, to the lack of simple and reproducible in vitro single-cell-migration models that can reliably mimic the in vivo microenvironment. Characterization of tumor dissemination at the single-cell level via in vivo imaging is extremely challenging.7–9 Moreover, ex vivo quantification with tissue explants tends to be laborious and problematic due to reproducibility issues.3,10 Simpler conventional in vitro assays,11–15 however, are often not physiologically relevant or are end-point assays that focus on the bulk behavior of highly heterogeneous cellular populations.
Recent studies have used micro- and nanoscale technologies to develop systems that can be utilized to analyze single-cell motility under more physiologically relevant conditions.2,3,7,16,17 Topographical and cell-confinement cues have been used to induce and mimic rapid and highly directional cell motility, which is a characteristic seen not only in invasive GBM but also in other types of cancer.2,3,7,18–20 Although such studies have provided great insight into the migratory behavior of a number of tumor-cell types, further analyses of single-cell biology aimed at explaining the observed motility and dissemination capabilities and developing therapeutic strategies remain a challenge due to the lack of proper and compatible tools for living-cell interrogation at the single-cell level.
Here, we introduce a new chip-supported strategy for in situ probing of patient-derived GSC populations, which is based on the implementation of single-clone biomimetic motility assays coupled with intracellular interrogation via nanochannel-based electroporation (NEP).21 Previous studies have looked into the development of cargo-delivery systems with single-cell resolution based, for example, on microelectroporation (MEP) or direct fluidic access into the cytosol.22,23 NEP, however, offers a number of advantages over existing technologies, including minimizing potential cell damage by confining the poration to a much-smaller (nanometer-sized) portion of the cell membrane (compared to MEP) and allowing almost instantaneous (μs- to ms-scale) dose-controlled cargo delivery compared to fluidic- and diffusion-based approaches, in which cargo uptake occurs over more-prolonged periods of time. Moreover, in contrast to our previous work on NEP-based systems,21,24,25 whose capabilities only focused on controlling cargo delivery for downstream applications, the novel platform technology described herein allowed us not only to monitor, in real time, single-cell motility but also to link specific migratory behaviors with distinct intracellular molecular signatures and therapy resistance, which could potentially be used for the development of improved therapeutic regimens as well as gaining a better understanding of tumor-cell biology.
Patient-Derived GSCs Exhibition of Distinct and Heterogeneous Motility Patterns
We compared the migratory behavior of two well-characterized GBM patient-derived GSC-enriched populations, GBM157 and GBM528, both of which were largely subclassified as proneural GSCs.26–28 Following short-term culture as neurospheres in serum-free medium, the cells were passaged as single clones into serum-containing medium (10%) and plated on the microtextured and biomimetic polydimethylsiloxane (PDMS) chip surface (Figure 1b). Such surfaces were fabricated via a simple replica-molding process from photolithographically fabricated Si masters and were designed to have arrays of parallel ridges (∼2 μm × 1 μm with 2 μm spacing) (Figure 1b, top) with dimensions that ranged within reported values for small blood vessels and white-matter tracts in the brain.2,3 Single-clone motility was monitored in real time by time-lapse microscopy for approximately 16 h.
Our results indicate that GBM528 clones displayed limited to no motility (Figure 1c,d and Supplementary Video 1). These cells demonstrated less-pronounced alignment with the topography compared to that of GBM157 clones (Figure 1b, top right) and had a tendency to exhibit erratic two-dimensional short-range motility that did not appear to be influenced or guided by the patterned surface. Conversely, GBM157 clones (Supplementary Video 2) showed significantly higher motility, which was one-dimensional and uniaxial in nature and was clearly guided by the underlying surface topography (Figure 1b–d). Such differences in motility were further corroborated in immunocompromized mouse-brain xenograft experiments (Supplementary Figure 1), which showed that GBM528 clones tended to form tumors with well-defined boundaries that span throughout the brain parenchyma, primarily by cell proliferation and volume expansion. In contrast, tumors formed from xenografted GBM157 clones showed clear signs of active single-clone migration and infiltration with diffuse and poorly defined boundaries.
Patient-derived GBM spheres are composed of heterogeneous cell populations with distinct GSC subtypes. As expected, we observed clear intertumoral differences across patients (e.g., GBM157 versus GBM528) and substantial interclonal migratory disparities within the same population, especially in GBM157 dissociates. Although some clones showed fast and highly unidirectional migration in the absence of chemoattractants, other clones presented low motility, bidirectional motility, or both. To further examine interclonal variations, we conducted on-chip 2D nanochannel electroporation (2D NEP)-based measurements of intracellular markers by in situ hybridization (Figure 1e–g). The NEP system allowed us to controllably and benignly inject cargo (e.g., molecular probes and gene silencers) into single clones,21 thus helping us establish potential correlations between the expression levels of certain intracellular molecules and a given migratory phenotype. The 2D NEP component was fabricated by PDMS replica molding from a photolithographically fabricated Si master and was stably integrated into the chip platform via oxygen plasma bonding. An optical tweezers system was used to selectively manipulate single clones within the chip (Supplementary Video 3). A first set of experiments was then conducted in an effort to evaluate the levels of vimentin mRNA in individual GBM157 clones that exhibited high versus low on-chip motility. Vimentin is a cytoskeletal protein that is known to be up-regulated in highly motile cancer cells.20 Fast- and slow-moving GBM157 clones were then randomly identified from a ∼16 h time-lapse experiment on our chip (Figure 1e) and subsequently transported into the microchannels of the NEP component using optical tweezers. The juxtaposing array of microchannels was filled with molecular beacons (MBs) against vimentin. MBs against GAPDH mRNA were used for comparison purposes. The MBs were then NEP-injected into the preselected clones by applying a ms-duration electric field across the microchannel arrays (Figure 1f). Beacon hybridization with the target molecules was then analyzed by fluorescence microscopy (Figure 1g). Our results indicate that from the heterogeneous GBM clones, velocity differences of approximately 5-fold between clones correlated with a change of ∼2.5 ± 0.3 (average ± standard error)−fold in vimentin loading. Such results confirm the chip’s ability to not only analyze clonal motility but also to identify potentially novel intracellular markers, at the single-clone level, associated with aggressive tumor phenotypes.
Additional cell-motility experiments (Supplementary Figures 2 and 3) provided further insights into the migratory behavior of GBM157 and GBM528. Notably, and in contrast to what we observed on textured and biomimetic surfaces, single -clones from both populations exhibited virtually no motility when plated as monodispersed cultures on tissue-culture polystyrene (TCPS) (Supplementary Figure 2). Although both GBM157 and GBM528 clones were able to successfully adhere and spread on the surface, they remained stagnant for the most part, with only some clones showing very-short-range and disorganized motility patterns (Supplementary Videos 4 and 5). In addition, when single-cell motility was further probed on softer and flat PDMS surfaces, GBM528 clones continued to show no significant signs of cell motility, and GBM157 clones exhibited substantial migratory activity (Supplementary Figure 2; Supplementary Videos 6 and 7). Similar results were seen when monitoring GBM157 and GBM528 neurosphere spreading under different conditions (Supplementary Figure 3; Supplementary Videos 8–13), with GBM157 neurospheres showing enhanced spreading on softer, textured and biomimetic PDMS surfaces, and GBM528 exhibited relatively similar spreading capabilities across all surfaces. Interestingly, however, although no single-cell motility was detected on TCPS, GBM157 and GBM528 spheroids both showed dissemination capabilities on such surface. The implications of these observations are 2-fold. First, it suggests that the neurosphere-spreading behavior observed for GBM157 and GBM528 is not necessarily representative of the dissemination capabilities of GSCs at the single-cell level. Second, it indicates that the inherent migratory potential of some GSC populations (e.g., GBM157) can be potentially restrained or enhanced by the physical or chemical properties (e.g., chemistry, stiffness, and topography) of the microenvironment on which they migrate, while other GSC populations (e.g., GBM528) appeared to be significantly less-sensitive to such cues.
Large-Scale Intracellular Screening Revealing of Clonal Subpopulations with More-Aggressive Phenotypes
Transcriptome microarray analysis of GBM157 versus GBM528 (Figure 2a) confirmed the underlying molecular differences between these two GBM neurosphere samples, with GBM157 cells harboring higher expression of migration-associated genes, including vimentin. Among these, however, CD44 exhibited the strongest contrast across populations, thus potentially highlighting the relevance of this gene in modulating the overall invasive behavior of the GBM157.29–33
Figure 2.
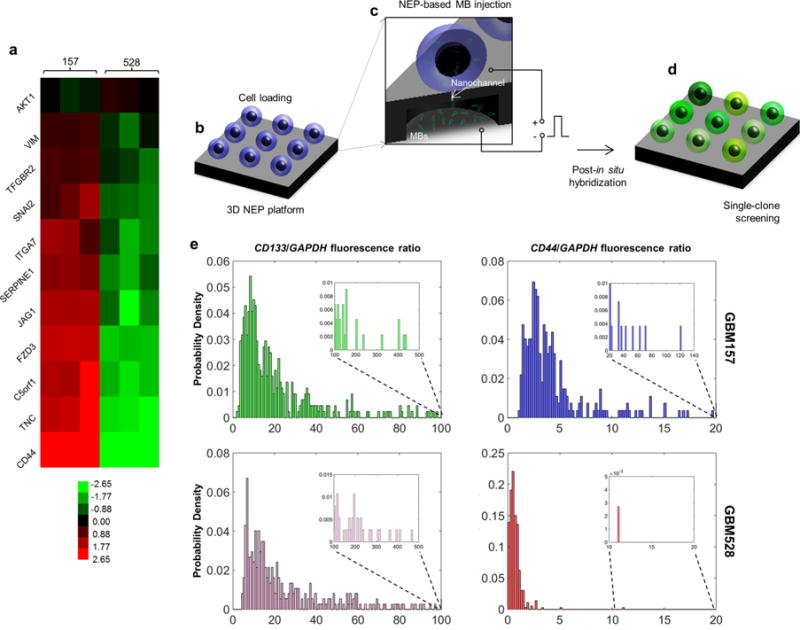
Large-scale screening of GSCs revealing of significant variations across and within (single-cell resolution) populations. (a) Microarray data showing that GBM157 tend to overexpress migration-related genes compared to GBM528. (b–d) Large-scale single-cell screening based on 3D NEP. (b) GSCs are first loaded on the chip surface. (c) Molecular beacons are then delivered into each cell by applying a focused, pulsed electric field through arrayed nanochannels. (d) Following in situ hybridization, the cells show varying levels of fluorescence depending on the expression level of the target gene. (e) Single-cell fluorescence is then quantified via fluorescence microscopy. The results show that both the GBM157 and GBM528 populations reveal similar levels of expression for CD133, which is a proneural GSC marker. CD44 expression was more pronounced for the GBM157 population, with a number of clones showing markedly higher expression (inset) compared to the rest of the population. Such clones are presumed to exhibit more aggressive behavior.
To further dissect the expression profile of CD44 in GBM157 and GBM528 with single-cell resolution, we implemented a large-scale 3D NEP approach to probe thousands of cells in a benign and controlled manner (Figure 2b–d).24 Although 3D NEP-based delivery of MBs against CD133, a signature proneural GSC gene,34 showed relatively similar single-cell expression spectra across GBM157 and GBM528 (Figure 2c, left), in situ hybridization with CD44 MBs shows a clear spectrum shift to the right for the GBM157 population compared to GBM528 (Figure 2c, right). Moreover, interclonal analysis showed significant variations in CD44 expression within the GBM157 population, with a subset of clones exhibiting significantly higher CD44 expression compared with the rest of the population (Figure 2c, right, inset). Altogether, these observations point to the presence of clonal subsets of GSCs within the GBM157 population that are prone to expressing more aggressive mesenchymal-like markers.29–35
On-Chip Therapy Efficacy Studies Revealing of the Existence of a Clonal Subset with High Antiapoptotic and Highly Migratory Capabilities
Additional on-chip studies were then conducted to examine how the more-aggressive GBM157 population reacted to the current first-line chemotherapeutic agent for GBM,36 temozolomide (TMZ), and a potential novel therapeutic agent, anti-miR363, which has attracted a great deal of attention recently as a potential antiapoptotic target in cancer therapy.37–39 Briefly, single GBM157 clones were treated with different doses of TMZ (Supplementary Figure 4) and anti-miR363 (i.e., two pulses versus five pulses) on the 3D NEP setup (Figure 3a,b). Cell viability was then monitored for up to 96 h post-treatment via live- and dead-cell staining and fluorescence microscopy (Figure 3c,d). Our results show that TMZ alone was able to cause a significant decrease in cell viability by almost 60% at 96 h. Conversely, the combined effect of TMZ and anti-miR363 led to a decrease in viability of 80–90% at 96 h. Notably, treatment with anti-miR363 alone also resulted in a decrease in cell viability close to 80% at 96 h. Dosed delivery of anti-miR363 by 3D NEP only showed pronounced differences at early time points (i.e., 48 h), at which point five NEP pulses led to increased cell death compared to results from two pulses (p = 0.014, Student t test). By 96 h, however, both conditions resulted in similar levels of cell survival.
Figure 3.
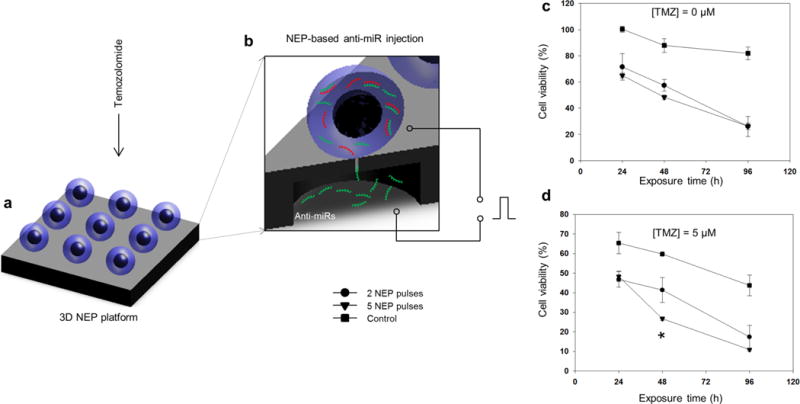
Therapy efficacy studies on a 3D NEP platform revealing the existence of a subset of clones exhibiting high antiapoptotic capabilities. Schematic diagram of the experimental setup in which GBM157 were exposed to (a) chemotherapy via direct-contact delivery of temozolomide (TMZ) or (b) oligo-RNA therapy via NEP-based delivery of anti-miR 363. (c) NEP-based delivery of anti-miR 363 shows a dose-dependent (two vs five pulses) decrease in cell viability over time, compared to the control. (d) Direct and concomitant exposure to temozolomide increased the proportion of cell death, with approximately 10% of the population still showing a high degree of resistance to drug- and oligo-RNA-induced apoptosis.
To test whether this pro-apoptotic therapy influenced the motility patterns of GBM157 at the single-clone level, we conducted on-chip migration assays after exposing the cells to TMZ, anti-miR363, or both. Interestingly, although TMZ alone caused significant cell death, we were not able to detect any major changes in cell motility for the surviving cell population (Figure 4a). However, 3D NEP-based delivery of anti-miR363 led to a marked decrease in single-clone motility within the first 48 h. Continued monitoring of cell motility (Figure 4b) revealed that clones that died within 48 h after anti-miR363 treatment showed the most-pronounced decrease in cell motility. However, clones that were able to survive past 48 h exhibited motility capabilities comparable to control and untreated cells. This is thus suggestive of the presence of a subset of GBM157 clones that not only show strongly antiapoptotic behavior but also enhanced dissemination capabilities. To phenotypically identify this subset, we conducted 3D NEP-based delivery of MBs for CD44 and CD133. Our findings clearly show that the surviving cell population after anti-miR363 delivery tended to have more pronounced CD44 expression and attenuated CD133 expression, which is indicative of a more mesenchymal-like aggressive phenotype. Multigene- and oligo-RNA-detection experiments (Supplementary Figure 5) showed increased expression of additional mesenchymal markers40 and an overall decrease in miR363 following anti-miR363 delivery.
Figure 4.
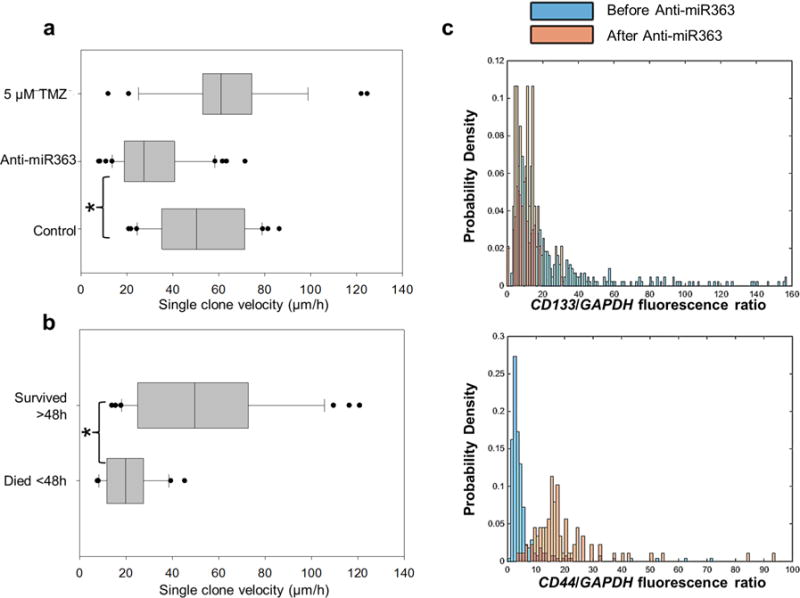
Therapy-resistant GSC exhibition of enhanced motility capabilities. (a) Single-cell motility assays showing that although both TMZ and anti-miR363 therapy had a significant negative effect on cell viability, only the anti-miR363 caused a significant and concomitant decrease in cell motility. (b) Continuous monitoring of single-cell motility following anti-miR363 delivery revealed that only the cells that were prone to undergoing apoptosis within the first 48 h showed a significant decrease in single-clone motility. Cells that had not undergone apoptosis past 48 h still showed marked motility, comparable to untreated and control cells. (c) In situ hybridization experiments coupled with drug-resistance studies by 3D NEP allowed us to identify the surviving and highly migratory cell population as being high in CD44 expression. Asterisks indicate p < 0.05 (Dunn’s method).
Here, we demonstrated a powerful micro-/nanotechnology-based chip platform for living-cell interrogation of cancer cell populations with single-cell resolution in terms of motility, intracellular marker expression, and therapy resistance. The platform incorporates two major components: a cell-migration stage in which single-clone motility can be monitored under guided migration conditions, which closely resembles the in vivo dissemination patterns of GSCs, and a biointerrogation stage that is supported by a unique NEP technology and can be used to selectively inject specific clones with molecular probes to identify certain markers, potentially associated with aggressive tumor phenotypes, and to controllably deliver a wide variety of agents that can be used to up- and downregulate key genes for subsequent analysis. The chip platform described herein could conceivably be used to better understand inherent heterogeneities within and across different types of cancer-initiating cells and thus to potentially develop novel and better-targeted therapeutics for this disease. Further in vitro and in vivo studies, however, need to be conducted to better define optimum time windows for ex vivo micro-/nanochip-based experiments, in which disparities between in vitro and in vivo behaviors, potentially introduced by isolation and culture procedures, are minimized to the greatest extent possible.
Supplementary Material
Acknowledgments
Funding for this work was provided by the National Institutes of Health (R21EB017539, R01NS083767, R01NS087913, R01CA183991, and R01CA201402) and the National Science Foundation (EEC-0914790).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nanolett.6b00902.
Image showing patient-derived GSC exhibition of different dissemination capabilities in a xenograft mouse model, single-cell motility of GSCs on flat surfaces, GSC motility analysis at the spheroid level, expanded data on therapy-efficacy studies at different timepoints, NEP- and MB-based detection of multi-gene-expression and oligo RNA, and fabrication schematics. (PDF)
Additional details on chip-platform fabrication, cell culture and analysis, and statistical analysis. (PDF) Time-lapse videos of cell motility and optical tweezers-based cell manipulation. (ZIP)
Author Contributions
D.G.-P. and L.C. contributed equally. Platform design and implementation was performed by D.G.P., D.J.H., and L.J.L. Platform fabrication, testing and analysis were conducted by D.G.P., L.C., J.S., J.M., S.-H.K., X.Z, V.M., X.W., M.M., K.J.K., Y.W., W.L., I.N., and L.J.L. G.L. provided guidance with the optical tweezers experiment. Xenograft experiments were conducted by S.-H.K., M.M., and I.N. The manuscript was written by D.G.P., D.J.H, I.N., and L.J.L.
Notes
The authors declare no competing financial interest.
References
- 1.Bellail AC, Hunter SB, Brat DJ, Tan C, Van Meir EG. Int J Biochem Cell Biol. 2004;36(6):1046–69. doi: 10.1016/j.biocel.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 2.Gallego-Perez D, Higuita-Castro N, Denning L, DeJesus J, Dahl K, Sarkar A, Hansford DJ. Lab Chip. 2012;12(21):4424–32. doi: 10.1039/c2lc40726d. [DOI] [PubMed] [Google Scholar]
- 3.Johnson J, Nowicki MO, Lee CH, Chiocca EA, Viapiano MS, Lawler SE, Lannutti JJ. Tissue Eng, Part C. 2009;15(4):531–40. doi: 10.1089/ten.tec.2008.0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giese A, Bjerkvig R, Berens ME, Westphal M. J Clin Oncol. 2003;21(8):1624–36. doi: 10.1200/JCO.2003.05.063. [DOI] [PubMed] [Google Scholar]
- 5.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, Frank A, Bayazitov IT, Zakharenko SS, Gajjar A, Davidoff A, Gilbertson RJ. Cancer Cell. 2007;11(1):69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 6.Ghotra VP, Puigvert JC, Danen EH. Int J Radiat Biol. 2009;85(11):955–62. doi: 10.3109/09553000903242164. [DOI] [PubMed] [Google Scholar]
- 7.Irimia D, Toner M. Integr Biol (Camb) 2009;1(8–9):506–12. doi: 10.1039/b908595e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Condeelis J, Segall JE. Nat Rev Cancer. 2003;3(12):921–30. doi: 10.1038/nrc1231. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz MP, Fairbanks BD, Rogers RE, Rangarajan R, Zaman MH, Anseth KS. Integr Biol (Camb) 2010;2(1):32–40. doi: 10.1039/b912438a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palfi S, Swanson KR, De Bouard S, Chretien F, Oliveira R, Gherardi RK, Kros JM, Peschanski M, Christov C. Br J Cancer. 2004;91(4):745–52. doi: 10.1038/sj.bjc.6602048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyden S. J Exp Med. 1962;115:453–66. doi: 10.1084/jem.115.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albini A, Benelli R. Nat Protoc. 2007;2(3):504–11. doi: 10.1038/nprot.2006.466. [DOI] [PubMed] [Google Scholar]
- 13.Rao JS. Nat Rev Cancer. 2003;3(7):489–501. doi: 10.1038/nrc1121. [DOI] [PubMed] [Google Scholar]
- 14.Yamada KM, Cukierman E. Cell. 2007;130(4):601–10. doi: 10.1016/j.cell.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 15.Liang CC, Park AY, Guan JL. Nat Protoc. 2007;2(2):329–33. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 16.Doyle AD, Wang FW, Matsumoto K, Yamada KM. J Cell Biol. 2009;184(4):481–90. doi: 10.1083/jcb.200810041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petrie RJ, Doyle AD, Yamada KM. Nat Rev Mol Cell Biol. 2009;10(8):538–49. doi: 10.1038/nrm2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sidani M, Wyckoff J, Xue C, Segall JE, Condeelis J. J Mammary Gland Biol Neoplasia. 2006;11(2):151–63. doi: 10.1007/s10911-006-9021-5. [DOI] [PubMed] [Google Scholar]
- 19.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. BMC Med. 2006;4(1):38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong IY, Javaid S, Wong EA, Perk S, Haber DA, Toner M, Irimia D. Nat Mater. 2014;13(11):1063–71. doi: 10.1038/nmat4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boukany PE, Morss A, Liao WC, Henslee B, Jung H, Zhang X, Yu B, Wang X, Wu Y, Li L, Gao K, Hu X, Zhao X, Hemminger O, Lu W, Lafyatis GP, Lee LJ. Nat Nanotechnol. 2011;6(11):747–54. doi: 10.1038/nnano.2011.164. [DOI] [PubMed] [Google Scholar]
- 22.Khine M, Lau A, Ionescu-Zanetti C, Seo J, Lee LP. Lab Chip. 2005;5(1):38–43. doi: 10.1039/b408352k. [DOI] [PubMed] [Google Scholar]
- 23.VanDersarl JJ, Xu AM, Melosh NA. Nano Lett. 2012;12(8):3881–6. doi: 10.1021/nl204051v. [DOI] [PubMed] [Google Scholar]
- 24.Chang L, Gallego-Perez D, Zhao X, Bertani P, Yang Z, Chiang CL, Malkoc V, Shi J, Sen CK, Odonnell L, Yu J, Lu W, Lee LJ. Lab Chip. 2015;15(15):3147–53. doi: 10.1039/c5lc00553a. [DOI] [PubMed] [Google Scholar]
- 25.Gallego-Perez D, Otero JJ, Czeisler C, Ma J, Ortiz C, Gygli P, Catacutan FP, Gokozan HN, Cowgill A, Sherwood T, Ghatak S, Malkoc V, Zhao X, Liao WC, Gnyawali S, Wang X, Adler AF, Leong K, Wulff B, Wilgus TA, Askwith C, Khanna S, Rink C, Sen CK, Lee LJ. Nanomedicine. 2016;12(2):399–409. doi: 10.1016/j.nano.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu C, Banasavadi-Siddegowda YK, Joshi K, Nakamura Y, Kurt H, Gupta S, Nakano I. Stem Cells. 2013;31(5):870–81. doi: 10.1002/stem.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guvenc H, Pavlyukov MS, Joshi K, Kurt H, Banasavadi-Siddegowda YK, Mao P, Hong C, Yamada R, Kwon CH, Bhasin D, Chettiar S, Kitange G, Park IH, Sarkaria JN, Li C, Shakhparonov MI, Nakano I. Clin Cancer Res. 2013;19(3):631–42. doi: 10.1158/1078-0432.CCR-12-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyazaki T, Pan Y, Joshi K, Purohit D, Hu B, Demir H, Mazumder S, Okabe S, Yamori T, Viapiano M, Shin-ya K, Seimiya H, Nakano I. Clin Cancer Res. 2012;18(5):1268–80. doi: 10.1158/1078-0432.CCR-11-1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoshida T, Matsuda Y, Naito Z, Ishiwata T. Pathol Int. 2012;62(7):463–70. doi: 10.1111/j.1440-1827.2012.02823.x. [DOI] [PubMed] [Google Scholar]
- 30.Hiraga T, Ito S, Nakamura H. Cancer Res. 2013;73(13):4112–22. doi: 10.1158/0008-5472.CAN-12-3801. [DOI] [PubMed] [Google Scholar]
- 31.Pietras A, Katz AM, Ekstrom EJ, Wee B, Halliday JJ, Pitter KL, Werbeck JL, Amankulor NM, Huse JT, Holland EC. Cell Stem Cell. 2014;14(3):357–69. doi: 10.1016/j.stem.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanaka S, Nakada M, Yamada D, Nakano I, Todo T, Ino Y, Hoshii T, Tadokoro Y, Ohta K, Ali MA, Hayashi Y, Hamada J, Hirao A. J Neuro-Oncol. 2015;121(2):239–50. doi: 10.1007/s11060-014-1630-z. [DOI] [PubMed] [Google Scholar]
- 33.Fu J, Yang QY, Sai K, Chen FR, Pang JC, Ng HK, Kwan AL, Chen ZP. Neuro Oncol. 2013;15(10):1353–65. doi: 10.1093/neuonc/not079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakano I. J Neurosurg. 2015;122(2):324–30. doi: 10.3171/2014.9.JNS132253. [DOI] [PubMed] [Google Scholar]
- 35.Mao P, Joshi K, Li J, Kim SH, Li P, Santana-Santos L, Luthra S, Chandran UR, Benos PV, Smith L, Wang M, Hu B, Cheng SY, Sobol RW, Nakano I. Proc Natl Acad Sci U S A. 2013;110(21):8644–9. doi: 10.1073/pnas.1221478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedman HS, Kerby T, Calvert H. Clin Cancer Res. 2000;6(7):2585–97. [PubMed] [Google Scholar]
- 37.Floyd DH, Zhang Y, Dey BK, Kefas B, Breit H, Marks K, Dutta A, Herold-Mende C, Synowitz M, Glass R, Abounader R, Purow BW. PLoS One. 2014;9(5):e96239. doi: 10.1371/journal.pone.0096239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sana J, Hajduch M, Michalek J, Vyzula R, Slaby O. J Cell Mol Med. 2011;15(8):1636–44. doi: 10.1111/j.1582-4934.2011.01317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Babashah S, Soleimani M. Eur J Cancer. 2011;47(8):1127–37. doi: 10.1016/j.ejca.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 40.Kim SH, Ezhilarasan R, Phillips E, Gallego-Perez D, Sparks A, Taylor D, Ladner K, Furuta T, Sabit H, Chhipa R, Cho JH, Mohyeldin A, Beck S, Kurozumi K, Kuroiwa T, Iwata R, Asai A, Kim J, Sulman EP, Cheng SY, Lee LJ, Nakada M, Guttridge D, DasGupta B, Goidts V, Bhat KP, Nakano I. Cancer Cell. 2016;29(2):201–13. doi: 10.1016/j.ccell.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.