

Abstract
The focus of our laboratory’s investigation is to study the direct-type DNA damage mechanisms resulting from γ-ray and ion-beam radiation-induced free radical processes in DNA which lead to molecular damage important to cellular survival. This work compares the results of low LET (γ−) and high LET (ion-beam) radiation to develop a chemical track structure model for ion-beam radiation damage to DNA. Recent studies on protonation states of cytosine cation radicals in the N1-substituted cytosine derivatives in their ground state and 5-methylcytosine cation radicals in ground as well as in excited state are described. Our results exhibit a radical signature of excitations in 5-methylcytosine cation radical. Moreover, our recent theoretical studies elucidate the role of electron-induced reactions (low energy electrons (LEE), presolvated electrons (epre−), and aqueous (or, solvated) electrons (eaq−)). Finally DFT calculations of the ionization potentials of various sugar radicals show the relative reactivity of these species.
Keywords: γ-radiation, ion-beam radiation, electron spin resonance (ESR), density functional theory (DFT), low energy electrons (LEE), DNA-radicals
1. Introduction
It has long been recognized that DNA is the principal target of ionizing radiation in the cell. The initial effect of ionizing radiation on DNA is to produce holes, electrons and excited states on the DNA itself, in its water of solvation, and in the bulk water surrounding the molecule. Irradiation of the bulk water produces water radicals, principally •OH, H•, and eaq−, which may diffuse to and damage nearby DNA in a process known as the indirect effect. However, owing to the scavenging of these radical species in the DNA environment the indirect effect has been estimated to contribute only ca. 50% of the radiation damage in cells. Direct-type effects, i.e., ionizations on the DNA itself and its water of solvation contribute the remaining 50% of damage.
Ionization events from direct type effects lead to the formation of DNA-cation radicals (DNA•+) and DNA-anion radicals (DNA•−) (Adhikary et al., 2012a; Adhikary et al., 2014a). Electron Spin Resonance (ESR) studies of the trapped radicals in irradiated hydrated DNA samples at low temperatures (4 K, 77 K) have established that rapid charge and spin transfer processes lead to hole localization on guanine, the base with the lowest ionization energy of the four bases (scheme 1); simultaneously, localization of the radiation-produced excess electrons occurs on the two most electron-affinic bases, thymine and cytosine (scheme 1) (Adhikary et al., 2014a; Becker et al., 2007; Bernhard, 2009; Close, 2008; Sagstuen and Hole, 2009). Before thermalizing and localization on the bases, a population of the radiation-produced excess electrons known as low energy electrons (LEE) have been shown to induce direct bond cleavage via dissociative electron attachment (DEA) processes resulting in both single and double-strand breaks (Alizadeh and Sanche, 2012; Kumar and Sevilla, 2012a), which are biologically important DNA-lesions. A variety of processes induce the formation of free radical on the dexoyribose sugar moieties of the DNA (vide infra). Many radiation chemical studies have established that sugar radicals are precursors of radiation-induced prompt strand breaks. These radiation chemical effects lead, eventually, to the biological damage that follows ionizing radiation.
Scheme 1.

Radiation-induced ionization-mediated DNA-radical formation by (A) the oxidative electron-loss path and by (B) the reductive electron-gain path along with the subsequent reactions of these DNA-radicals. SSB denotes single strand break in both (A) and (B). Adapted from Adhikary et al. 2012a, with permission.
Scheme 1 illustrates the direct-type effect processes that occur in DNA irradiated at 77 K.
In this review, we provide a summary of recent results which describe the major radiation chemical induced processes from direct-type effects that lead to DNA damage. In section 1, we describe a radiation chemistry track structure model that delineates the spatial orientation of radical formation in the high linear energy transfer tracks of ion-beams. The role of LEE and possible excited states from ion-beam irradiation are explored here. In section 2, we provide a short overview of DNA•+ reactions by taking G•+ as an example and how the protonation state of DNA•+ affects these reactions. Also, the reactions of C•+ and its prototropic equlibria are examined. Lastly, ground state base-to-backbone hole transfer in a 2′-deoxycytidine derivative anti-cancer drug, gemcitabine as well as the ground and excited state reactions of 5-methyl cytosine cation radical are commented on. In section 3, theoretical studies that detail electron-induced reactions are explored including effects for LEE, presolvated electrons (epre−), and aqueous (or, solvated) electrons (eaq−). Also included are recent theoretical investigations into the ionization potentials of various sugar radicals, and some implications that arise from these potentials.
2. The Radiation Chemistry Track Structure of Ion-Beam Irradiated DNA
As ion-beam therapy for cancer becomes more prevalent and the likelihood of a manned mission to Mars moves closer to reality, the need to understand the details of the radiation chemistry that follows ion-beam irradiation of DNA becomes more pressing. According to the National Association for Proton Therapy, there are now 22 Proton-Therapy Centers now in operation in the US and 14 new centers in-development (National Association of proton therapy website, 2016). Carbon ion-beam facilities are now becoming more common around the world (Ohno, 2013). For these reasons, understanding the radiation effects of ion-beams has become a focus of considerable research around the globe.
The underlying chemical reactions that occur in cellular DNA after irradiation ultimately determine the biological effects of the radiation. The chemical processes that occur after γ-irradiation and ion-beam irradiation reflect these differences in spatial energy deposition. Therefore, characterizing ion-beam radiation chemistry track structure, in three dimensions has been a focus of our research.
This section of our review will focus on ion-beam irradiation of DNA under conditions for which direct-type effects predominate. This is accomplished by hydrating the DNA to levels at which no bulk water exists, i.e., < 14 waters/nucleotide (Swarts et al., 1992; Adhikary et al., 2014a).
2.1 The Track Structure of Ion-Beams
γ-irradiation results in sparse ionization, largely in spurs, with an average of one or two ionizations per spur in liquid water (Turner et al. 1983, Figure 1A). Spurs are formed by the cascade of electrons that results from the initial photon interactions with target molecules (Turner et al. 1983). With an ion-beam, however, energy is deposited by the ion along its path by relatively frequent interactions of the ion with target molecules, resulting in a concentrated deposition of energy along the ion path (Hatano and Mozumder, 2004; Muroya et al., 2006; Adhikary et al., 2014a). The energy deposition is characterized by the Linear Energy Transfer (LET) of the ion in the material being irradiated. The LET of an ion is an average quantity that depends on the ion velocity (energy) and charge, as well as on the properties of the material being irradiated. Collisions both with large impact parameters (glancing collisions) and with small impact parameters (knock-on collisions) occur. As a result of the ion interactions with the material, a relatively small, nearly cylindrical volume of high energy deposition exists, designated as the “core” of the ion track (LaVerne, 2004; Muroya et al., 2006). Electrons are ejected from the core, sometimes at fairly high angles from the core. These so called δ-rays, form a large penumbra, which is very similar in its radiation chemistry characteristics to a γ-irradiated target (Muroya et al., Figure 1B). As can be seen in Figure 1B, the energy deposition is quite heterogeneous, with a high energy density core and low energy density penumbra.
Figure 1.

A. Track of electron in water. Reprinted with permission from Turner et al. 1983, Radiat. Res., copyright (1983), Radiation Research Society. B. Track structure of 20Ne10+ ion (97.5 MeV/nucleon, LET = ca. 70 keV/μm) in water. Ions are generated at the bottom. The point of energy deposition in the track is represented by each dot. Reprinted with permission from Muroya et al. Radiat. Res., Copyright (2006), Radiation Research Society.
The chemical reactions that follow irradiation, are, as might be expected, spatially heterogeneous.
2.2 Radicals in Ion-Beam Irradiated DNA
Electron Spin Spectroscopy (ESR) has been used successfully in identifying a variety of radicals stabilized and trapped at 77 K in ion beam irradiated DNA. Once the location and identity of trapped radicals are known, the chemistry that leads to them can be posited (see section 3.1). Figure 2 shows the ESR spectra (77 K) of closely matched salmon sperm DNA samples irradiated with an Ar-36 beam and with γ-irradiation, also at 77 K (Becker et al., 2003). Beam descriptions and hydration levels (Γ, waters per nucleotide) are shown in the figure. It is notable that the spectrum resulting from ion-beam irradiation is qualitatively different from that of the γ-irradiated sample, even though both are at similar hydration levels and have been irradiated at quite similar dose. This indicates that the populations of radicals which are responsible for the two spectra are not the same. Each of the spectra shown in Figure 2A is a composite of the spectra from at least seven radicals. Using carefully constructed benchmark spectra for the radicals, shown in the Figure 2B, it is possible to determine what underlying radicals are responsible for the composite DNA spectrum found after irradiation (Figure 3).
Figure 2.

A. ESR spectra of argon-ion irradiated and γ-irradiated, hydrated (Γ = 13±3 D2O/nucleotide) DNA at 77 K. Adapted from Becker et al., 2003, with permission. B. Benchmark ESR spectra used to analyze argon ion-beam irradiated DNA sample spectra. Adapted from Becker et al., 2003, with permission.
Figure 3.

Radicals which are responsible for the ESR spectra shown in Figure 2.
This analysis allows for preparation of a dose-response curve (Becker et al., 2003), and from that, G-values of radical formation and k-values for radical destruction by additional radiation (Wang et al., 1993). At this point, it has not been possible to quantitate G-values for the individual sugar radicals (Figure 3) but composite values for the sum of the sugar radicals (ΣdR•=C1′• + C3′• + C5′• + C3′•dephos) have been determined.
Table 1 shows selected G-values for Argon and Krypton ion-beams as a function of LET and hydration (Becker et al. 2003, 2012). From the yields of radicals, it has been possible to develop a model of the radiation chemical track structure in irradiated DNA that includes a description of the spatial arrangement of the radicals in the track and that allows for determination of the energy partition between the core and penumbra.
Table 1.
G-values for Radicals in DNA Hydrated to ca. 12 H2O/nucleotide (Γ = ca. 12)
| Irradiation | Hydrationa (Γ) | LET (keV/μm) | G(−H)•b | C(N3)H•b | T•− b | ΣdR• b |
|---|---|---|---|---|---|---|
| γ c | 14 | 0.2 | 0.095 | 0.050 | 0.045 | 0.030 |
| Ar-36d | 12 | 350 | 0.046 | 0.036 | 0.028 | 0.021 |
| Ar-36d | 12 | 600 | 0.043 | 0.032 | 0.034 | 0.032 |
| Kr-86e | 12 | 1080 | 0.033 | 0.013 | 0.0097 | 0.037 |
| Kr-86e | 12 | 1460 | 0.031 | 0.011 | 0.0081 | 0.027 |
| Kr-86e | 12 | 2000 | 0.022 | 0.010 | 0.0058 | 0.027 |
| Kr-86e | 12 | 4000 | 0.0069 | 0.0031 | 0.0031 | 0.0075 |
H2O (or D2O)/nucleotide
G-values in μmol/J
Shukla et al., 2005
Becker et al., 2003
Becker et al., 2012
2.3 Spatial Arrangement of DNA-Radicals in Ion-Beam Track
A comparison of the G-values for γ-irradiated samples versus ion-beam irradiated samples (Table 1 and Adhikary et al. 2014a) reveals a large disparity in the yields of base radicals between the two forms of irradiation. For this comparison, it is profitable to compare the sums of the yields of base radicals. For example, for Kr-86 irradiation at LET = 1080 keV/μm, ΣG(base radicals) = 0.056 μmol/J. The same sum for γ-irradiated samples is 0.19 μmol/J. The difference between these values is most simply explained by postulating that base radicals are formed predominantly or entirely in the γ-like track penumbra. It is presumed that, in the core, the high density of ionizations results in formation of oppositely charged proximate base radicals in such that rapid recombination occurs due to Coulomb attractions; as a result, none of the base radicals stabilized and trapped at 77 K in the core. It is also possible that early holes on the bases (A•+, G•+, C•+, T•+) recombine with nearby electrons before chemical processes that would lead to stably trapped radicals have a chance to occur.
With this model, the difference in base radical yields is attributed to the partition of energy between the core and the penumbra, and only the energy (and ionizations) in the penumbra produce base radicals. This model now allows for determination of the partition of energy. If 100% of the energy were deposited in the penumbra, the G-value of base radicals would be the same as that found with γ-radiation, 0.19 μmol/J. Using Kr-86 ion beam irradiation at LET=1080 keV as an example, with a base radical G-value of 0.056 μmol/J, the actual fraction of energy in the penumbra is (0.056 μmol•J−1)/(0.19 μmol•J−1) = 0.29. The fraction in the core is the remainder, 0.71. For argon ion-beam irradiated samples, the fraction of energy in the core varied from 0.49 to 0.56, depending on Γ and on LET of the argon beam. In krypton irradiated samples, with the hydration of the DNA at 12±2 D2O/nucleotide, the fraction of energy in the core varied from 0.67 to 0.93, with LETs varying from 1080 keV/μm to 4000 keV/μm. Figure 4 shows the fraction of energy deposited in the core as a function of log(LET) for argon and krypton ion-beam irradiated DNA.
Figure 4.

Plot of log(LET), with LET in keV/μm, versus fraction energy in core for argon ion-beam irradiated DNA (triangles) and krypton ion beam irradiated DNA (squares). As expected, the percent energy in the core decreases as LET decreases. Reprinted with permission from Becker et al. Radiat. Res., Copyright (2012), Radiation Research Society.
This model now presents a picture of the ion-beam track in which there is a relatively high concentration of sugar radicals in the small volume of the track core, and, in contrast, somewhat sparsely distributed base radicals stabilized throughout the large volume of penumbra. It should be noted that theoretical calculations of the radius of the physical track core give values of a few nanometers to tens or hundreds of nanometers (Chatterjee and Holley, 1993; Nikjoo and Uehara, 2004) depending on the ion energy and LET, whereas the track penumbra has a radius measured in micrometers, thus the volume of the penumbra is considerably larger than that of the core. It should also be noted that ca. 15% of the radicals formed in the penumbra are sugar radicals, so even though sugar radical formation is concentrated in the core, it is not exclusive to the core.
2.4 Radicals Formed by LEE
Figure 5 shows the ESR spectra of two radicals formed in DNA irradiated with Argon and with Krypton ion-beams (Becker et al. 2003, 2012). Because of the hyperfine couplings from four protons, the C3′•dephos is characterized by a wide ESR spectrum of over 140 G, as shown in the Figure. The simulation shown, in which the outer line components match line components in the experimental spectrum (arrows) uses the following parameters for proton couplings: [−11, −23.4, −34] G (1 anisotropic H); 26.9 G (1H), 34.9 G (1H), 49.1 G (1H), gxx = 2.0036, gyy = 2.0023, gzz = 2.0044, linewidth = 6 G. The phosphorus-centered radical spectrum is typical for a radical of the form ROPO2•−. The simulated spectrum assumed axial symmetry and employed the following parameters: A⊥ = 775 G, A|| = 610 G, g⊥ = 2.00, g||= 2.01 and linewidth = 12 G. The origin of these radicals is suggested by the fact that LEEs are known to create strand breaks in DNA by dissociative electron attachment (DEA) (Boudaïffa et al., 2000). Thus, LEE can form both C3′•dephos and ROPO2•− by DEA at the sugar phosphate backbone (scheme 2) (Kumar and Sevilla, 2009, 2012; Adhikary et al., 2014a).
Figure 5.

A. Sugar radical spectrum isolated from the DNA ESR spectrum from argon ion beam irradiated DNA. The outer line components (arrows) closely match those of a simulated spectrum of C3′•dephos. Adapted from Becker et al., 2003, with permission. B. DNA spectrum of krypton irradiated DNA, using a 1600 G scan range to show line components from a phosphorus-centered radical. The simulated spectrum matches the experimental spectrum well. Reprinted with permission from Becker et al. Radiat. Res., Copyright (2003), Radiation Research Society.
Scheme 2.

LEE leads to strand break as well as C3′•dephos and ROPO2•− formation.
Thus, via DEA either the C3′—O bond is broken (path I, scheme 2) or the P–O bond is broken (path II, scheme 2). With path I, C3′•dephos is formed along with a diamagnetic phosphate entity. With path II, a phosphoryl radical results, along with a diamagnetic sugar based entity. For both pathways, a frank single strand break occurs as a result, in agreement with the experiments that show that LEE causes strand breaks in plasmid DNA. In experiments with both Ar and Kr beams, the approximate ratio of the C3′•dephos to ROPO2•− formed is approximately 20:1 (Becker et al., 2003, 2012; Adhikary et al., 2014a).
The yield of these LEE-induced strand break radicals in Ar and Kr ion-beam irradiated DNA is found to be ca. ten times that found in γ-irradiated samples irradiated to the same dose (Becker et al., 2003, 2012; Adhikary et al., 2014a). This suggests that the LEE reactivity is largely a core process in the ion-beam irradiated samples. Since LEEs are formed both in the core and penumbra, these results suggest that other factors, such as, excited electronic states or excited vibrational modes add to the effectiveness of LEE in the core. In this regard, we note that theoretical calculations show that stretching of the C-O bond in the sugar-phosphate backbone (as would occur in excited vibrational modes) leads to electron capture into a dissociative state leading to a strand break (Li et al., 2003, Kumar and Sevilla., 2008, 2009, Kumar and Sevilla., 2012).
2.5 Excited States in Ion-Beam Irradiated DNA
The high energy density in the ion-beam track core would be expected to lead to excited states in the core and to the possible subsequent reactions owing to these excited states (scheme 1). So far, there exists no direct experimental evidence that proves the existence of such excited state reactions from the oxidative path radiation chemistry, i.e., from reactions that originate with hole formation on the DNA. However, as pointed out in section 3.2.2 of this review, it has been well-established that sugar radicals are formed from excited state base radicals in DNA (Adhikary et al., 2005, 2014a, 2014b), and from nucleotides and oligomers as well (Adhikary et al., 2005, 2014a; Khanduri et al., 2011). Depending on the specific compounds and experimental conditions, C1′•, C3′•, and C5′• have all been observed to form from excited states of the DNA base cation radicals (Adhikary et al., 2012a, 2014a; Becker et al., 2010).
In Kr-86 irradiated DNA, the G-values for of sugar radical formation are the same at 0.026±.001 μmol/J as the LET changes from 1080 keV/μm to 2000 keV/μm (Becker et al., 2012). At 4000 keV/μm, it drops to 0.0069 μmol/J, indicating that DNA•+ (on the sugar backbone) is undergoing significant recombination with a proximate electron or DNA•− before deprotonation of DNA•+ can occur, at this higher LET. The question now arises of whether at the lower LETs, recombination is overcome to some extent by very fast deprotonation of DNA•+ (Adhikary et al., 2012b) to achieve the unchanging G-values. Further studies are required to answer this significant question.
2.6 An Ion-Beam Radiation Chemistry Track Structure Model
A full appreciation of radical formation and the consequences of that formation in heavy ion-beam irradiated DNA would demand knowledge of the radicals that are formed, the reactions that lead to them, their spatial orientation in the track, and their fate, especially with regard to the potential damage done to the cell (Adhikary et al., 2014a; Becker et al., 2012). Schemes 3 and 4 summarize this information for the core and penumbra of the ion track for argon and krypton irradiated DNA.
Scheme 3.

Penumbra reactions and radical formation/stabilization in Argon and Krypton ion-beam irradiated DNA.
Scheme 4.

Core reactions and radical formation/stabilization in Argon and Krypton ion beam irradiated DNA. Radicals shown in red recombine at some stage of their development and are not observed at 77 K.
In the penumbra (Scheme 3), the processes that occur are the same as those that occur in low LET irradiated DNA. After γ-irradiation at 77 K, oxidative and reductive paths of reaction develop as shown in scheme 1. In the oxidative path, ionization occurs at all moieties of the DNA, i.e., at all four bases and the dexoyribose sugars. Since the only electron-loss base radical that is observed at 77 K is the deprotonated guanine cation radical (as 40% of the radical population stabilized at 77 K) (Adhikary et al., 2006, 2009, 2010, 2014a; Khanduri et al., 2011), it is evident that extensive hole transfer occurs to guanine. This hole transfer is also required to explain the predominance of guanine damaged base yields (predominantly 8-oxoguanine (Swarts et al., 1996) and 8-oxo-guanine radical (Shukla et al., 2004)) observed after γ-irradiation. The three uncharged sugar radicals C1′•, C3′•, and C5′• also originate with the oxidative path, from deprotonation of deoxyribose radical cations that themselves originate from ionization (Adhikary et al., 2012b). In the reductive path, the observation of the protonated cytosine anion radical (C(N3)H•) and the pristine thymine anion radical (T•−) is evidence of electron attachment to these two bases (Adhikary et al., 2014a). The fact that cytosine and thymine have high electron affinities is consistent with this observation (Kumar and Sevilla, 2012a). The protonated cytosine anion makes up 38% of the total radical population stabilized at 77 K and the thymine anion radical, 18%. In low LET irradiated samples, and presumably in the penumbra as well, there is only a small population of radicals that result from low energy electrons (LEE). These are not inconsequential, however, for as indicated earlier, LEE cause immediate strand breaks to occur. It is pertinent to understanding the radiation chemistry of DNA to note that only ca. 30% of the ionizations that occur, end up as trapped radicals in our samples at low temperatures. Significant ion-radical recombination events (Bernhard, 2009; Adhikary et al., 2014a) account for the loss of ca. 70% of the radicals. These may be geminate or by long range electron/hole transfer.
In the model being described, the reaction paths in the track core are quite different from those in the penumbra (scheme 4 and Adhikary et al., 2014a). These reaction paths are driven by the presumption that none of the base radicals trapped at 77 K originates in the core. Thus, after ionization by an energetic ion (and the cascade of secondary electrons that result from the ionizations), the oxidative and reductive paths are initiated in a fashion that is identical to that in low LET regions. However, extensive recombinations occur that result in the elimination of base radical formation in the core (Adhikary et al. 2014a; Becker et al., 2010). The large number of ionizations in the small volume of core results in formation of proximate ionic radicals (or electrons) of opposite charge, and strong Coulomb attractions result in extensive recombination. However, in the oxidative path, a population of deoxyribose cation radicals escape recombination and eventually deprotonate to form the neutral C1′•, C3′•, and C5′• that are observed in relatively high concentration (relative to base radicals) in ion-beam irradiated samples. It is possible that this process is facilitated by excited states in which deprotonation occurs rapidly (Adhikary et al., 2014a; Khanduri et al., 2011) before recombinations can occur, even in the densely ionized core.
Another unique feature of core chemistry is suggested by the presence of the C3′•dephos and ROPO2•−, in concentrations ca. 10 times those in γ-irradiated samples (Adhikary et al., 2014a). As shown in schemes 2 and 4, these originate with LEE and dissociative electron attachment and are obviously augmented by track processes. Since these radicals are indicative of an immediate strand break, it is safe to assume that the core contains a significant population of strand breaks in a small volume core. This is especially significant near the end of the ion range, where track “pencil down” results in a small core volume.
In conclusion, the heterogeneity of energy deposition in ion beam irradiated DNA leads to a heterogeneous spatial distribution of radicals in the ion track at 77 K. Unique core processes, including fast recombinations, formation of a high yield of LEE and possible excited state reactions lead to a high number of sugar radicals in the small volume core. These radicals eventually form strand breaks at higher temperatures. LEE lead to frank strand breaks, again, in the low volume core. Track overlap as well as radicals formation in the core itself likely results in difficult to repair clustered damage sites, which may be lethal to the cell.
3. Recent experimental and theoretical studies of radiation-induced DNA-base and DNA-sugar radicals
3.1. General reactions of trapped ion-radicals (holes and anion radicals)
Experimental as well as theoretical studies have demonstrated that DNA•+ is a stronger proton donor and DNA•− is stronger proton acceptor than their parent compounds (Adhikary et al. 2014a; Kumar and Sevilla, 2010; Wagenknecht, 2005). For example, in aqueous solution and at room temperature the pKa of guanine for H+ loss from N1 is reported to be 9.6; the corresponding pKa for the guanine cation radical (G•+) is found to be 3.9 (Steenken, 1997; von Sonntag, 2006). The protonation states of G•+ have been experimentally studied employing various experimental techniques, e.g., pulse radiolysis and flash photolysis (Steenken, 1997; von Sonntag, 2006), low temperature ESR of X-ray irradiated single crystals (Bernhard, 2009; Close, 2008) and ESR of γ-irradiated homogeneous glassy solutions at 77 K (Adhikary et al., 2006; Adhikary et al., 2009; Adhikary et al., 2010; Adhikary et al., 2014a; Khanduri et al., 2011). The results obtained in aqueous solutions at ambient temperatures matched very well with those obtained in γ-irradiated homogeneous frozen glassy solutions at 77 K (Adhikary et al., 2009; Adhikary et al., 2010). Understanding the various protonation states of DNA•+ is important because reactions of DNA•+ (e.g., G•+) depend on its protonation state (Adhikary et al., 2009; Adhikary et al., 2010; Adhikary et al., 2014a). G•+ in dsDNA undergoes a variety of reactions, for example, intra-base pair proton transfer, proton transfer to the surrounding solvent, nucleophilic addition of water at C-8 of the guanine base, and sugar radical formation via excited G•+ etc. (scheme 1) (Adhikary et al., 2009; Adhikary et al., 2010; Adhikary et al., 2014a). Owing to the greatly increased lifetime of G•+ in dsDNA relative to that of a separate nucleotide cation radical, the nucleophilic addition of water at C-8 of the G•+ in dsDNA can occur before its deprotonation to the surrounding water molecules; this is evidenced by the increased yield of 8-oxo-G in high intensity laser-irradiated dsDNA relative to that in single stranded (ss) DNA or to that in the monomer (Angelov et al., 1997). Various reactions of G•+ including its prototropic equilibria (Scheme 1) in DNA as well as in model systems have recently been reviewed (Adhikary et al., 2012a; Adhikary et al., 2014a; Becker et al., 2010).
We note here that experiments and calculations employing density functional theory (DFT) show that π-type DNA-base cation radicals from a DNA base (G•+, A•+, C•+, T•+) or modified analog are almost always lower in energy than the σ-type radical (Adhikary et al., 2015; Kumar and Sevilla, 2013). However, the σ-states of C•+ and T•+ are predicted to be energetically closer to their lower-lying π-states than the σ-states of G•+ and A•+ are to their respective low lying π-states (Adhikary et al., 2015; Kumar and Sevilla, 2013).
Although cytosine (C) forms a π-cation radical (C•+) upon one-electron oxidation, prototropic equilibria of pyrimidine cation radicals – especially of C•+ are not as well-studied in comparison to those of G•+ (von Sonntag, 2006; Samson-Thibault et al., 2012; Adhikary et al. 2015). Our recent investigations on the reactions of excited one-electron oxidized pyrimidines have elucidated an area of little previous work (Adhikary et al., 2014b). Furthermore, our recent works have established that by deprotonation and by subsequent tautomerization, σ-type radicals are formed from C•+ in N1-substituted cytosine derivatives, e.g., 1-methylcytosine (1-MeC), 2′-deoxycytidine (dCyd) but not in 5-methylcytosine (5-MeC) and in its derivatives (Adhikary et al., 2014b; Adhikary et al., 2015). Below we summarize our recent studies on the reactions of C•+ and 5-MeC•+ in its ground state including its prototropic equilibria as well as on the reactions of 5-MeC•+ in its excited state.
3.2. Reactions of C•+ in its ground state
3.2.1a. Prototropic equilibria of C•+ in various cytosine derivatives
ESR and ENDOR studies on X-ray irradiated single crystal of cytosine have shown that C•+ deprotonates at N1 even at 10 K (Close, 2008; Bernhard, 2009). Owing to this very rapid deprotonation of C•+, the pKa of N1-H atom of this cation radical could not be experimentally determined (Close, 2008). However, any cytosine derivative in which the N1-H atom is substituted (e.g., 1-methylcytosine, dCyd) is appropriate as a model system of dsDNA. Employing pulse radiolysis (Naumov et al., 2001; von Sonntag, 2006) and ESR spectroscopy (Naumov et al., 2001; Close, 2013; Adhikary et al., 2015), experimental studies (Naumov et al., 2001; Close, 2013; Adhikary et al., 2015) have been performed to elucidate the prototropic equilibria of C•+ in DNA-models (Scheme 5). These works have been supported by DFT studies (Naumov et al., 2001; Close, 2013).
Scheme 5.

This scheme represents C•+ production via one-electron oxidation of cytosine derivatives and the prototropic equilibria of C•+ in these compounds. By deprotonation and subsequent tautomerization, the C•+ π-radical forms the iminyl σ-radical. The atom numbering scheme of cytosine base is presented. The relative stabilities of C(N4-H)•syn, C(N4-H)•anti and the iminyl σ-radical in kcal/mol as well as the pKa values (Close, 2013) of C•+, C(N4-H)•syn, C(N4-H)•anti are provided. The energy values were obtained with the DFT/B3LYP/6-31G* method using geometry optimized structures. Reprinted with permission from Adhikary et al. 2015, J. Phys. Chem. B, copyright (2015), American Chemical Society.
In aqueous solution near pH 7 and the site of deprotonation of C•+ in N1-substituted cytosines is at the exocyclic –NH2 group, as determined by pulse radiolysis and time-resolved ESR studies in aqueous solutions at room temperature, and by ESR studies in homogeneous glassy solutions at low temperature (von Sonntag, 2006; Close, 2013; Adhikary et al., 2015). Pulse radiolysis, time-resolved ESR, and theoretical studies have established that the pKa of C•+ in these DNA-models is ca. 4 (Close, 2013).
Deprotonation from the –NH2 group in C•+ produces the neutral π-aminyl radical, C(N4-H)• (Scheme 5). Recent ESR and theoretical studies have established that C(N4-H)•, undergoes rapid tautomerization to produce a surprisingly stable iminyl σ-radical (Scheme 5) (Adhikary et al., 2015). The iminyl σ-radical has a unique electronic structure for a DNA base radical. The radical site p-orbital at N4 is in the plane of the molecule and is perpendicular to the π-electron cloud. As a result, an anisotropic nitrogen hyperfine coupling from N4 (Axx = 0 G, Ayy = 0 G, Azz = 40 G) is observed; furthermore, the iminyl σ-radical exhibits a near-isotropic β-nitrogen hyperfine coupling of 9.7 G due to N3 (Adhikary et al., 2015). This is first observation of a ring beta nitrogen hyperfine coupling that is near-isotropic in a DNA-model system. These experimental results have been validated by theoretical studies (Adhikary et al., 2015). Our theoretical work (Adhikary et al., 2015) has further shown that the iminyl σ-radical can be produced in dsDNA by radiation-induced ionization of cytosine followed by deprotonation at its N4 site and subsequent tautomerization as described above. The resultant-iminyl σ-radical radical is only ca. 8 kcal/mol higher in energy than G•+C and 10 kcal higher than G(-H)• C(H)+.
In summary, the protonation state of C•+ has profound effects on the radical electronic structure and its subsequent reactivity.
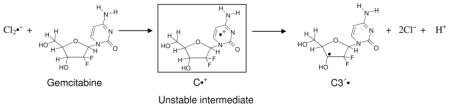
3.2.1b. Ground state base-to-backbone hole transfer processes found in gemcitabine
Gemcitabine (2′,2′-difluoro-2′-deoxycytidine), an anti-cancer drug that is widely used to treat pancreatic cancer patients, is a modified cytidine analog having two fluorine atoms in its 2′-deoxyribose moiety (reaction (1), Adhikary et al., 2014c). It has been hypothesized that inhibition of ribonucleotide reductase (RNR) activity by gemcitabine is one of the predominant processes involved in its anti-cancer activity. The radicals C3′• and C2′• produced from gemcitabine are the key intermediates that play an important role in its RNR inhibition (Adhikary et al., 2014c). However, these radicals had not been identified in gemcitabine or in its analogs in a non-enzymatic system until our recent ESR studies on one-electron oxidized gemcitabine and its various analogs such as dCyd, 2′-fluoro-2′-deoxycytidine (2′-F-dCyd), and 2′-deoxy-2′-fluoro-2′-C-methylcytidine (MeFdCyd) in which we investigated the effect of 2′-substitution on the formation of the radical site (Adhikary et al., 2014c).
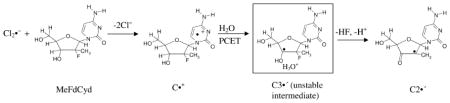
The ESR spectrum of one-electron oxidized gemcitabine at pH ca. 7 shows ESR line components owing to two anisotropic β 2′-F hyperfine couplings (Azz of ca. 105 and ca. 69 G) and one β–proton (C4′) hyperfine coupling of ca. 24 G. On increasing the pH to ca. 9 and above, the two anisotropic β 2′-F hyperfine couplings become identical (Azz of ca. 86 G) while the single β–proton (C4′) hyperfine coupling remains unchanged at ca. 24 G. DFT-predicted hyperfine couplings matched these experimental values well. Theoretical studies also demonstrate the feasibility of C3′• formation by one-electron oxidation of gemcitabine (reaction 1) (Adhikary et al., 2014c). However, the conversion of C3′• to C2′• is expected from gemcitabine studies with RNR but was not experimentally observed in one-electron oxidized gemcitabine. However, C3′• to C2′• conversion has been observed for the 2′-methyl-2′-F analog MeFdCyd (vide infra).
Based upon these results, we proposed a mechanism of C3′• formation in one-electron oxidized gemcitabine via a ground state base-to-backbone hole transfer process (reaction (1)). The first step of this mechanism involves formation of an unstable C•+ radical after one-electron oxidation of the cytosine base moiety. In gemcitabine; this intermediate is unstable even at ca. 155 K and undergoes rapid deprotonation at C3′ via a proton-coupled electron transfer (PCET) mechanism, producing C3′• (reaction (1)) (Adhikary et al., 2014c).
ESR studies show C2′• formation in one-electron oxidized MeFdCyd, unlike the results found for gemcitabine. Theoretical calculations confirm that formation of this radical is energetically favored. The mechanism suggested is that initially, on one-electron oxidation, a highly unstable C3′• intermediate is produced via rapid ground state base-to-backbone hole transfer (reactions (1) and (2)). This is followed by the subsequent rapid conversion of C3′• to C2′•, which the calculations suggest can occur via a barrierless proton-assisted HF loss in the ground state (reaction (2), Adhikary et al., 2014c). However, the fact that no experimentally observed C3′• to C2′• conversion is found in one-electron oxidized gemcitabine, is supported by DFT calculations which predict that the proton-assisted HF loss from C3′• has a low (5 kcal/mol), but still significant barrier at 155 K (Adhikary et al., 2014c). Thus, at low temperatures, C3′• formation is found from one-electron oxidized gemcitabine and C2′• is found in its derivative MeFdCyd in a homogeneous aqueous glassy system (reactions (1) and (2), Adhikary et al., 2014c).
 |
(1) |
 |
(2) |
Formation of only C•+/C(N4-H)• is observed in one-electron oxidized dCyd as well as 2′-F-dCyd (Adhikary et al., 2014c); whereas C3′• production is found in one-electron oxidation of gemcitabine and C2′• formation is observed in one-electron oxidized MeFdCyd. These results show that relatively small changes in structure exhibit significant effect on the radical chemistry, and the differences in reactivity are governed by both thermodynamic and kinetic factors.
3.3. Reactions of the 5-methylcytosine pi-cation radical (5-MeC•+) in its ground and in its excited state
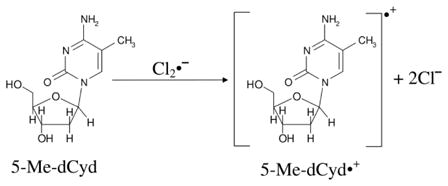
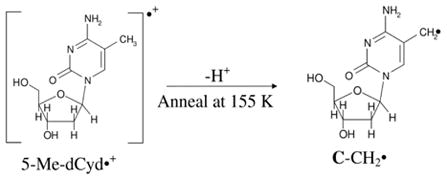
5-methylcytosine (5-MeC) is a modified cytosine base in which the C5-H atom of the pyrimidine ring is substituted by methyl group. 5-MeC is abundant in the mammalian genome; it is involved in the transcriptional regulation of genes and, thereby, is essential for normal development (Adhikary et al., 2014b). Employing ESR and theoretical calculations, we investigated production of 5-methyl-2′-deoxycytidine cation radical (5-Me-dCyd•+) and its reactions in nucleosides, in fully ds DNA-oligomers, and in dsDNA-oligomers with a 5-MeC/A mismatch (Adhikary et al., 2014b). 5-Me-dCyd•+ is produced by one-electron oxidation of 5-Me-dCyd with Cl2•− – by annealing in the dark at 150 K (reaction (3)). Raising the pH up to ca. 9 did not lead to the formation of the iminyl σ-radical, scheme 5. 5-Me-dCyd•+, when annealed further at 155 K, produced the allylic radical (C-CH2•) (reaction (4)). On the other hand, 5-Me-dCyd•+, when photoexcited, at 143 K, with a 405 nm laser, produced only C3′• (reaction (5)). In a similar manner, photoexcitation of 5-Me-2′, 3′-ddCyd•+ (here the 3′-OH is replaced by an H-atom) still produced a radical at the 3′-site, C3′dephos• (reaction (6), Adhikary et al., 2014b, also see scheme 2). These results establish that: (a) 5-Me-dCyd•+ led to C-CH2• formation owing to ground state deprotonation from C5-methyl group in the base. (b) Spin and charge localization at C3′ in the excited 5-Me-dCyd•+ ((5-Me-dCyd•+)*) and in (5-Me-2′,3′-ddCyd•+)* along with subsequent fast deprotonation from C3′ produced a C-centered radical at C3′. It is significant that substitution of the 3′-OH by an H-atom in (5-Me-2′,3′-ddCyd•+)* does not hinder deprotonation from C3′. This work concludes that sugar radical formation via an excited base cation radical is kinetically controlled and, in these deprotonation events of excited cation radicals, the sugar C-H bond energies do not determine the site of deprotonation and associated radical formation (Adhikary et al., 2014b).
 |
(3) |
 |
(4) |
One-electron oxidation of either a fully dsDNA-oligomer d[GCxGCxGCxGCx]2 (Cx = 5-MeC) or of a dsDNA-oligomer with a 5-MeC/A mismatch (d[GGACxAAGC:CCTAATCG], (Cx = 5-MeC)) results in one-electron oxidation of guanine and a fast deprotonation the guanine cation radical to a base-paired cytosine. In this manner, (G(N1-H)•:C(+H+)) is formed. In these systems, ESR studies showed no observable formation of 5-Me-dCyd•+ (Adhikary et al., 2014b). These results establish that hole trapping at guanine in a dsDNA-oligomer is not affected by substitution of C by 5-MeC. This is in keeping with the order of IPs of G < 5-MedCyd < C (Close, 2008, Adhikary et al., 2014b).
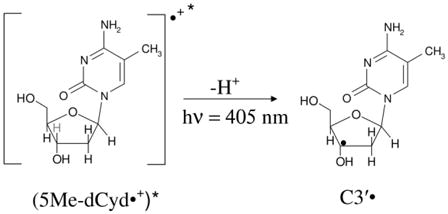 |
(5) |
Photoexcitation of (G(N1-H)•:C(+H+)) in d[GCxGCxGCxGCx]2 produced C1′• only and did not result in formation of the expected photoproducts from (5-Me-dCyd•+)* (Adhikary et al., 2014b). On the other hand, C5′• and C1′• were formed via photoexcitation of (G(N1-H)•:C(+H+)) in d[GGACxAAGC:CCTAATCG]. Since C5′• is produced from excitation of one-electron oxidized ssDNA-oligomers (Becker et al., 2010; Kahnduri et al., 2011), it is likely that the 5-MeC/A mismatched region in the excited one-electron oxidized d[GGACxAAGC:CCTAATCG] led to the C5′• formation. It is well-established in the literature that C5′• is a frank strand break precursor radical (von Sonntag, 2006). Therefore, the mismatch region should lead to a strand break formation via photolysis. Employing photolyzed oligomers with and without 5-Me-C mismatches in aqueous solutions at ambient temperature, Joseph et al. (Joseph and Schuster, 2012) showed that the mismatched regions of the oligomer act a “hotspot” for strand break formation upon hot piperidine treatment. Thus, there is substantial evidence that 5-Me-C sites are potential “mutational hot spots” in photolyzed DNA (Adhikary et al., 2014b) (Joseph and Schuster, 2012).
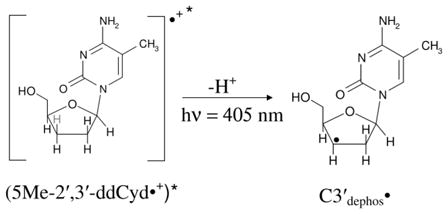 |
(6) |
4. Electron Reaction with DNA
The reaction of electrons with DNA and its constituents is of fundamental importance to DNA radiation damage (Adhikary et al., 2014a; Alizadeh and Sanche, 2012; Kumar and Sevilla, 2010, 2012a; von Sonntag, 2006). The interaction of ionizing radiation with DNA and the surrounding bulk water molecules produce “holes” (electron deficient sites), secondary electrons and excited states (scheme 1). Within 100 femtoseconds, the hole on water, H2O•+ deprotonates to form •OH and H3O+. Secondary electrons produced by the ionizing radiation lose their kinetic energy and after thermalization, are captured by the pyrimidines, T and C, owing to their high electron affinity in comparison to the purines, G and A (Sevilla et al., 1991). Secondary electrons having high kinetic energy before thermalization can lead to DNA damage (Alizadeh and Sanche, 2012; Kumar and Sevilla, 2012a). Those electrons having kinetic energy below 20 eV are recognized as low energy electrons (LEE) (Kumar and Sevilla, 2012a). As these electrons lose energy due to collisions with atoms, presolvated electrons (epre−), which are in the water conduction band (0 to −0.2 eV), are produced. On the picosecond timescale, epre− is solvated by the surrounding water molecules and leads to the formation of solvated electron (eaq−, −1.6 eV) (Mozumder, 1999; Paik, 2004; Pimblott and Laverne, 1998). The reactivity of these radiation-produced electrons (LEE, epre−, and eaq−) with DNA is discussed below, and their relative energies compared to DNA in solution are shown in Figure 6.
Figure 6.

Schematic diagram showing the addition of electrons of various energies to DNA including LEE, epre−, and eaq−. LEE can be captured into one of the UMOs (unoccupied molecular orbital) of DNA (shown as SOMO after the capture) creating the TNI. A TNI formed in liquid water is solvated quickly resulting in the adiabatic DNA anion radical. The energy of this adiabatic anion radical must be below the energy of eaq− to be stable to electron loss to water. For clarity, the MOs of DNA below the HDMO (highest doubly occupied MOs) and above the SOMO are not shown in the figure. The blue arrows surrounding epre− show four unbound water molecules while blue arrows surrounding eaq− show the binding of solvated electron (eaq−) in the cavity formed by four waters in a tetrahedral arrangement (Kumar et al., 2015).
4.1. Reactivity of radiation-produced electrons (LEE, epre−, and eaq−) with DNA and its model systems
4.1a. Low energy electron (LEE)
LEEs are produced in significant amounts (4 x 104 per MeV energy deposited) along ionizing radiation tracks and these are found to be potent DNA damaging agent (Boudaïffa et al., 2000). In fact, LEE creates more damage than the photons of similar energy. Sanche and his group first discovered that LEE can induce single- and double-strand breaks (SSB and DSB) in plasmid DNA (Boudaïffa et al., 2000). In a number of experiments, they showed that LEEs below ca. 4 eV can induce SSB in plasmid DNA through dissociative electron attachment (DEA). In this DEA process, LEEs (0 – 4 eV) initially attach to the unoccupied molecular orbitals of the neutral molecule which is termed as a “shape resonance”. This results in the formation of a transient negative ion (TNI); TNI immediately leads to a variety of chemical changes (Alizadeh and Sanche, 2012; Boudaïffa et al., 2000; Kumar and Sevilla, 2007, 2008, 2012a). Since its discovery, the LEE has been widely used as an “electron scissor” to dissociate chemical bonds in a molecule and has been studied extensively using experiment and theory (Alizadeh and Sanche, 2012; Boudaïffa et al., 2000; Gu et al., 2012; Kumar and Sevilla, 2007, 2008, 2012a; Ptasińska et al., 2006; Simons, 2006).
4.1b. Pre-hydrated electron (epre−)
In an aqueous environment, ionizing radiation initially produces epre− in the water conduction band (Figure 6). The energy of epre− lies in between ca. 0 to −0.2 eV and epre− has a lifetime of several hundred femtoseconds (ca. 100 – 550 fs) (Alizadeh and Sanche, 2012; Kumar and Sevilla, 2012a). epre− is the precursor of the solvated electron (eaq−) and has been suggested to react with DNA and other molecules to cause fragmentation.
Bond breaking in nucleotides due to epre− was first reported by Lu and coworkers (Wang et al., 2009). In their experiment, electrons were generated by water excitation using two UV photon laser pulses. They proposed that epre− thus generated were captured by the nucleotides to form a TNI which subsequently leads to strand breaks via the DEA process. Though this work is very interesting, since the solutes did not have high enough solute concentrations to scavenge extremely short lived (0.3 ps) epre−, the role of epre− is an open question. Very recently, the reactions of epre− with glycine methyl ester and N-acetylalanylalanine methyl ester have been investigated using ESR at 77 K and density functional theory (DFT) (Kheir et al., 2013). In this study, it was found that epre− adds to the peptide bonds initially at 77 K and subsequently cleaves the carboxylic ester group to produce methyl radical. In another study (Petrovici et al., 2014), the reaction of epre− with methyl acetoacetate was investigated at 77 K employing ESR and in this case, it was found that epre− added initially to the methyl acetoacetate to form an anion radical which was subsequently protonated from the water solvent.
4.1c. Solvated electron (eaq−)
Solvation of epre− by the surrounding medium (water) is completed within picoseconds of epre− formation, and solvated electrons (eaq−) are produced (Alizadeh and Sanche, 2012; Kumar and Sevilla, 2012a). The broad absorption spectrum of eaq− with a maximum absorbance around 720 nm was first reported using pulse radiolysis of irradiated water (Hart and Boag, 1962). Owing to its fundamental importance in biology, radiation chemistry, photochemistry and nanotechnology, knowledge about its physical and chemical properties using experiment and theory has been the locus of considerable debate (Bartels et al., 2005; Coe et al., 2008; Feng and Kevan, 1980; Kumar et al., 2015; Larsen et al., 2010; Siefermann et al., 2010; von Sonntag, 2006; Uhlig et al., 2012; Young and Neumark, 2012).
Based on ESR experiments on electron solvation in NaOH aqueous glasses at 77 K, Kevan and coworkers proposed a structure for the solvated electron. In their model, the solvated electron resides in a cavity surrounded by six water molecules arranged in an octahedral fashion (Feng and Kevan, 1980). Using quantum chemical calculations and molecular dynamics simulation the cavity model of the solvated electron has been elaborated in detail (Kumar et al., 2015; Shkrob, 2007; Uhlig et al., 2012; Young and Neumark, 2012). Recently, the cavity model of the solvated electron has been challenged by Larson et al. (Larsen et al., 2010) who proposed a delocalized picture of the solvated electron. In their picture the solvated electron resides in a 10 Å diameter region of enhanced water density instead of residing in a cavity void. Subsequently, the delocalized model of solvated electron of Larson et al. was found to be inadequate in reproducing certain physical observables such as the radius of gyration and the vertical detachment energy (VDE) (Jacobson and Herbert, 2011; Turi and Madarasz, 2011). Very recently, using ab initio and DFT methods incorporating the effect of solvation via the polarized continuum model, we have treated the cavity model of the solvated electron (Kumar et al., 2015). From this study, we found that the solvated electron is localized at the center of a cavity enclosed by four water molecules arranged in a tetrahedral fashion. The 4-H2O cluster model of the solvated electron is embedded in a polarized continuum dielectric produces a robust model of the solvated electron that successfully reproduced each of the experimental observations, such as, resonance Raman properties, the radius of gyration derived from the optical spectrum, the vertical detachment energy, and the thermodynamic properties. This model also successfully predicted the EPR g-factor shift and low temperature ice hyperfine couplings (Kumar et al., 2015).
In addition to the structure of eaq− (discussed above), the reaction of eaq− with DNA is crucial to radiation chemistry. Earlier pulse radiolysis experiments reported that the solvated electron was a strong reducing agent and reduced all the DNA/RNA bases with near diffusion-controlled reaction rates, with rate constants of 3.8 x 109 M−1 s−1 to 1.8 x 1010 M−1 s−1 (von Sonntag, 2006). ESR experiments using γ-irradiated frozen aqueous solutions of DNA and model systems show that the electron adds to the bases to form anionic radicals which are then protonated from the solvent water (Wang and Sevilla, 1994). This well-established experimental fact has been recently ignored by Abel and coworkers (Siefermann et al., 2010; Siefermann and Abel, 2011). They proposed that fully solvated electrons are energetically restricted from reacting with DNA bases. Their argument is based on their measurement of VDE (3.3 eV) of the solvated electron which they interpreted as the binding energy (electron affinity) of the solvated electron. The theoretical electron affinities of DNA bases lie between ca. 1.7 – 2.2 eV which is smaller than the VDE (3.3 eV) of solvated electron. The VDE and the electron affinity of the electron are two different quantities and well defined in the literature (Gu et al., 2012; Kumar et al., 2016; Kumar and Sevilla, 2010, 2012a). The VDE is the energy required to remove the electron vertically from liquid water into the gas phase, without structure relaxation, whereas, the adiabatic electron affinity (AEA) is the binding energy of the gas phase electron to the liquid water after full nuclear and electronic relaxation of the surrouinding water molecules. The VDE for water is ca. 1.8 eV larger than the AEA because the cavity remains after electron ejection for the VDE but is relaxed in the AEA. The AEA is the criteria to use to determine if the electron can react with the base.
Very recently, we used an ab initio (Gaussian-4 (G4) level calculation) and the density functional B3LYP method, including the full solvent effect via PCM, to calculate the free energy of solvation (ΔGo) of a solvated electron bound in a cavity enclosed by 4-H2O (our proposed model of the solvated electron, discussed above) (Kumar et al., 2015) and the ΔGo for eaq− binding with the DNA/RNA bases, guanine (G), adenine (A), thymine (T), cytosine (C) and uracil (U). The calculation of ΔGo directly leads to the evaluation of one-electron reduction potentials (Eo vs. NHE) of eaq− and nucleobases. The procedures for calculation of ΔGo and Eo vs. NHE are provided in our recent work (Kumar et al., 2016).
Our G4-calculated Eo vs. NHE of eaq− is −2.88 V which is in excellent agreement with the reported experimental values −2.87 V and −2.95 V, respectively (Kumar et al., 2016). The G4-calculated Eo vs. NHE of G, A, C, T and U in DMF agree with the experimental values well with a mean unsigned error (MUE) with respect to experiment is 0.22 V (Kumar et al., 2016). The G4-calculated Eo vs. NHE of G, A, C, T and U in water are −2.84 V, −2.78 V, −2.44 V, −2.42 V and −2.33 V, respectively. From a comparison of the Eo vs. NHE of nucleobases with that of eaq−, it is very clear that eaq− is able to reduce all the nucleobases and contradicts the recent proposal of Abel and coworker (Siefermann et al., 2010; Siefermann and Abel, 2011).
In addition, we also modeled the reaction of eaq− with nucleobases using an approximate ab initio molecular dynamics (MD) simulation (Kumar et al., 2016). In our MD simulations, we found that an eaq− residing in a cavity near to U, T and C is transferred within 30 fs and in adenine transfer occurs within 120 fs. For guanine, the solvated electron does not transfer until a water hydrogen-bond forms to the O6 atom of guanine (Kumar et al., 2016). Also, we note that eaq− does not lead to prompt DNA-strand breaks (Kumar and Sevilla, 2007, 2009; Nabben et al., 1982; Rezaee et al., 2013; von Sonntag, 2006). But, eaq− does cause fragmentation of halogenated nucleobases. In 5-halogenated uracil, for instance, eaq− forms a transient anion radical that results in dissociation of the carbon-halogen bond (Wieczór et al., 2014) and formation of an uracilyl radical and halogen anion. A schematic diagram of energetics and reactions of the electron in the gas phase and in solution are presented in Figure 6.
4.2. One-Electron Oxidation of Neutral Sugar Radicals
The direct-type and indirect effects of radiation can produce neutral sugar radicals (C1′•, C2′•, C3′•, C4′•, and C5′•) at each carbon site of the deoxyribose sugar in DNA (see scheme 1, sections 2 and 3). The irradiation of hydrated DNA (Γ = 12 D2O/nucleotide) by a high linear energy transfer (LET) argon ion-beam produced higher sugar radical to base radical ratio relative to that found in γ-irradiated samples (section 2).
Since these excess sugar radicals were formed in the track core, where excitations and ionizations are in proximity, it was proposed that excited-state cation radicals could be the direct precursors of the neutral sugar radicals (Becker et al., 2003). Based on this hypothesis, the radical cations of nucleosides, nucleotides, DNA and RNA oligomers were excited using UV–visible light and found to produce neutral sugar radicals, which were further characterized by the ESR experiment as the C1′•, C3′•, and C5′• (Adhikary et al., 2005, 2012a, 2014a; Becker et al., 2010).
In the indirect effect, the •OH produced by ionization of surrounding water molecules may diffuse to the DNA, where they react with the DNA with near diffusion controlled rates. Most •OH radicals add to the unsaturated bonds in the DNA bases (von Sonntag, 2006). However, the data from model systems (nucleosides, nucleotides, and poly (U)) suggest that a few (ca. <7%) of the •OH radicals attack the sugar backbone and efficiently abstracts hydrogen atoms to produce sugar radicals. The rate and extent of the hydrogen abstraction by •OH from different sites of the sugar ring depends on the accessibility of the hydrogen atoms to the solvent. The proposed order for reaction is, H5′ > H4′ > H3′ ≈ H2′ ≈ H1′ (Balasubramanian et al., 1998). In contrast, for oxidatively damaged DNA, C1′• was found to be preferred (Roginskaya et al., 2005a, 2005b; Xue and Greenberg, 2007). In general, the relative stability of neutral sugar radicals, calculated using theory, follow the order C1′• > C4′• > C5′• > C3′• > C2′• (Kumar et al., 2012b; Li et al., 2006; Miaskiewicz and Osman, 1994).
The neutral sugar radicals are highly reactive and are known to cause DNA/RNA damage such as strand breaks, unaltered base release, and C5′-C8-cyclization (von Sonntag, 2006). Experimentally, it has also been found that neutral sugar radicals are the locus for further reaction with O2 which reacts with C1′• by one-electron oxidation to produce a carbocation, which then reacts with water resulting in base release and the formation of 2′-deoxyribonolactone (von Sonntag, 2006). Thus, the neutral sugar radical formation (first oxidation) and subsequently its one-electron oxidation (second oxidation) is overall a “double oxidation” process. These double oxidized sugars (C1′+, C2′+, C3′+, C4′+, C5′+) are non-radical carbocations and may cause DNA strand breaks as proposed by Bernhard and coworkers (Bernhard, 2009). Therefore, it is obvious that the values of the ionization potentials (IP) of the neutral sugar radicals are indicative of their reactivity. In this context, we used B3LYP/6-31++G(d) and ωB97x/6-31++G(d) density functional methods and calculated the vertical and adiabatic ionization potentials of neutral sugar radicals of 2′-deoxyguanosine (dGuo) and thymidine (Thd), in the gas phase and in water, considering the effect of full aqueous phase via the polarized continuum model with ε = 78. The outcome of our study is described below.
4.2a. Ionization potential
The calculated vertical and adiabatic ionization potentials (IPvert and IPadia) of various sugar radicals of dGuo and Thd are presented in Table 2. From Table 2, it is evident that C1′• from dGuo and Thd have the lowest IP (vertical and adiabatic) in the gas phase as well as in solution, and the C2′• radicals have the highest IP as predicted by both methods (B3LYP/6-31++G(d) and ωB97x/6-31++G(d)). From these IP values, we infer that the C1′• of the sugar ring is easiest to oxidize while the C2′• is the most difficult to oxidize. The order of the vertical IPs, calculated for different sites of the sugar radicals of dGuo in the gas phase, is C1′• < C4′• < C3′• < C5′• < C2′•, while, in the case of Thd, the vertical IPs in the gas phase are in the order C1′• < C3′• < C4′• < C5′• < C2′•. The B3LYP and ωB97x calculated gas phase IP of C2′• and of dGuo lie in the range 8.02 eV – 8.72 eV which is comparable to the calculated gas phase IP (7.99 – 8.14 eV) of guanine, however, for Thd the gas phase IP of C2′• is lower than the thymine base by ca. 0.2 –0.5 eV (Kumar et al., 2012b).
Table 2.
B3LYP/6-31++G(d) and ωB97×/6-31++G(d) calculated vertical and adiabatic ionization potentials (IPvert and IPadia) in eV of neutral sugar radicals of dGuo and Thd in gas phase and in aqueous solution (Kumar et al., 2012b).
| Sugar radicals | B3LYP/6-31++G(d) | ωB97×/6-31++G(d) | ||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Gas phase | aqueous phasea | gas phase | aqueous phasea | |||||
|
| ||||||||
| IPvert | IPadia | IPvert | IPadia | IPvert | IPadia | IPvert | IPadia | |
| dGuo | ||||||||
| C1′• | 6.33 | 5.34 | 4.71 | 3.82 | 6.56 | 5.33 | 4.97 | 3.83 |
| C2′• | 8.02b | c | 6.41 | c | 8.71b | c | 6.75 | C |
| C3′• | 7.28 | 6.44 | 5.29 | 4.31 | 7.65 | 6.47 | 5.43 | 4.29 |
| C4′• | 7.17 | c | 5.31 | 4.33 | 7.44 | c | 5.43 | 4.33 |
| C5′• | 7.50 | 6.86 | 5.44 | 4.63 | 7.97 | 6.84 | 5.52 | 4.67 |
| Thd | ||||||||
| C1′• | 6.59 | 5.60 | 4.83 | 3.85 | 6.90 | 5.60 | 5.20 | 3.85 |
| C2′• | 8.43 | 6.50 | 8.83 | 6.69 | ||||
| C3′• | 7.38 | 6.56 | 5.26 | 4.37 | 7.70 | 6.22 | 5.38 | 4.34 |
| C4′• | 7.65 | 5.42 | 7.94 | 5.53 | 4.44 | |||
| C5′• | 7.77 | 5.32 | 4.55 | 8.07 | 5.43 | 4.61 | ||
Calculated using IEFPCM model with ε = 78.38.
Restricted open shell B3LYP/6-31++G(d) and ω 97×/6-31++G(d) calculated values are 7.98 and 8.66 eV, respectively.
Optimized structures underwent significant rearrangements that did not correspond to their parent sugar radical structure, see supporting information of reference (Kumar et al., 2012b).
4.2b. 1′, 2′-Hydride Shift of C1′-H to C2′ in C2′+ Singlet State
C2′• has the highest IP among all the sugar radicals (see, Table 1) and the formation of C2′+ from one-electron oxidation of C2′• is experimentally found to readily occur. C2′+ has been generated from the photoreaction of 2′-iododeoxyuridine which then underwent a 1′,2′-hydride shift of C1′-H to C2′ to produce C1′+ (Sugiyama et al., 1995). This experimental observation was supported by our calculation of the energy of C2′+ within dGuo sing the ω 97x/6-31++G(d) method with PCM. Our calculation showed that geometry optimization of C2′+ proceeds on a barrierless surface to produce C1′+ by transferring a hydrogen from C1′ to C2′, see Figure 2d in reference Kumar et al., 2012b.
4.2c. Cyclization of C5′• and C8 and spin transfer
Experimentally, it has been found that the C5′• of dAdo or dGuo forms a bond with the C8 atom with a cyclization rate constant of 1.6 × 105 s−1 (Boussicault et al., 2008; Huang et al., 2011; Romieu et al., 1999). The C5′-C8 cycloguanine structure exists in two diastereoisomeric forms: (i) 5′(S),8-cyclo-2′-deoxyguanosin-7-yl and (ii) 5′ (R),8-cyclo-2′-deoxyguanosin-7-yl. Using B3LYP/6-31++G(d) and ωB97x/6-31++G(d) methods, we optimized the C5′-C8-cycloguanine structures in these two diastereoisomeric forms in their neutral radical and in their oxidized cationic states. Our calculations showed that the spin density, which is localized on the C5′ site of the sugar ring before cyclization transfers to the guanine base after the cyclization, see Figure 7. The calculated IPadia values of these two isomers in the gas phase and in solution lie in the range ca. 4.3–6.4 eV which is lower than the corresponding IPadia values of canonical DNA bases. Thus, from the IPadia values, it is evident that these cyclic radical structures in DNA would be the preferred site for one-electron oxidation compared to the unreacted DNA bases (Kumar et al., 2012b). Oxidation of this species may result in the cyclized 5′,8-cyclo-dGuo molecular product found in irradiated DNA.
Figure 7.

B3LYP/6-31++G(d) calculated spin density distribution in C5′•, and the C5′-C8 cyclic structures of 2′-doxyguanosine (Kumar et al., 2012b).
5. Conclusion
This short review discusses our recent investigations into the study of the mechanisms involved in DNA-radical formation via direct-type effects by γ- (low LET) and ion-beam radiation. These DNA-base and sugar radicals, through subsequent reactions, lead to the formation of stable DNA damage products. This comparative work between γ- and ion-beam radiation-induced DNA-damage has elucidated the radiation chemical track structure model for ion-beam radiations. This model includes contributions of physicochemical reactions occurring in both core and penumbra regions of the ion-beam track, and our studies provide support for the concept that penumbra-driven processes in an ion-beam radiation track resembles those in γ-radiation. Our work aids our understanding of processes after the initial ionization event in which electrons (LEE, epre−, and eaq−), the hole ground and excited states and their prototropic processes play important roles in the production of DNA damage including mutagenic lesions, strand breaks and release of unaltered bases.
Highlights.
The Radiation Chemistry Track Structure of Ion-Beam Irradiated DNA.
Recent experimental and theoretical studies of radiation-induced DNA-base and DNA-sugar radicals.
Electron Reactions with DNA including low energy electrons (LEE), prehydrated and solvated electrons.
Acknowledgments
The authors thank the National Cancer Institute of the National Institutes of Health (Grant RO1CA045424) for support. We also thank and gratefully acknowledge Dr. Reginald Ronningen, Dr. R. Anantaraman, Dr. Thomas Baumann, and the laboratory staff of the National Superconducting Cyclotron Laboratory at Michigan State University for their assistance with the heavy-ion-beam irradiations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adhikary A, Becker D, Palmer BJ, Heizer AN, Sevilla MD. Direct Formation of The C5′-radical in The Sugar-phosphate Backbone of DNA By High Energy Radiation. J Phys Chem B. 2012b;116:5900–5906. doi: 10.1021/jp3023919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A, Becker D, Sevilla MD. Electron Spin Resonance of Radicals in Irradiated DNA. In: Lund A, Shiotani M, editors. Applications of EPR in radiation research. Springer-Verlag; Berlin, Heidelberg: 2014a. pp. 299–352. [Google Scholar]
- Adhikary A, Khanduri D, Sevilla MD. Direct observation of the protonation state and hole localization site in DNA-oligomers. J Am Chem Soc. 2009;131:8614–8619. doi: 10.1021/ja9014869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A, Kumar A, Becker D, Sevilla MD. The Guanine Cation Radical: Investigation of Deprotonation States by ESR and DFT. J Phys Chem B. 2006;110:24171–24180. doi: 10.1021/jp064361y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A, Kumar A, Becker D, Sevilla MD. Understanding DNA Radicals Employing Theory and Electron Spin Resonance Spectroscopy. In: Chatgilialoglu C, Struder A, editors. Encyclopedia of Radicals in Chemistry, Biology and Materials. John Wiley & Sons Ltd; Chichester, UK: 2012a. pp. 1371–1396. [Google Scholar]
- Adhikary A, Kumar A, Bishop CT, Wiegand TJ, Hindi RM, Adhikary A, Sevilla MD. π-Radical to σ-Radical Tautomerization in One-Electron-Oxidized 1-Methylcytosine and Its Analogs. J Phys Chem B. 2015;119:11496–11505. doi: 10.1021/acs.jpcb.5b05162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A, Kumar A, Munafo SA, Khanduri D, Sevilla MD. Prototropic Equilibria in DNA Containing One-electron Oxidized GC: Intra-duplex vs. Duplex to Solvent Deprotonation. Phys Chem Chem Phys. 2010;12:5353–5368. doi: 10.1039/B925496J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A, Kumar A, Palmer BJ, Todd AD, Heizer AN, Sevilla MD. Reactions of 5-methylcytosine cation radicals in DNA and model systems: thermal deprotonation from the 5-methyl group vs. excited state deprotonation from sugar. In: Adhikary A, Cadet J, O’Neill P, editors. Int J Radiat Biol. Vol. 90. 2014b. pp. 433–445. Clemens von Sonntag Memorial issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A, Kumar A, Palmer BJ, Todd AD, Sevilla MD. Formation of S-C1 Phosphorothioate Adduct Radicals in dsDNA-S-oligomers: Hole Transfer to Guanine vs. Disulfide Anion Radical Formation J Am Chem Soc. 2013;135:12827–12838. doi: 10.1021/ja406121x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A, Kumar A, Rayala R, Hindi RM, Adhikary A, Wnuk SF, Sevilla MD. One-electron oxidation of Gemcitabine and analogs: Mechanism of formation of C3′ and C2′ sugar radicals. J Am Chem Soc. 2014c;136:15646–15653. doi: 10.1021/ja5083156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary A, Malkhasian AYS, Collins S, Koppen J, Becker D, Sevilla MD. UVA-Visible Photo-Excitation of Guanine Radical Cations Produces Sugar Radicals in DNA and Model Structures. Nucleic Acids Res. 2005;33:5553–5564. doi: 10.1093/nar/gki857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh E, Sanche L. Precursors of Solvated Electrons in Radiobiological Physics and Chemistry. Chem Rev. 2012;112:5578–5602. doi: 10.1021/cr300063r. [DOI] [PubMed] [Google Scholar]
- Angelov D, Spassky A, Berger M, Cadet J. High-intensity UV laser photolysis of DNA and purine 20-deoxyribonucleosides: formation of 8-oxopurine damage and oligonucleotide strand cleavage as revealed by HPLC and gel electrophoresis studies. J Am Chem Soc. 1997;119:11373–11380. doi: 10.1021/ja971728r. [DOI] [Google Scholar]
- Balasubramanian B, Pogozelski WK, Tullius TD. DNA strand breaking by the hydroxyl radical is governed by the accessible surface areas of the hydrogen atoms of the DNA backbone. Proc Natl Acad Sci U S A. 1998;95:9738–9743. doi: 10.1073/pnas.95.17.9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels DM, Takahashi K, Cline JA, Marin TW, Jonah CD. Pulse Radiolysis of Supercritical Water. 3 Spectrum and Thermodynamics of the Hydrated Electron. J Phys Chem A. 2005;109:1299–1307. doi: 10.1021/jp0457141. [DOI] [PubMed] [Google Scholar]
- Becker D, Adhikary A, Sevilla MD. The Role of Charge and Spin Migration in DNA Radiation Damage. In: Chakraborty T, editor. Charge Migration in DNA: Physics, Chemistry and Biology Perspectives. Springer-Verlag; Berlin, Heidelberg, New York: 2007. pp. 139–175. [Google Scholar]
- Becker D, Adhikary A, Sevilla MD. Physicochemical mechanisms of radiation induced DNA damage. In: Hatano Y, Katsumura Y, Mozumder A, editors. Charged Particle and Photon Interactions with Matter - Recent Advances, Applications, and Interfaces. CRC Press, Taylor & Francis Group; Boca Raton, London, New York: 2010. pp. 503–541. [Google Scholar]
- Becker D, Adhikary A, Tetteh ST, Bull AW, Sevilla MD. Kr-86 Ion-Beam Irradiation of Hydrated DNA: Free Radical and Unaltered Base Yields. Radiat Res. 2012;178:524–537. doi: 10.1667/RR3066.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker D, Bryant-Friedrich A, Trzasko C, Sevilla MD. Electron spin resonance study of DNA irradiated with an argon-ion beam: Evidence for formation of sugar phosphate backbone radicals. Radiat Res. 2003;160:174–185. doi: 10.1667/rr3037. [DOI] [PubMed] [Google Scholar]
- Bernhard WA. Radical Reaction Pathways Initiated by Direct Energy Deposition in DNA by Ionizing Radiation. In: Greenberg MM, editor. Radical and Radical Ion Reactivity in Nucleic Acid Chemistry. John Wiley & Sons, Inc; New Jersey: 2009. pp. 41–68. [Google Scholar]
- Boudaïffa B, Cloutier P, Hunting D, Huels MA, Sanche L. Resonant formation of DNA strand breaks by low-energy (3 to 20 eV) electrons. Science. 2000;287:1658–1660. doi: 10.1126/science.287.5458.1658. [DOI] [PubMed] [Google Scholar]
- Boussicault F, Kaloudis P, Caminal C, Mulazzani QG, Chatgilialoglu C. The Fate of C5′ Radicals of Purine Nucleosides under Oxidative Conditions. J Am Chem Soc. 2008;130:8377–8385. doi: 10.1021/ja800763j. [DOI] [PubMed] [Google Scholar]
- Chatterjee A, Holley WR. Computer-simulation of Initial Events in the Biochemical-mechanisms of DNA-damage. Adv Radiat Biol. 1993;17:181–226. doi: 10.1016/b978-0-12-035417-7.50007-6. [DOI] [PubMed] [Google Scholar]
- Close DM. From the Primary Radiation Induced Radicals in DNA Constituents to Strand Breaks: Low Temperature EPR/ENDOR studies. In: Shukla MK, Leszczynski J, editors. Radiation-induced Molecular Phenomena in Nucleic Acids: A Comprehensive Theoretical and Experimental Analysis. Springer-Verlag; Berlin, Heidelberg, New York: 2008. pp. 493–529. [Google Scholar]
- Close DM. Calculated pKa’s of the DNA ase Radical Ions. J Phys Chem A. 2013;117:473–480. doi: 10.1021/jp310049b. [DOI] [PubMed] [Google Scholar]
- Coe JV, Williams SM, Bowen KH. Photoelectron spectra of hydrated electron clusters vs. cluster size: connecting to bulk. Int Rev Phys Chem. 2008;27:27–51. doi: 10.1080/01442350701783543. [DOI] [Google Scholar]
- Feng DF, Kevan L. Theoretical models for solvated electrons. Chem Rev. 1980;80:1–20. doi: 10.1021/cr60323a001. [DOI] [Google Scholar]
- Gu J, Leszczynski J, Schaefer HF. Interactions of Electrons with Bare and Hydrated Biomolecules: From Nucleic Acid Bases to DNA Segments. Chem Rev. 2012;112:5603–5640. doi: 10.1021/cr3000219. [DOI] [PubMed] [Google Scholar]
- Hart EJ, Boag JW. Absorption Spectrum of the Hydrated Electron in Water and in Aqueous Solutions. J Am Chem Soc. 1962;84:4090–4095. doi: 10.1021/ja00880a025. [DOI] [Google Scholar]
- Huang H, Das RS, Basu AK, Stone MP. Structure of (5′ S)-8,5′-Cyclo-2′-deoxyguanosine in DNA. J Am Chem Soc. 2011;133:20357–20368. doi: 10.1021/ja207407n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson LD, Herbert JM. Comment on “Does the Hydrated Electron Occupy a Cavity? Science. 2011;331:1387–1387. doi: 10.1126/science.1198191. [DOI] [PubMed] [Google Scholar]
- Joseph J, Schuster GB. Oxidatively generated damage to DNA at 5-methylcytosine mispairs. Photochem Photobiol Sci. 2012;11:998–1003. doi: 10.1039/c2pp05379a. [DOI] [PubMed] [Google Scholar]
- Khanduri D, Adhikary A, Sevilla MD. Highly Oxidizing Excited States of One-Electron Oxidized Guanine in DNA: Wavelength and pH Dependence. J Am Chem Soc. 2011;133:4527–4537. doi: 10.1021/ja110499a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheir J, Chomicz L, Engle A, Rak J, Sevilla MD. Presolvated Low Energy Electron Attachment to Peptide Methyl Esters in Aqueous Solution: C–O Bond Cleavage at 77 K. J Phys Chem B. 2013;117:2872–2877. doi: 10.1021/jp400176c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Adhikary A, Shamoun L, Sevilla MD. Do Solvated Electrons (e aq–) Reduce DNA Bases? A Gaussian 4 and Density Functional Theory- Molecular Dynamics Study. J Phys Chem B. 2016;120:2115–2123. doi: 10.1021/acs.jpcb.5b11269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Pottiboyina V, Sevilla MD. One-Electron Oxidation of Neutral Sugar Radicals of 2′-Deoxyguanosine and 2′-Deoxythymidine: A Density Functional Theory (DFT) Study. J Phys Chem B. 2012b;116:9409–9416. doi: 10.1021/jp3059068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Sevilla MD. Low-Energy Electron Attachment to 5′-Thymidine Monophosphate: Modeling Single Strand Breaks Through Dissociative Electron Attachment. J Phys Chem B. 2007;111:5464–5474. doi: 10.1021/jp070800x. [DOI] [PubMed] [Google Scholar]
- Kumar A, Sevilla MD. The Role of πσ* Excited States in Electron-Induced DNA Strand Break Formation: A Time-Dependent Density Functional Theory Study. J Am Chem Soc. 2008;130:2130–2131. doi: 10.1021/ja077331x. [DOI] [PubMed] [Google Scholar]
- Kumar A, Sevilla MD. Role of Excited States in Low-Energy Electron (LEE) Induced Strand Breaks in DNA Model Systems: Influence of Aqueous Environment. ChemPhysChem. 2009;10:1426–1430. doi: 10.1002/cphc.200900025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Sevilla MD. Proton-Coupled Electron Transfer in DNA on Formation of Radiation-Produced Ion Radicals. Chem Rev. 2010;110:7002–7023. doi: 10.1021/cr100023g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Sevilla MD. Handbook of Computational Chemistry. Springer; 2012a. Low-Energy Electron (LEE)-Induced DNA Damage: Theoretical Approaches to Modeling Experiment; pp. 1215–1256. [Google Scholar]
- Kumar A, Sevilla MD. π- vs. σ-Radical States of One-electron-oxidized DNA/RNA Bases: A Density Functional Theory Study. J Phys Chem B. 2013;117:11623–11632. doi: 10.1021/jp407897n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Walker JA, Bartels DM, Sevilla MD. A Simple ab Initio Model for the Hydrated Electron That Matches Experiment. J Phys Chem A. 2015;119:9148–9159. doi: 10.1021/acs.jpca.5b04721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen RE, Glover WJ, Schwartz BJ. Does the Hydrated Electron Occupy a Cavity? Science. 2010;329:65–69. doi: 10.1126/science.1189588. [DOI] [PubMed] [Google Scholar]
- LaVerne J. Radiation Chemical Effects of Heavy Ions. In: Hatano Y, Mozumder A, editors. Charged Particle and Photon Interactions with Matter: Chemical, Physicochemical, and Biological Consequences with Applications. Macel Dekkar; New York: 2004. pp. 403–429. [Google Scholar]
- Li MJ, Liu L, Wei K, Fu Y, Guo QX. Significant Effects of Phosphorylation on Relative Stabilities of DNA and RNA Sugar Radicals: Remarkably High Susceptibility of H-2′ Abstraction in RNA J. Phys. Chem B. 2006;110:13582–13589. doi: 10.1021/jp060331j. [DOI] [PubMed] [Google Scholar]
- Li X, Sevilla MD, Sanche L. Can zero eV electrons induce Strand Breaks? J Am Chem Soc. 2003;125:13668–13669. doi: 10.1021/ja036509m. [DOI] [PubMed] [Google Scholar]
- Miaskiewicz K, Osman R. Theoretical study on the deoxyribose radicals formed by hydrogen abstraction. J Am Chem Soc. 1994;116:232–238. doi: 10.1021/ja00080a027. [DOI] [Google Scholar]
- Mozumder A. Fundamentals of radiation chemistry. Academic Press; San Diego: 1999. [Google Scholar]
- Muroya Y, Plante I, Azzam EI, Meesungnoen J, Katsumura Y, Jay-Gerin JP. High-LET Ion Radiolysis of Water: Visualization of the Formation And Evolution of Ion Tracks And Relevance to The Radiation-induced By-stander Effect. Radiat Res. 2006;165:485–491. doi: 10.1667/rr3540.1. [DOI] [PubMed] [Google Scholar]
- Nabben FJ, Karman JP, Loman H. Inactivation of Biologically Active DNA by Hydrated Electrons. Int J Radiat Biol. 1982;42:23–30. doi: 10.1080/09553008214550881. [DOI] [PubMed] [Google Scholar]
- National Association of proton therapy website. ( http://www.proton-therapy.org/), as on 11th April 2016.
- Naumov S, Hildenbrand K, von Sonntag C. Tautomers of the N-centered Radical Generated by Reaction of SO4•– With N1-substituted Cytosines in Aqueous Solution. Calculation of Isotropic Hyperfine Coupling Constants by a Density Functional Method. J Chem Soc, Perkin Trans. 2001;2:1648–1653. [Google Scholar]
- Nikjoo H, Uehara S. Track Structure Studies of Biological Systems. In: Hatano Y, Mozumder A, editors. Charged Particle and Photon Interactions with Matter Chemical, Physico-chemical and Biological Consequences with Applications. Macel Dekkar; New York: 2004. pp. 491–531. [Google Scholar]
- Ohno T. Particle radiotherapy with carbon ion beams. EPMA J. 2013;4:9. doi: 10.1186/1878-5085-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik DH. Electrons in Finite-Sized Water Cavities: Hydration Dynamics Observed in Real Time. Science. 2004;306:672–675. doi: 10.1126/science.1102827. [DOI] [PubMed] [Google Scholar]
- Petrovici A, Adhikary A, Kumar A, Sevilla M. Presolvated Electron Reactions with Methyl Acetoacetate: Electron Localization, Proton-Deuteron Exchange, and H-Atom Abstraction. Molecules. 2014;19:13486–13497. doi: 10.3390/molecules190913486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimblott SM, LaVerne JA. On the Radiation Chemical Kinetics of the Precursor to the Hydrated Electron. J Phys Chem A. 1998;102:2967–2975. doi: 10.1021/jp980496v. [DOI] [Google Scholar]
- Ptasińska S, Denifl S, Gohlke S, Scheier P, Illenberger E, Märk TD. Decomposition of Thymidine by Low-Energy Electrons: Implications for the Molecular Mechanisms of Single Strand Breaks in DNA. Angew Chem Int Ed. 2006;45:1893–1896. doi: 10.1002/anie.200503930. [DOI] [PubMed] [Google Scholar]
- Rezaee M, Sanche L, Hunting DJ. Cisplatin Enhances the Formation of DNA Single-and Double-Strand Breaks by Hydrated Electrons and Hydroxyl Radicals. Radiat Res. 2013;179:323–331. doi: 10.1667/RR3185.1. [DOI] [PubMed] [Google Scholar]
- Roginskaya M, Bernhard WA, Marion RT, Razskazovskiy Y. The release of 5-methylene-2-furanone from irradiated DNA catalyzed by cationic polyamines and divalent metal cations. Radiat Res. 2005a;163:85–89. doi: 10.1667/rr3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roginskaya M, Razskazovskiy Y, Bernhard WA. 2-Deoxyribonolactone Lesions in X-ray-Irradiated DNA: Quantitative Determination by Catalytic 5-Methylene-2-furanone Release. Angew Chem Int Ed. 2005b;44:6210–6213. doi: 10.1002/anie.200501956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romieu A, Gasparutto D, Cadet J. Synthesis and Characterization of Oligonucleotides Containing 5′,8-Cyclopurine-2′-Deoxyribonucleosides: (5′ R)-5′,8-Cyclo-2′-deoxyadenosine, (5′ S)-5′,8-Cyclo-2′-deoxyguanosine, and (5′R)-5′,8-Cyclo-2′-deoxyguanosine. Chem Res Toxicol. 1999;12:412–421. doi: 10.1021/tx9802668. [DOI] [PubMed] [Google Scholar]
- Sagstuen E, Hole EO. Radiation Produced Radicals. In: Brustolon Giamello., editor. Electron Paramagnetic Resonance. John Wiley & Sons, Inc; New Jersey: 2009. pp. 325–381. [Google Scholar]
- Samson-Thibault F, Madugundu GS, Gao S, Cadet J, Wagner JR. Profiling Cytosine Oxidation in DNA by LC-MS/MS. Chem Res Toxicol. 2012;25:1902–1911. doi: 10.1021/tx300195f. [DOI] [PubMed] [Google Scholar]
- Sevilla MD, Becker D, Yan M, Summerfield SR. Relative abundances of primary ion radicals in gamma.-irradiated DNA: cytosine vs thymine anions and guanine vs adenine cations. J Phys Chem. 1991;95:3409–3415. doi: 10.1021/j100161a080. [DOI] [Google Scholar]
- Shkrob IA. The Structure of the Hydrated Electron. Part 1 Magnetic Resonance of Internally Trapping Water Anions: A Density Functional Theory Study. J Phys Chem A. 2007;111:5223–5231. doi: 10.1021/jp068278m. [DOI] [PubMed] [Google Scholar]
- Shukla LI, Adhikary A, Pazdro R, Becker D, Sevilla MD. Formation of 8-oxo-7,8-dihydroguanine-Radicals in γ–irradiated DNA by Multiple One-Electron Oxidations. Nucleic Acids Res. 2004;32:6565–6574. doi: 10.1093/nar/gkh989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla LI, Pazdro R, Becker D, Sevilla MD. Sugar Radicals in DNA: Isolation of Neutral Radicals in Gamma-Irradiated DNA by Hole and Electron Scavenging. Radiat Res. 2005;163:591–602. doi: 10.1667/rr3347. [DOI] [PubMed] [Google Scholar]
- Siefermann KR, Abel B. The Hydrated Electron: A Seemingly Familiar Chemical and Biological Transient. Angew Chem Int Ed. 2011;50:5264–5272. doi: 10.1002/anie.201006521. [DOI] [PubMed] [Google Scholar]
- Siefermann KR, Liu Y, Lugovoy E, Link O, Faubel M, Buck U, Winter B, Abel B. Binding energies, lifetimes and implications of bulk and interface solvated electrons in water. Nat Chem. 2010;2:274–279. doi: 10.1038/nchem.580. [DOI] [PubMed] [Google Scholar]
- Simons J. How Do Low-Energy (0.1–2 eV) Electrons Cause DNA-Strand Breaks? Acc Chem Res. 2006;39:772–779. doi: 10.1021/ar0680769. [DOI] [PubMed] [Google Scholar]
- Steenken S. Electron Transfer in DNA? Competition by Ultra-Fast Proton Transfer? Biol Chem. 1997;378:1293–1297. [PubMed] [Google Scholar]
- Sugiyama H, Fujimoto K, Saito I. Stereospecific 1,2-Hydride Shift in Ribonolactone Formation in the Photoreaction of 2′-Iododeoxyuridine. J Am Chem Soc. 1995;117:2945–2946. doi: 10.1021/ja00115a037. [DOI] [Google Scholar]
- Swarts SG, Becker D, Sevilla MD, Wheeler KT. Radiation-induced DNA damage as a function of hydration. II Base damage from electron-loss centers. Radiat Res. 1996;145:304–314. [PubMed] [Google Scholar]
- Swarts SG, Sevilla MD, Becker D, Tokar CJ, Wheeler KT. Radiation-induced DNA damage as a function of hydration. I Release of unaltered bases. Radiat Res. 1992;129:333–344. [PubMed] [Google Scholar]
- Turi L, Madarasz A. Comment on “Does the Hydrated Electron Occupy a Cavity? Science. 2011;331:1387–1387. doi: 10.1126/science.1197559. [DOI] [PubMed] [Google Scholar]
- Turner JE, Magee JL, Wright HA, Chatterjee A, Hamm RN, Ritchie RH. Physical and Chemical Development of Electron Tracks in Liquid Water. Radiat Res. 1983;96:437–449. [Google Scholar]
- Uhlig F, Marsalek O, Jungwirth P. Unraveling the Complex Nature of the Hydrated Electron. J Phys Chem Lett. 2012;3:3071–3075. doi: 10.1021/jz301449f. [DOI] [PubMed] [Google Scholar]
- von Sonntag C. Free-radical-induced DNA Damage and Its Repair. Springer-Verlag; Berlin, Heidelberg: 2006. pp. 213–482. [Google Scholar]
- Wagenknecht H-A, editor. Charge Transfer in DNA: From Mechanism to Application. Willey-VCH Verlag GmbH & Co. KGaA; Weiheim: 2005. [Google Scholar]
- Wang CR, Nguyen J, Lu QB. Bond Breaks of Nucleotides by Dissociative Electron Transfer of Nonequilibrium Prehydrated Electrons: A New Molecular Mechanism for Reductive DNA Damage. J Am Chem Soc. 2009;131:11320–11322. doi: 10.1021/ja902675g. [DOI] [PubMed] [Google Scholar]
- Wang W, Becker D, Sevilla MD. The Influence of Hydration on the Absolute Yields of Primary Ionic Free-Radicals in γ-irradiated DNA at 77 K.1. Total Radical Yields Radiat Res. 1993;135:146–154. [PubMed] [Google Scholar]
- Wang W, Sevilla MD. Protonation of Nucleobase Anions in Gamma-Irradiated DNA and Model Systems. Which DNA Base Is the Ultimate Sink for the Electron? Radiat Res. 1994;138:9–17. doi: 10.2307/3578841. [DOI] [PubMed] [Google Scholar]
- Wieczór M, Wityk P, Czub J, Chomicz L, Rak J. A first-principles study of electron attachment to the fully hydrated bromonucleobases. Chem Phys Lett. 2014;595–596:133–137. doi: 10.1016/j.cplett.2014.01.052. [DOI] [Google Scholar]
- Xue L, Greenberg MM. Use of Fluorescence Sensors To Determine that 2-Deoxyribonolactone Is the Major Alkali-Labile Deoxyribose Lesion Produced in Oxidatively Damaged DNA. Angew Chem Int Ed. 2007;46:561–564. doi: 10.1002/anie.200603454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RM, Neumark DM. Dynamics of Solvated Electrons in Clusters. Chem Rev. 2012;112:5553–5577. doi: 10.1021/cr300042h. [DOI] [PubMed] [Google Scholar]