

Abstract
Objective
HIV-infected patients are at an increased risk of developing atherosclerosis, in part due to downmodulation and functional impairment of ATP-Binding Cassette A1 (ABCA1) cholesterol transporter by the HIV-1 protein Nef. The mechanism of this effect involves Nef interacting with an endoplasmic reticulum (ER) chaperone calnexin and disrupting calnexin binding to ABCA1, leading to ABCA1 retention in ER, its degradation and resulting suppression of cholesterol efflux. However, molecular details of Nef-calnexin interaction remained unknown, limiting translational impact of this finding.
Approach and results
Here, we used molecular modeling and mutagenesis to characterize Nef-calnexin interaction and to identify small molecule compounds that could block it. We demonstrated that interaction between Nef and calnexin is direct and can be reconstituted using recombinant proteins in vitro with a binding affinity of 89.1 nM measured by surface plasmon resonance. The cytoplasmic tail of calnexin is essential and sufficient for interaction with Nef, and binds Nef with affinity of 9.4 nM. Replacing lysine residues in positions 4 and 7 of Nef with alanines abrogates Nef-calnexin interaction, prevents ABCA1 downregulation by Nef, and preserves cholesterol efflux from HIV-infected cells. Through virtual screening of the NCI library of compounds, we identified a compound, 1[(7-Oxo-7H-benz[de]anthracene-3-yl)amino]anthraquinone, which blocked Nef-calnexin interaction, partially restored ABCA1 activity in HIV-infected cells, and reduced foam cell formation in a culture of HIV-infected macrophages.
Conclusion
This study identifies potential targets that can be exploited to block the pathogenic effect of HIV infection on cholesterol metabolism and prevent atherosclerosis in HIV-infected subjects.
Keywords: HIV-1, cholesterol efflux, calnexin, Nef, molecular modeling, mutagenesis, virtual screening
Subject terms: Basic science research, Cell biology/structural biology, Computational biology, Lipids and cholesterol, Mechanisms
INTRODUCTION
HIV-1 infection, via activity of viral protein Nef, impairs cholesterol efflux mediated by the cholesterol transporter ATP-Binding Cassette A1 (ABCA1) 1. ABCA1 is the main cellular cholesterol transporter regulating delivery of cellular cholesterol to extracellular acceptor, apolipoprotein A-I. Studies in animal models demonstrated that this activity of Nef may be responsible for hypoalphalipoproteinemia and high risk of atherosclerosis observed in HIV-infected subjects 2–4. Our recent study identified calnexin, an integral endoplasmic reticulum (ER) membrane lectin-like chaperone, as a key player in the mechanism of Nef-mediated inhibition of ABCA1 and cholesterol efflux 5. Calnexin (CNX) and its homologue calreticulin (CRT) regulate folding and maturation of newly synthesized glycoproteins by engaging them in a CNX/CRT cycle 6.
ABCA1 is a highly glycosylated protein 7. Although no evidence for the role of CNX in ABCA1 biogenesis is available, two well-studied ABC transporters, ABCC7 (also known as cystic fibrosis transmembrane conductance regulator, CFTR) and ABCB1 (also known as multidrug resistance protein 1 or P-glycoprotein 1), interact with CNX, and folding mutants of these transporters are retained within the ER by CNX and eventually degraded 8, 9. Importantly, ABCC7 and ABCB1 mutants that escape CNX binding do not achieve mature glycosylation and these mutations result in reduced transporter function 8, 9. Our recently published study demonstrated that ABCA1 interacts with CNX, and reduction of CNX expression by RNAi resulted in a significant decrease in functional activity of ABCA1, evidenced by reduced cholesterol efflux to ABCA1-specific acceptor apoA-I 5. We also showed that Nef impairs interaction between ABCA1 and CNX, and this effect of Nef is essential for inactivation and downregulation of ABCA1 5. Importantly, inhibition of ABCA1-CNX interaction by Nef is specific, as interaction between ABCA1 and two other proteins, dystrophin and serine palmitoyltransferase, shown previously to bind ABCA1 10, was not affected. Also not affected was the interaction between calnexin and HIV-1 envelope glycoprotein, gp160; in fact this interaction was even enhanced by Nef 5. These findings suggested that Nef modulates activity of CNX, but the mechanism of this effect and molecular details of Nef/CNX interaction remained unknown. Moreover, it was unclear whether the interaction between Nef and CNX is direct, making screen for inhibitory compounds difficult.
Calnexin is a 592-amino acid Type I transmembrane protein composed of three parts: a lumenal fragment consisting of a globular β-sandwich domain responsible for the interaction with carbohydrates and a proline-rich tandem sequence repeat domain (the P domain) involved in protein-protein interactions, a transmembrane domain, and a cytoplasmic domain of 90 residues 11, 12. The cytoplasmic tail of CNX can undergo phosphorylation and palmitoylation which regulate CNX association with a number of proteins and protein complexes that influence functional activity of this chaperone 13–18. For example, palmitoylation of the C-tail of CNX mediates its association with the ribosome-translocon complex, which is essential for the ability of CNX to capture its client proteins as they emerge from the translocon 18. Ribosome association of CNX is also regulated by phosphorylation on Ser534 and Ser544 by casein kinase 2 and on Ser563 by protein kinase C/proline-directed kinase 11. In addition, phosphorylation at Ser563 has been shown to play essential role in quality control function of CNX 15. Therefore, the C-tail of CNX may play a functional role regulating activity of the chaperone both directly, by affecting ER lumenal events involving CNX, and indirectly, via modification of CNX localization in the ER.
Here, we demonstrate that the C-tail of CNX is targeted by the HIV-1 protein Nef, which uses this interaction to disrupt CNX-assisted maturation of ABCA1 and impair cholesterol efflux. We characterize important structural features of the Nef/CNX interaction and identify a small molecule compound that blocks this interaction and reverses negative effects of HIV infection on cellular cholesterol metabolism.
MATERIALS AND METHODS
Materials and Methods are available in the online-only Data Supplement.
RESULTS
Cytoplasmic domain of calnexin is necessary for interaction with Nef
In our previous study we have shown that HIV-1 Nef interacts with the ER chaperone CNX 5. To test which region of CNX is necessary for binding to Nef, we used CNX constructs that had deletion of the lumenal repeat segment (aa 276-409) or truncation of the C-terminal cytoplasmic domain (aa 504-586) (Fig. 1A). We co-transfected HEK293T cells with Nefcons-expressing vector and HA-tagged variants of wild-type (WT) calnexin or the deletion mutants and performed co-immunoprecipitation. Figure 1B shows that WT CNX interacted strongly with Nef, whereas CNX construct with internal repeat motif deletion (CNXΔ276-409) exhibited partially reduced binding (40% reduction). However, binding of Nef to CNX construct carrying the truncation of the C-terminal cytoplasmic tail (CNXΔ504-586) was reduced dramatically (70% reduction). This finding highlights the importance of the cytoplasmic region of CNX in interaction with Nef. The role of CNX cytoplasmic tail in the interaction with Nef is consistent with Nef’s predominant localization to the cytoplasm 19, and with the charges of the interacting domains: the cytoplasmic domain of CNX is composed mainly of negatively charged amino acids, which may promote interaction with the N-terminal region of Nef (see below) enriched in positively charged residues. The modest effect that deletions in the lumenal repeat motif of CNX had on Nef binding (40% reduction of binding), and residual binding (30%) of Nef to CNX construct with deleted cytoplasmic domain may be due to artefacts caused by overexpression. In addition, mutations in the lumenal domain may affect binding properties of the cytoplasmic domain via reverse signal transduction mechanism (see Discussion). These results provided an initial lead, which was followed in subsequent experiments.
FIGURE 1. Nef interacts with the cytoplasmic domain of calnexin.

(A) HA-tagged full-length and mutant CNX constructs used for transfection. Green, blue, brown and black boxes represent globular domain, tandem repeat motif (P domain), TM domain and C-terminal cytoplasmic tail, respectively. Red boxes represent deleted fragments. CNXWT represents full length calnexin, CNXΔ504-586 has 81 out of the 89 cytoplasmic tail residues deleted while maintaining the ER localization sequence RKPRRE; CNXΔ276-409 construct has 132 residues of the P domain deleted. (B) HEK293T cells were co-transfected with vectors expressing Nef and HA-tagged CNXWT, CNXΔ504-586 or CNXΔ276-409 and blotted for HA, Nef and GAPDH (lysate). Calnexin variants were immunoprecipitated 48 h post-transfection using anti-HA coupled agarose beads (upper panel) and resulting immunoprecipitates were immunoblotted for calnexin and Nef. Numbers under the lanes show relative amounts of co-precipitated Nef obtained by gel densitometry.
Computational model of Nef-CNX interaction
Experimentally solved molecular structure of CNX is available only for the lumenal domain 12, and to obtain three-dimensional structure of CNX cytoplasmic domain we performed its modeling with several modeling servers implementing different methods, which produced a number of models ranging from the fully folded structures to structures that included natively disordered regions. The models have been assessed for accuracy and final round of modeling performed with the server QA-RecombineIt. The final model had a loosely folded structure (Fig. 2A, panel a). Computational prediction of Nef-CNX complexes showed Nef N-terminal alpha-helix forming the interaction interface with CNX cytoplasmic domain (Fig. 2A, panels b and c).
FIGURE 2. Intermolecular interactions between Nef and calnexin.

(A) Schematic representation of Nef-CNX binding. (a) Schematic representation of the CNX structure. Lumenal domain is represented by the model structure; the transmembrane region is shown as a helical domain according to the Uniprot (calnexin, P27824) domain classification. Calnexin cytoplasmic domain and Nef are represented by models built as described in Experimental Procedures, and Nef-CNX binding is shown according to the results of docking. (b) Docking model of the CNX cytoplasmic domain (green) – Nef (magenta) interaction. The binding interface is formed by the Nef N-terminal alpha-helix, with Lys 7 and Arg 8, 19, and 22 forming interactions with CNX. (c) Lys7 in Nef (magenta) displays strong interaction with Glu533 in CNX (green) formed by the hydrogen and ionic bonds. (d) Lys4 plays a key role in the N-terminal region of Nef structure model (magenta). It forms a strong intramolecular interaction with Nef Asp90 with hydrogen and ionic bonds, supporting structural rigidity of the Nef N-terminal alpha-helix relative to the rest of Nef structure. (B) Interactions in Nef-CNX docking models mapped on Nef and CNX sequences. Bars show the number of interactions, with the numbers for each maximum showing sequence number. In the Nef sequence, there are three distinct interaction clusters centered on residues 7, 22, 75 and 109, with sharp maxima for lysine 7, and arginines in positions 8, 19, 22, 75 and 109. Two interaction clusters in the CNX sequence are formed by amino acids 528 – 533 and 545 – 557; they include glutamic acid residues in positions 529, 532, 533.
In comparison with the CNX cytoplasmic domain model, the model of Nef was based on a number of experimental structures 20–24 and thus had better accuracy. Nef-CNX interaction has been modeled by global docking using four different docking servers, Cluspro, HEX, SwarmDock, and Zdock. Combined set of the best Nef-CNX docking models produced with these servers contained 80 models. The advantage of this approach is that the resulting models represented Nef-CNX interaction modeled by four different, unrelated methods and therefore it was more reliable than using a single server. From these, 49 models have been filtered out as possibly interfering with interaction of Nef with ER membrane. Intermolecular interactions in the remaining subset of 31 models have been identified. There are several distinct clusters of interactions, with sharp maxima for Lys7 and Arg in positions 8, 19, 22, 75 and 109 (Fig. 2B). Notably, similar analysis of interactions carried out on the full initial dataset of 80 docking models showed similar clustering and maxima (not shown). We can therefore hypothesize that the identified residues represent the overall favorable Nef-CNX interaction sites. All these residues, except Lys7 and Arg8, have been also identified as participating in interactions in the experimental structures of complexes which included Nef (Table 1). A representative model of Nef-CNX binding is shown in Figure 2A, panels b and c. Analysis of the conserved residues in Nef performed with ConSurf 25 revealed several such conserved positions in the N-terminal region, including Lys4, Ser6, Lys7 and Arg19. Multiple sequence alignment of the human HIV Nef sequences from Uniprot showed that Lys7 is highly conserved across the spectrum of HIV-1 and HIV-2 sequences. Conserved residues indicate structurally and functionally important positions, including interaction sites. Therefore, Lys7 represents a new interaction site predicted by us, which was not previously identified in Nef interactions with other proteins (Table 1).
Table 1. Comparison of Nef predicted interacting residues with interactions identified in experimentally determined structures.
Nef amino acid residues predicted in this study to form interactions with calnexin are compared with the interactions identified in experimental structures of complexes which include Nef. Comparison is done on the basis of structure/sequence alignments of the Nef model used to predict interactions with calnexin, and PDB experimental structures. Nef interacting partners in the experimental structures are listed as they are given in the corresponding PDB files.
| Predicted interaction clusters | PDB structures | UniProt code HIV virus subtype (isolate) |
||
|---|---|---|---|---|
| Matching Residues | PDB ID | Nef interacting partner | ||
| Lys7 | No hits | |||
| Arg8 | No hits | |||
| Arg19 | Arg19 | 4emzC | AP-1 COMPLEX SUBUNIT MU-1 |
Q90VU7_9HIV1 Human immunodeficiency virus 1 |
| Arg22 | Arg22 | 4emzC | AP-1 COMPLEX SUBUNIT MU-1 |
Q90VU7_9HIV1 Human immunodeficiency virus 1 |
| Arg75 | Thr71 | 4emzC, 4en2B | AP-1 COMPLEX SUBUNIT MU-1 |
Q90VU7_9HIV1 Human immunodeficiency virus 1 |
| Arg71 | 1efnB | FYN SH3 DOMAIN |
P03406 (NEF_HV1BR) Human immunodeficiency virus type 1 group M subtype B (isolate BRU/LAI) |
|
| Arg71 | 4d8dB | FYN SH3 DOMAIN |
P03406 (NEF_HV1BR) Human immunodeficiency virus type 1 group M subtype B (isolate BRU/LAI) |
|
| Arg75 | 3rebA, 3rbbA, 3reaA | TYROSINE-PROTEIN KINASE HCK |
P03407 (NEF_HV1A2) Human immunodeficiency virus type 1 group M subtype B (isolate ARV2/SF2) |
|
| Arg75 | 4orzB | FYN SH3 DOMAIN |
P03407 (NEF_HV1A2) Human immunodeficiency virus type 1 group M subtype B (isolate ARV2/SF2) |
|
| Arg109 | Arg105 | 4neeC | AP-2 COMPLEX SUBUNIT ALPHA-2 |
P04601 (NEF_HV1H2) Human immunodeficiency virus type 1 group M subtype B (isolate HXB2) |
Lysine residues of Nef in positions 4 and 7 are critical for Nef-CNX interaction
From the perspective of drug design, targeting viral proteins has lower potential for side effects than targeting cellular partners. We thus focused our efforts on Nef. According to docking modeling and sequence conservation results, Lys7 possibly represents a new binding site in Nef and accordingly it has been selected for mutagenesis experiments. Lys4 has been also selected since it is a Lys7 near neighbor and, as demonstrated in the Nef model, it plays a key structural role for the N-terminus (Fig. 2A, panel d). Therefore, mutation of both Lys4 and Lys7 was predicted to invoke structural rearrangement in the Nef N-terminal region thus disrupting the interaction between Nef and calnexin. Alanine substitution of basic residues at the N-terminus of Nef has previously been shown to preserve membrane association and CD4 down-regulation by Nef 26, and intracellular localization of the mutant Nef was indistinguishable from that of Nef WT 27.
To verify the role of these residues in Nef interaction with CNX, we used the mutant HIV-1 NL4-3 clone carrying Nef with Lys4 and Lys7 changed to valine and alanine, respectively (NefK) 27. Calnexin was immunoprecipitated from HEK293T cells transfected with WT or mutant HIV-1 clones and the precipitate was immunoblotted for Nef. As shown in Figure 3A, interaction with CNX was evident for Nef WT, but not for NefK. Interaction with the double mutant was reduced by 95%, indicating that the lysine residues in positions 4 and 7 are essential for Nef interaction with CNX.
FIGURE 3. Mutation of Nef lysine residues 4 and 7 blocks interaction with calnexin.

(A) HEK293T cells transfected with HIV-1 molecular clones encoding for Nef WT or Nef K4VK7A (NefK), or (B) HeLa ABCA1 cells transfected with pcDNA plasmids expressing Nef WT or mutants Nef K4A, Nef K7A or NefK4,7A were lysed 48 h post-transfection. Endogenous CNX was immunoprecipitated using monoclonal anti-CNX antibody and immunoprecipitates were blotted for Nef and CNX (top panels). Whole cell lysates were analyzed for expression of CNX, Nef and GAPDH (bottom panels). Numbers under the Nef panels show relative amounts of co-precipitated Nef obtained by gel densitometry. (C) HeLa-ABCA1-GFP cells were transfected with Nef WT or mutants Nef K4A, Nef K7A or Nef K4,7A. Cells were lysed 48 h post-transfection and lysates were analyzed for expression of ABCA1, Nef and GAPDH. Numbers under the ABCA1 panel show relative amounts of ABCA1 obtained by gel densitometry. (D) HEK293T cells were co-transfected with ABCA1-FLAG and Nef WT or NefK4,7A and were lysed 48 h post-transfection. ABCA1 was immunoprecipitated using anti-FLAG beads and precipitates were blotted for ABCA1 and Nef (top panel). Whole cell lysates were analyzed for expression of ABCA1-FLAG, Nef and GAPDH (bottom panel). Numbers under the Nef panel show relative amounts of co-precipitated Nef obtained by gel densitometry. (E) HEK293T cells were transfected with Nef WT (left panels) or NefK4,7A (right panels), stained with Alexa Fluor®647 conjugated antibody for Nef (green), DyLight 550 conjugated antibody for calnexin (red), and DAPI for nuclei (blue), and analyzed by confocal fluorescent microscopy. Large panels show overlay of all three channels, whereas small panels show red channel (calnexin, top panels), green channel (Nef, middle panels), and magenta staining for co-localization obtained using Volocity software (bottom panels). Scale bar equals 26.7 μm.
In order to look at the individual contribution of the two lysine residues to the interaction with CNX, we mutagenized the Nefcons plasmid to create single and double lysine mutant constructs. To minimize variables introduced by co-transfection, we used HeLa cells that stably express ABCA1-GFP, transfected them with WT or mutant Nef constructs and analyzed the amount of Nef that co-immunoprecipitates with CNX. Based on densitometric analysis, interaction of CNX with NefK4A was reduced by 80%, whereas interaction with NefK7A was reduced by 70% when compared to interaction with WT Nef (Fig. 3B). Interaction of CNX with double mutant NefK4,7A was undetectable. The reduced interaction observed with the Nef single mutants was sufficient to down-regulate ABCA1, as shown in Figure 3C. NefK4A and NefK7A mutants reduced ABCA1 abundance as much as the wild-type Nef, whereas near control level of ABCA1 was observed when both lysine residues were mutated (Fig. 3C). This result highlights the importance of both residues in ABCA1 down-regulation, and suggests that under Nef overexpression conditions even reduced interaction with CNX observed for NefK4A and NefK7A mutants is sufficient for ABCA1 downregulation. It also supports conclusions of the previous study that demonstrated that Nef-CNX interaction is essential for Nef-mediated retention of ABCA1 in ER and subsequent degradation 5.
To rule out the possibility that mutation of these residues grossly affected the behavior of the N-terminal domain of Nef, we tested the interaction of the mutant Nef with ABCA1. Previous studies demonstrated that interaction between Nef and ABCA1 also involves the N-terminal domain 1, although the specific residues involved have not been identified. Co-precipitation analysis revealed about a 30% reduction in NefK4,7A interaction with ABCA1 as compared to ABCA1 interaction with wild-type Nef (Fig. 3D). The reduction, however, remains in stark contrast to the >95% loss of interaction observed in the Nef-CNX interaction studies (Fig. 3A).
To visualize the effect of mutations on Nef-calnexin interaction, HEK 293T cells were transfected with vectors expressing wild-type or mutant (NefK4,7A) Nef, stained with a combination of anti-Nef rabbit polyclonal and anti-CNX mouse monoclonal antibody followed by a combination of Alexa Fluor®647 anti-rabbit IgG (green) and DyLight 550 anti-mouse IgG (red), counterstained with DAPI (blue), and analyzed by confocal fluorescent microscopy. Images were processed using Volocity software to identify co-localizing pixels (magenta staining). Consistent with our previous report 5, wild-type Nef co-localized with CNX (Fig. 3E, left panels). However, the co-localization between CNX and NefK4,7A was dramatically reduced (Fig. 3E, right panels), confirming the key role of these residues in Nef-CNX binding.
Functional analysis of Nef mutants
In our previous study, we reported that Nef plays a central role in the down-modulation of ABCA1 expression and function 1. This phenotype was associated with Nef’s ability to interact with CNX and disrupt CNX interaction with ABCA1 5. Identification of Nef residues required for interaction with CNX provided an opportunity to verify the critical role of this interaction for the effects of Nef on cellular cholesterol metabolism. To assess the functional consequence of losing the Nef/CNX interaction for ABCA1 functionality, we co-transfected HEK293T cells with ABCA1 and HIV-1 NL4-3 infectious clones that express either Nef WT or Nef K4VK7A (NefK) 27. Lysates were immunoblotted for ABCA1 (Fig. 4A). Consistent with results obtained with Nef-expressing vector (Fig. 3C), total ABCA1 abundance was significantly reduced in the presence of Nef WT, however, expression of ABCA1 in the presence of NefK was comparable to that of the control sample, which was transfected with an empty vector. This result is consistent with conclusions of the previous study that identified Nef as the key viral factor responsible for ABCA1 downregulation 1.
FIGURE 4. HIV-1 clones expressing mutant Nef K4VK7A do not down-regulate ABCA1 or cholesterol efflux.
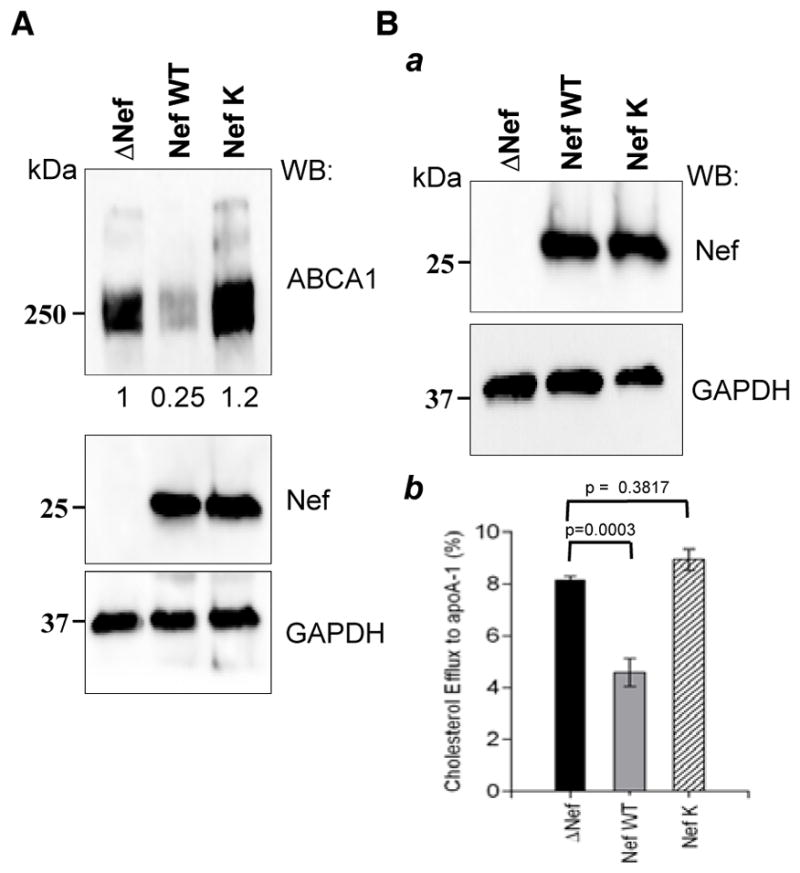
(A) HEK 293T cells were co-transfected with ABCA1 and HIV-1 molecular clones encoding Nef WT or Nef K4VK7A (NefK). HIV-1 clone with a Nef deletion (DNef) was used as control. Cells were lysed 48 h post-transfection and immunoblotted for ABCA1, Nef and GAPDH. (B) THP-1 cells were infected with VSV-G pseudotyped HIV-1 molecular clones used in A. Western blot shows expression of Nef in cell lysates (a). Cholesterol efflux was measured 7 days after infection (b). Results show apoA-I specific cholesterol efflux as mean ± SEM of quadruplicates.
We further analyzed the effect of mutations disrupting Nef/CNX interaction on the ability of Nef to downregulate apoA-1 specific cholesterol efflux. THP-1 cells were differentiated into macrophages using PMA and were infected with HIV-1 expressing either wild-type Nef or NefK. Given that the virus used in this experiment was the X4-tropic strain NL4-3, we pseudotyped it with VSV-G to ensure one-cycle replication. Both Nef WT and NefK viruses successfully infected the cells, establishing similar levels of Nef expression (Fig. 4Ba). Seven days after infection, cholesterol efflux assay was performed. In agreement with previous reports 1, 5, 28, cells infected with the wild-type virus had significantly reduced cholesterol efflux relative to mock-infected cells (Fig. 4Bb). However, infection with the virus carrying NefK did not lead to efflux decrease.
Interaction between Nef and CNX is direct
To test whether Nef and CNX interact directly with each other, we expressed CNX and the cytoplasmic tail of CNX (CNX-CT) in E coli and purified recombinant proteins by column chromatography. For purification of full-length calnexin, we have developed and implemented a novel purification system based on the ultra-high affinity (Kd ~ 10−14–10−17M) small protein complex of genetically inactivated colicin 7 DNAse (CL7) and its inhibitor, immunity protein 7 (Im7) 29–32. We have attached a CL7 variant, which possesses no DNAse activity but retains full Im7 affinity, as a C-terminal tag on His-tagged CNX construct (Fig. 5A, left side). A cleavage site for the pre-scission protease (PSC) inserted between CNX and CL7 allowed for elution of CNX from the Im7 column through cleavage by PSC. A single purification step provided an excellent yield of ~90% pure protein (Fig. 5A), in which major contamination represented CNX molecules (confirmed by mass-spec), most likely, truncated from the N-terminus. The CNX-CT construct was designed with a single N-terminal His-tag and was purified using the standard procedure (Fig. 5A, right side).
FIGURE 5. Nef directly binds to CNX and its cytoplasmic tail.

(A) Purification of CNX and CNX-CT. Steps of purification of full-length CL7-tagged calnexin (CNX-CL7) are shown in detail. Whole cell lysate (WCL) was centrifuged to remove cell debris, the supernatant (SN) was treated with 0.07% polyethylene-emine (PE) to precipitate DNA, the pellet (PL), which contained most of CNX protein, was washed with detergent-containing buffer to release CNX into solution, centrifuged and the resulting supernatant was loaded on Immunity protein 7 (Im7) column. Bound proteins were eluted by treating the column with pre-scission protease (PSC) (EL lane), whereas flow-through (FT) lane shows unbound proteins. DCNX – truncated CNX fragment; SUMO – SUMO domain; P(PSC), P(SUMOP) – cleavage sites for the PSC and SUMO proteases, respectively; H8 – 8-Histidine tag. (B) Surface plasmon resonance experiments were done in a Biacore T-200 by using a CM5 chip. CNX (left panel) and CNX-CT (right panel) were captured by amine coupling, and myristoylated NefSF2 protein was injected over the chip surface at 6 different concentrations (6.25 nM – 200 nM range) in triplicates. Colored lines represent actual data and black lines represent curve fit to a monovalent analyte binding model in BiaEvaluation software.
Binding of myristoylated NefSF2 33 to CNX and its cytoplasmic domain was analyzed using surface plasmon resonance (Fig. 5B). CNX and CNX-CT were immobilized on microchip surfaces and myristoylated Nef was injected over the surface. NefSF2 directly bound to calnexin with an affinity (KD) of 89.1 nM (ka = 1.338E5 M−1s−1, kd = 0.01192 s−1, Chi2 = 2.77 RU) (Fig. 5B, left panel). Binding to CNX-CT was observed to have higher affinity of KD=9.4 nM (ka = 9.083E5 M−1s−1, kd = 0.008569 s−1, Chi2 = 0.474 RU) (Fig. 5B, right panel). Taken together, these experiments demonstrate that Nef/CNX interaction is direct and involves the cytoplasmic domain of calnexin.
Virtual screening for compounds interfering with Nef-CNX interaction
Docking-based virtual screening has been performed on compounds from the Zinc NCI Plated 2007 dataset with docking program Vina 34. Nef model described in Figure 2 has been used, with the interaction site for ligand docking selected to cover amino acid residues Lys4 and Lys7. The dataset consisted of 139,735 compounds. Ten putative ligands were identified and prioritized according to the Vina ranking, and structural alignment of these compounds to the Nef-CNX complex is shown in Figure 6A (panel a). The model shows that these compounds can block Nef/CNX interaction at the CNX residues Glu529, Glu532 and Glu533. Docking of NSC 13987, which turned out in the later studies to be the most effective inhibitor of the Nef-CNX interaction, is shown in panel b (Fig. 6A). Interactions of the compound with Nef include two hydrogen bonds with Nef amino acid residues Ser6 and Tyr124. Three of the 10 compounds, NSC 1758, NSC 13987, and NSC 92938 have been submitted for experimental testing. The chemical names and molecular structures of these compounds are shown in Figure 6B.
FIGURE 6. Characterization of compounds targeting Nef-CNX interaction.

(A) a - Ten compounds with the best score (grey) from virtual screening performed on the Zinc NCI Plated 2007 dataset. Their overall location in the structural alignment with the model of Nef (magenta) - CNX (green) complex is shown as translucent molecular surface (cyan). The model shows that these compounds can block the Nef/CNX interaction at calnexin residues Glu529, Glu532 and Glu533. The set of compounds includes NSC 1758, NSC 13987, and NSC 92938 selected for experimental testing. b - Compound NSC 13987 docked to Nef binding site, which is centered on Lys4 and Lys7. Interactions of the compound with Nef include two hydrogen bonds with Nef amino acid residues Ser6 and Tyr124. (B) Chemical structures and names of compounds NSC 1758, NSC 13987, and NSC 92938. (C) Dose-response effect of NSC 1758, NSC 13987 and NSC 92938 on viability of THP-1 cells. THP-1 cells were treated with indicated compounds for 5 days and cytotoxicity was measured using MTT assay. (D) HEK 293T cells were transfected with HA-tagged Nef, treated with compounds NSC 1758, NSC 13987 or NSC 92938, and lysed. Nef was immunoprecipitated using anti-HA agarose beads and bound complexes were immunoblotted for Nef and CNX (left panels labeled IP: αHA). Densitometric quantification of calnexin co-immunoprecipitated with Nef is presented in the right panel. Results are presented as mean ± SD of 3 independent experiments, and p values are shown above the bars. Whole cell lysates were analyzed for amount of CNX, Nef and GAPDH (left panels labeled Lysate). (E) HEK293T cells were co-transfected with ABCA1-FLAG and Nefcons and treated with compound NSC 13987. Cells were lysed 48 h post-transfection and ABCA1-FLAG was immunoprecipitated using anti-FLAG beads. Precipitated complexes were blotted for ABCA1 and Nef (top panels). Input amount was analyzed from whole cell lysates by immunoblotting for ABCA1, Nef and GAPDH (bottom panels).
Testing the compounds’ activity
To test whether the compounds identified in our virtual screen can interfere with Nef-CNX interaction, we first performed co-immunoprecipitation assay. HEK293T cells were transfected with plasmid encoding for Nefcons and 6 h post-transfection were treated with NSC 1758 (4 μM), NSC 13987 (5 μM), or NSC 92938 (5 μM). These concentrations of the compounds were determined by the MTT assay to reduce cell metabolism by less than 10% during 5-day incubation (Fig. 6C). Among the 3 compounds tested, one compound, NSC 13987, inhibited co-immunoprecipitation of Nef and CNX by over 50%, whereas the effect of NSC 1758 and NSC 92938 showed a partial inhibition of Nef/CNX binding, which did not reach statistical significance (Fig. 6D). We have previously shown that membrane localization of Nef is important for interaction of Nef with CNX 5. In order to rule out the possibility that the compound interferes with membrane localization of Nef, we tested whether NSC 13987 affects interaction between Nef and ABCA1, as ABCA1-Nef interaction also requires membrane localization of Nef 1. As shown in Fig. 6E, ABCA1-Nef interaction remained unaffected in the presence of compound indicating that the inhibition was specific for the molecular interaction of Nef and CNX.
We next tested whether the three compounds could prevent impairment of cholesterol efflux by Nef. THP-1 cells were transfected with a Nef encoding plasmid and drug treatment was started 6 h after transfection. The following day, cells were activated with PMA after which cholesterol efflux assay was performed. Drug treatment was continued throughout the duration of the experiment. Figure 7Aa shows cholesterol efflux measured in untreated cells or cells treated with DMSO or each of the 3 compounds. Cholesterol efflux in Nef-transfected untreated or DMSO-treated cells was reduced by over 2-fold relative to mock-transfected cells. NSC 13987, which showed inhibition of Nef-CNX interaction (Fig. 6D), significantly increased cholesterol efflux as compared to DMSO-treated Nef expressing cells, although the rescue was partial and did not completely reverse the inhibition. Two other compounds did not significantly rescue Nef-suppressed cholesterol efflux. Treating untransfected THP-1 cells with the compounds did not lead to any changes in cholesterol efflux (Fig. 7Ab). This result implies that the impact in efflux capacity observed in the presence of NSC 13987 was specific to the compound’s activity in Nef-expressing cells. It also demonstrates that the compounds were not toxic to cells.
FIGURE 7. Compound NSC 13987 prevents impairment of cholesterol efflux by HIV-1 and Nef.

(A) THP-1 cells were transfected with Nef (or mock-transfected, control) and incubated with compounds NSC 1758, NSC 13987 and NSC 92938 or DMSO, and cholesterol efflux was measured 5 days post-transfection (panel a). Cholesterol efflux was measured in THP-1 cells treated only with compounds NSC 1758, NSC 13987 and NSC 92938 or DMSO (panel b). Results are presented as mean ± SEM (n=4; one-way ANOVA with post-hoc Tukey HSD test). (B) Primary MDM were infected in triplicates with HIV-1 ADA. Compound NSC 13987 (5 μM) or DMSO was added 3 days after infection and maintained thereafter, and virus replication was monitored by measuring RT activity in the supernatant over a 14-day period. Results of 3 measurements are shown as mean ± SEM, (n=3, * p< 0.05; ** p< 0.07 by Student’s unpaired t-test) (panel a). Fold change in RT activity was calculated from three independent experiments performed using macrophages from three donors. Error bars show SD (n=3, p value was calculated using Student’s unpaired t-test) (panel b). (C) MDMs were infected with HIV-1 ADA or mock-infected and treated with DMSO or NSC 13987 as in B. Cholesterol efflux was measured 14 days post-infection in quadruplicate wells. Results are presented as mean ± SEM (n=4; one-way ANOVA with Tukey’s HSD test) (panel a). Cholesterol efflux in MDM obtained from two donors was measured independently and fold change in efflux activity in the presence of drug is shown (panel b). Results are presented as mean ± SEM (n=2, student’s unpaired t-test). (D) a - MDMs were infected with VSV-G-pseudotyped HIV-1 NL4-3 (upper row), mock-infected (middle row), or infected with VSV-G-pseudotyped Nef-deficient HIV-1ΔNef NL4-3 (bottom row). NSC 13987 (5 μM) was added on day 5 post-infection and maintained thereafter. Ten days post-infection, cells were stained with ORO and analyzed by bright field microscopy at 100x magnification. b – Analysis of images in panel a was performed using Image J program. Histograms in the left panel represent pixel distribution along the 256 RGB shades, averaged from 55 cells in each condition. Pie charts in the right panel show percentage of cells in each condition with ORO-stained area over or under an arbitrarily selected value of 20%. c - MDMs were infected with HIV-1 ADA or mock-infected. NSC 13987 was added on day 5 and maintained thereafter. On day 14 post-infection, cells were stained with ORO, counterstained with DAPI and analyzed by fluorescent microscopy at 100x magnification.
To test the effect of NSC 13987 in the context of natural infection, we infected monocyte-derived macrophages (MDM) with HIV-1 ADA, treated them with NSC 13987 and measured cholesterol efflux. Viral replication in the presence of the compound was reduced (Fig. 7Ba), and fold change analysis showed on average a 2-fold reduction in reverse transcriptase (RT) activity measured in three independent experiments with cells from different donors (Fig. 7Bb), consistent with demonstrated rescue by the compound of Nef-inhibited cholesterol efflux (Fig. 7A) and previous studies demonstrating anti-HIV activity of ABCA1 and ABCA1-stimulated cholesterol efflux 28, 35, 36. Consistent with previous studies 1, 5, 36, cholesterol efflux from HIV-infected cells was decreased by 60%, whereas HIV-infected cells treated with NSC 13987 showed cholesterol efflux not significantly different from that of mock-infected cells (Fig. 7Ca). Fold change in cholesterol efflux from two independent experiments with cells from different donors showed consistent efflux rescue by the compound (Fig. 7Cb). A more potent reversal by NSC 13987 of cholesterol efflux inhibited by HIV-1 infection (Fig. 7C) than by Nef transfection (Fig. 7Aa) is likely due to higher levels of Nef expression in transfected cells, and a combined effect of reduced virus replication and inhibition of the Nef-CNX interaction in HIV-infected cells.
Lack of a small animal model of HIV-associated atherosclerosis prevented us from testing the anti-atherogenic potential of NSC 13987 in vivo. We therefore used the best available in vitro approximation of HIV-associated pro-atherogenic changes, conversion of HIV-infected macrophages into the foam cells 1. In this experiment, we employed the vesicular stomatitis virus (VSV) G protein-pseudotyped HIV-1 NL4-3 and its Nef-deficient mutant (HIV-1ΔNef), which can go through only one cycle of replication in MDM cultures. This approach allowed us to eliminate the drug’s effect on viral replication, leaving changes in cholesterol metabolism as the only cause of potential effects on lipid droplets accumulation. Indeed, virus replication, as measured by RT activity in culture supernatants, was 400–500 cpm/μl for both wild-type and ΔNef viruses treated or untreated with NSC 13987. As revealed by bright field microscopy, MDM cultures infected with wild-type HIV-1 (Fig. 7Da, top left panel) were enriched in Oil Red O (ORO) stained lipid droplets compared to mock-infected cells (middle left panel), and treatment with NSC 13987 (top right panel) reduced the number of stained cells and decreased intensity of staining to the level observed in mock-infected cells. Importantly, infection with the Nef-deficient HIV-1 did not increase the number of lipid droplets (Fig. 7Da, bottom left panel), indicating that the observed effect was mediated by Nef expression. Of note, treatment with NSC 13987 of mock-infected MDM or MDM infected with the ΔNef virus did not decrease lipid droplets, indicating the compound’s effect was dependent on Nef. Quantitative analysis of the images performed on 55 cells from each condition is presented in Fig. 7Db. Histograms in the left panel demonstrate a shift of RGB pixel distribution to low-intensity area (indicative of reduction of ORO-stained lipid droplets) in MDMs infected with wild-type HIV-1 and treated with NSC 13987 relative to HIV-infected untreated culture, whereas treatment of mock-infected cultures or cultures infected with the ΔNef virus did not significantly affect pixel distribution. Importantly, histograms obtained with HIV-infected cells treated with NSC 13987 were similar to those with mock-infected cells, indicating that the drug fully reversed HIV-induced accumulation of lipid droplets. The pie-chart graphs on the right show the distribution of cells according to ORO-stained area. 65% of HIV-infected cells had over 20% of cell area stained with ORO, whereas such cells constituted only 42% and 44% in mock- and HIV-1ΔNef-infected MDMs, respectively. NSC 13987 reduced the percentage of such cells in HIV-infected MDM to 22%, which was even smaller than in drug-treated uninfected cells.
To better visualize lipid droplets, we used fluorescent microscopy. We also employed macrophage-tropic HIV-1 isolate ADA to better mimic natural conditions. The diameter of ORO-stained lipid droplets in HIV-infected cells varied from 0.1 to 3 μm (Fig. 7Dc, middle panel). The size of lipid droplets accumulated in HIV-infected MDMs treated with NSC 13987 was visibly reduced and did not exceed 0.5 μm (right panel); in fact, it was similar to the size of the droplets in mock-infected cells (left panel).
Taken together, these results provide a proof of concept for the idea that HIV-induced impairment of cholesterol efflux can be reversed pharmacologically by blocking the Nef/CNX interaction.
DISCUSSION
Highly active anti-retroviral therapy (HAART) has transformed treatment of the HIV disease changing prognosis from acutely lethal to chronic illness, and lifespan of HIV-infected subjects approximates that of uninfected individuals. However, HAART does not cure HIV, and chronic HIV infection is associated with a number of co-morbidities, such as premature atherosclerosis and cardio-vascular disease 37. An essential component in pathogenesis of cardio-vascular disease in HIV-infected subjects is HIV-associated dyslipidemia, which is caused both by drugs used to treat HIV infection and by the effects of HIV itself on cholesterol metabolism 38. In this report, we identify a small-molecule compound that blocks HIV-mediated impairment of cellular cholesterol metabolism. Excitingly, this compound also inhibited replication of HIV, suggesting that, if developed into a drug, it can target both HIV infection and virus-induced metabolic co-morbidities.
Our previous studies demonstrated that HIV critically depends on interaction with host cholesterol metabolism and modifies it for optimization of viral replication 1, 2, 28, 35, 36. Specifically, HIV, through viral protein Nef, reduces abundance and impairs functional activity of ABCA1, a key transporter in cholesterol efflux pathway 1. As a result, host cells accumulate excessive cholesterol promoting formation of plasma membrane lipid rafts, which are sites of HIV entry, assembly and budding 39. Recently, we demonstrated that an important mechanism of down-regulation and/or functional impairment of ABCA1 by HIV is Nef-mediated inhibition of the interaction between ABCA1 and the ER chaperone, CNX 5. The current study provides the first characterization of the exact molecular structures involved in Nef-CNX interaction.
First, we established that interaction between Nef and CNX involves the cytoplasmic domain of CNX. While this finding is consistent with demonstrated localization of Nef to the cytoplasmic side of membranes 27 and lack of evidence for Nef localization to ER, it is surprising given that the C-tail of CNX is not involved in the interaction between CNX and ABCA1, which is disrupted by Nef 5. Indeed, CNX interactions with glycosylated proteins are mediated by its lumenal domains 12. Therefore, Nef interaction with the C-tail alters activity of the lumenal domains of CNX. How Nef is doing it is unknown and several possibilities can be considered. Binding of Nef may prevent post-translational modifications of the C-tail of CNX, such as phosphorylation on Ser563 that has been shown to regulate CNX interaction with α1-antitrypsin and a number of other glycoproteins 15. However, docking analysis did not reveal Ser563 as a likely site for interaction with Nef (Fig. 5B). The same argument can be applied to SUMOylation at Lys506, which has been shown to regulate CNX interaction with another ER protein, protein tyrosine phosphatase 1B 40: Lys506 is not among the preferred sites for Nef binding. It is likely that Nef binding to the cytoplasmic domain of CNX results in signal transduction from the cytoplasmic to the lumenal domain, e.g., via a conformational change in CNX. This explanation is consistent with the partial reduction of Nef binding to CNX carrying deletion in the lumenal domain (Fig. 1B), which may be due to reverse signaling from lumenal to cytoplasmic domain. Mechanistic details of such an effect await careful structural analysis. Regardless of the mechanism, this finding provides the first example of a pathogen utilizing the CNX C-tail to regulate functional activity of this chaperone.
Second, we identified the Nef residues critical for interaction with CNX: mutation of lysine residues in positions 4 and 7 of Nef abrogated Nef-CNX binding, prevented ABCA1 downregulation, and restored cholesterol efflux in cells infected with HIV-1. Our finding that Nef-CNX interaction involves the flexible N-terminal region of Nef was surprising, as this region has not been implicated before in protein-protein interactions (Table 1). However, molecular modeling (Fig. 2) suggests that Lys4 of Nef forms a hydrogen bond with Asp90 located in an alpha-helix, thus contributing to stabilization of the structure of the N-terminal region, and therefore acts as a structural anchor for the Nef Lys7 interaction with CNX. Nef Lys7 is predicted to form a strong interaction with Glu533 in CNX through the hydrogen and ionic bonds. Thus, mutation of both lysine residues destabilizes the structure of Nef, and cancels the strong interaction with CNX provided by Lys7, which explains the dramatic effect of these mutations on Nef-CNX interaction. The N-terminal region of Nef has not been involved in protein-protein interactions, but its basic and hydrophobic residues were shown to be essential for membrane association of Nef 41. Interestingly, lysine residues at positions 4 and 7, which participate in interaction with CNX, were not essential for the membrane association of Nef 42. Therefore, our study identified a novel epitope on Nef involved in the interaction with the cytoplasmic tail of CNX.
Using this information, we performed virtual screening for compounds that can potentially disrupt Nef-CNX interaction, and identified a number of candidates. One of these compounds, 1[(7-Oxo-7H-benz[de]anthracene-3-yl)amino]anthraquinone (NSC 13987), prevented co-precipitation of CNX with Nef, reversed Nef-mediated effect on ABCA1 abundance, and restored cholesterol efflux impaired by Nef, thus effectively reversing the effects of Nef on host cholesterol metabolism. In addition, the compound resulted in a near 2-fold inhibition of viral replication (Fig. 7B). This latter effect may have two main explanations. First, the compound prevents ABCA1 downregulation by Nef, and ABCA1 has been shown to inhibit HIV-1 replication by reducing lipid rafts abundance on the plasma membrane and affecting production and infectivity of nascent virions 3, 28, 35, 36. Second, previous reports presented evidence that anthraquinone derivatives inhibit the ribonuclease H function of HIV-1 reverse transcriptase 43, 44. Therefore, the action of compound NSC 13987 in HIV-1-infected cells may be a combination of inhibiting Nef-CNX interaction and a separate antiviral activity. This, together with differences in Nef expression, could explain why the rescue of the cholesterol efflux by the compound was only partial in Nef-transfected cells (Fig. 7Aa), but almost complete in HIV-infected macrophages (Fig. 7C). Our findings provide basis for using NSC 13987 as a foundation for development of novel treatment approaches for HIV-associated atherosclerosis and other Nef-dependent metabolic co-morbidities. Indeed, the effects of Nef secreted from HIV-infected cells may be responsible for many lipid-related complications of HIV disease, such as atherosclerosis, diabetes, lipodistrophy and neurodegeneration, so the compounds similar to the one identified in this study may reverse HIV-induced impairment of cholesterol metabolism in uninfected cells mitigating lipid-related complications of HIV infection.
Unfortunately, no small animal model is available to test in vivo whether NSC 13987 reverses development of atherosclerosis associated with HIV infection. Indeed, mice do not develop atherosclerosis unless certain genes (apoE or ldlr) are knocked out, and humanization of such mutant mice to make them susceptible to HIV infection has not been attempted. Injection of Nef into mice reproduces only some features of the disease and it remains uncertain whether these effects of Nef involve CNX 4. Here, we demonstrated that NSC 13987 inhibits accumulation of lipid droplets in HIV-infected macrophage cultures (Fig. 7D), which is a characteristic feature of foam cells, and foam macrophages are a hallmark of the development of atherosclerosis 45. Our findings indicate that the effect of NSC 13987 is specific for Nef-expressing cells, as the compound did not affect lipid droplets in mock-infected macrophages or cells infected with Nef-deficient HIV-1 (Fig. 7D). This result supports our conclusion that the protective effect of NSC 13987 is due to inhibition of the Nef-CNX interaction, rather than to an off-target effect on cholesterol metabolism.
Calnexin is an ER-integral membrane protein and is responsible for the folding of several glycoproteins. Depletion of CNX has been shown to result in the elevation of several other ER-folding factors minimizing aberrant protein folding and expression 46. This is mainly true for glycoproteins which are common substrates of other soluble ER chaperones like CRT. However, solubility and oligosaccharide variability impose a limit on this commonality, making CNX vital for expression and function of proteins like ABCA1 and several others 5, 47, 48. Nef’s ability to target several host factors, such as CD4, MHC I, CXCR4, may in part be due to the limitation Nef imposes on the access of these proteins to CNX. Therefore, the protective effect of compound NSC 13987 may well extend to restoring the expression and function of other proteins targeted by Nef.
In conclusion, in this study we identified the molecular mechanisms and structural epitopes involved in interaction between HIV-1 Nef and host CNX and characterized a compound capable of reversing the effects of Nef, thus presenting potential utility in treatment of HIV-1 infection and its metabolic side effects.
HIGHLIGHTS.
HIV-1 Nef interacts directly with the cytoplasmic tail of the endoplasmic reticulum chaperone calnexin
Mutation of lysine residues in positions 4 and 7 of Nef disrupts Nef-calnexin interaction and prevents Nef-mediated inhibition of ABCA1 and impairment of cholesterol efflux
Anthraquinone derivative NSC 13987 blocks Nef-calnexin interaction, reverses impairment of cholesterol efflux and reduces accumulation of lipid droplets in HIV-infected macrophages
Acknowledgments
Sources of Funding: This study was supported by RFBR grant 13-04-91458; by NIH grants HL093818, HL101274, and AI108533; by the District of Columbia Center for AIDS Research (DC CFAR), an NIH-funded program (5P30 AI055019); and by the Molecular and Cellular Biology Program of the Russian Academy of Sciences. We thank the Biacore Molecular Interaction shared resources at the Lombardi Comprehensive Cancer Center (Georgetown University), which is supported by a grant P30 CA51008 (PI Louis Weiner) from the National Cancer Institute. We would like to thank Dr. Mark Harris for kindly providing the pCG-NL4-3 and pCG-NL4-3NefK plasmids encoding for HIV-1 viruses with WT and mutant Nef, respectively, and Dr. Mathias Geyer from University of Bonn Institute of Innate Immunity for myristoylated recombinant Nef protein. The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: pT7consnefhis6 from Dr. Ron Swanstrom; Catalog #2949, anti-HIV-1 Nef polyclonal from Dr. Ronald Swanstrom; pHEF-VSVG from Dr. Lung-Ji Chang; p83-10 and p210-8 from Dr. Ronald Desrosiers. Ruth Hunegnaw is a pre-doctoral student in the Microbiology and Immunology Program of the Institute for Biomedical Sciences at the George Washington University. This work is from a dissertation to be presented to the above program in partial fulfillment of the requirements for the Ph.D. degree.
Abbreviations
- HIV
Human Immunodeficiency Virus
- ORO
Oil Red O
- ABCA1
ATP-binding cassette A1
- ER
endoplasmic reticulum
- CNX
calnexin
- CRT
calreticulin
- GFP
green fluorescent protein
- WT
wild type
- PSC
pre-scission protease
- Im7
immunity protein 7
- CL7
colicin 7
- MDM
monocyte-derived macrophages
- RT
reverse transcriptase
Footnotes
Other contributors: None.
Disclosures: None.
References
- 1.Mujawar Z, Rose H, Morrow MP, Pushkarsky T, Dubrovsky L, Mukhamedova N, Fu Y, Dart A, Orenstein JM, Bobryshev YV, Bukrinsky M, Sviridov D. Human immunodeficiency virus impairs reverse cholesterol transport from macrophages. PLoS Biol. 2006;4:e365. doi: 10.1371/journal.pbio.0040365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asztalos BF, Mujawar Z, Morrow MP, Grant A, Pushkarsky T, Wanke C, Shannon R, Geyer M, Kirchhoff F, Sviridov D, Fitzgerald ML, Bukrinsky M, Mansfield KG. Circulating nef induces dyslipidemia in simian immunodeficiency virus-infected macaques by suppressing cholesterol efflux. J Infect Dis. 2010;202:614–623. doi: 10.1086/654817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dubrovsky L, Van Duyne R, Senina S, Guendel I, Pushkarsky T, Sviridov D, Kashanchi F, Bukrinsky M. Liver x receptor agonist inhibits hiv-1 replication and prevents hiv-induced reduction of plasma hdl in humanized mouse model of hiv infection. Biochem Biophys Res Commun. 2012;419:95–98. doi: 10.1016/j.bbrc.2012.01.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cui HL, Ditiatkovski M, Kesani R, Bobryshev YV, Liu Y, Geyer M, Mukhamedova N, Bukrinsky M, Sviridov D. Hiv protein nef causes dyslipidemia and formation of foam cells in mouse models of atherosclerosis. FASEB J. 2014;28:2828–2839. doi: 10.1096/fj.13-246876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jennelle L, Hunegnaw R, Dubrovsky L, Pushkarsky T, Fitzgerald ML, Sviridov D, Popratiloff A, Brichacek B, Bukrinsky M. Hiv-1 protein nef inhibits activity of atp-binding cassette transporter a1 by targeting endoplasmic reticulum chaperone calnexin. J Biol Chem. 2014;289:28870–28884. doi: 10.1074/jbc.M114.583591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Helenius A, Aebi M. Roles of n-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 7.Tanaka AR, Ikeda Y, be-Dohmae S, Arakawa R, Sadanami K, Kidera A, Nakagawa S, Nagase T, Aoki R, Kioka N, Amachi T, Yokoyama S, Ueda K. Human abca1 contains a large amino-terminal extracellular domain homologous to an epitope of sjogren’s syndrome. Biochem Biophys Res Commun. 2001;283:1019–1025. doi: 10.1006/bbrc.2001.4891. [DOI] [PubMed] [Google Scholar]
- 8.Pind S, Riordan JR, Williams DB. Participation of the endoplasmic reticulum chaperone calnexin (p88, ip90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1994;269:12784–12788. [PubMed] [Google Scholar]
- 9.Loo TW, Clarke DM. Prolonged association of temperature-sensitive mutants of human p-glycoprotein with calnexin during biogenesis. J Biol Chem. 1994;269:28683–28689. [PubMed] [Google Scholar]
- 10.Okuhira K, Fitzgerald ML, Sarracino DA, Manning JJ, Bell SA, Goss JL, Freeman MW. Purification of atp-binding cassette transporter a1 and associated binding proteins reveals the importance of beta1-syntrophin in cholesterol efflux. J Biol Chem. 2005;280:39653–39664. doi: 10.1074/jbc.M510187200. [DOI] [PubMed] [Google Scholar]
- 11.Chevet E, Wong HN, Gerber D, Cochet C, Fazel A, Cameron PH, Gushue JN, Thomas DY, Bergeron JJ. Phosphorylation by ck2 and mapk enhances calnexin association with ribosomes. EMBO J. 1999;18:3655–3666. doi: 10.1093/emboj/18.13.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schrag JD, Bergeron JJ, Li Y, Borisova S, Hahn M, Thomas DY, Cygler M. The structure of calnexin, an er chaperone involved in quality control of protein folding. Mol Cell. 2001;8:633–644. doi: 10.1016/s1097-2765(01)00318-5. [DOI] [PubMed] [Google Scholar]
- 13.Roderick HL, Lechleiter JD, Camacho P. Cytosolic phosphorylation of calnexin controls intracellular ca(2+) oscillations via an interaction with serca2b. J Cell Biol. 2000;149:1235–1248. doi: 10.1083/jcb.149.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myhill N, Lynes EM, Nanji JA, Blagoveshchenskaya AD, Fei H, Carmine SK, Cooper TJ, Thomas G, Simmen T. The subcellular distribution of calnexin is mediated by pacs-2. Mol Biol Cell. 2008;19:2777–2788. doi: 10.1091/mbc.E07-10-0995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cameron PH, Chevet E, Pluquet O, Thomas DY, Bergeron JJ. Calnexin phosphorylation attenuates the release of partially misfolded alpha1-antitrypsin to the secretory pathway. J Biol Chem. 2009;284:34570–34579. doi: 10.1074/jbc.M109.053165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lynes EM, Bui M, Yap MC, Benson MD, Schneider B, Ellgaard L, Berthiaume LG, Simmen T. Palmitoylated tmx and calnexin target to the mitochondria-associated membrane. EMBO J. 2012;31:457–470. doi: 10.1038/emboj.2011.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lynes EM, Raturi A, Shenkman M, Ortiz Sandoval C, Yap MC, Wu J, Janowicz A, Myhill N, Benson MD, Campbell RE, Berthiaume LG, Lederkremer GZ, Simmen T. Palmitoylation is the switch that assigns calnexin to quality control or er ca2+ signaling. J Cell Sci. 2013;126:3893–3903. doi: 10.1242/jcs.125856. [DOI] [PubMed] [Google Scholar]
- 18.Lakkaraju AK, Abrami L, Lemmin T, Blaskovic S, Kunz B, Kihara A, Dal Peraro M, van der Goot FG. Palmitoylated calnexin is a key component of the ribosome-translocon complex. EMBO J. 2012;31:1823–1835. doi: 10.1038/emboj.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ranki A, Lagerstedt A, Ovod V, Aavik E, Krohn KJ. Expression kinetics and subcellular localization of hiv-1 regulatory proteins nef, tat and rev in acutely and chronically infected lymphoid cell lines. Arch Virol. 1994;139:365–378. doi: 10.1007/BF01310798. [DOI] [PubMed] [Google Scholar]
- 20.Grzesiek S, Bax A, Hu JS, Kaufman J, Palmer I, Stahl SJ, Tjandra N, Wingfield PT. Refined solution structure and backbone dynamics of hiv-1 nef. Protein Sci. 1997;6:1248–1263. doi: 10.1002/pro.5560060613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akgun B, Satija S, Nanda H, Pirrone GF, Shi X, Engen JR, Kent MS. Conformational transition of membrane-associated terminally acylated hiv-1 nef. Structure. 2013;21:1822–1833. doi: 10.1016/j.str.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jung J, Byeon IJ, Ahn J, Gronenborn AM. Structure, dynamics, and hck interaction of full-length hiv-1 nef. Proteins. 2011;79:1609–1622. doi: 10.1002/prot.22986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grzesiek S, Bax A, Clore GM, Gronenborn AM, Hu JS, Kaufman J, Palmer I, Stahl SJ, Wingfield PT. The solution structure of hiv-1 nef reveals an unexpected fold and permits delineation of the binding surface for the sh3 domain of hck tyrosine protein kinase. Nature Struct Biol. 1996;3:340–345. doi: 10.1038/nsb0496-340. [DOI] [PubMed] [Google Scholar]
- 24.Singh P, Yadav GP, Gupta S, Tripathi AK, Ramachandran R, Tripathi RK. A novel dimer-tetramer transition captured by the crystal structure of the hiv-1 nef. PLoS One. 2011;6:e26629. doi: 10.1371/journal.pone.0026629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Celniker GN, Ashkenazy H, Glaser F, Martz E, Mayrose I, Pupko T, Ben-Tal N. Consurf: Using evolutionary data to raise testable hypotheses about protein function. Isr J Chem. 2013;53:199–206. [Google Scholar]
- 26.Aiken C, Konner J, Landau NR, Lenburg ME, Trono D. Nef induces cd4 endocytosis: Requirement for a critical dileucine motif in the membrane-proximal cd4 cytoplasmic domain. Cell. 1994;76:853–864. doi: 10.1016/0092-8674(94)90360-3. [DOI] [PubMed] [Google Scholar]
- 27.Bentham M, Mazaleyrat S, Harris M. Role of myristoylation and n-terminal basic residues in membrane association of the human immunodeficiency virus type 1 nef protein. J Gen Virol. 2006;87:563–571. doi: 10.1099/vir.0.81200-0. [DOI] [PubMed] [Google Scholar]
- 28.Cui HL, Grant A, Mukhamedova N, Pushkarsky T, Jennelle L, Dubrovsky L, Gaus K, Fitzgerald ML, Sviridov D, Bukrinsky M. Hiv-1 nef mobilizes lipid rafts in macrophages through a pathway that competes with abca1-dependent cholesterol efflux. J Lipid Res. 2012;53:696–708. doi: 10.1194/jlr.M023119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ko TP, Liao CC, Ku WY, Chak KF, Yuan HS. The crystal structure of the dnase domain of colicin e7 in complex with its inhibitor im7 protein. Structure. 1999;7:91–102. doi: 10.1016/s0969-2126(99)80012-4. [DOI] [PubMed] [Google Scholar]
- 30.Kleanthous C, Kuhlmann UC, Pommer AJ, Ferguson N, Radford SE, Moore GR, James R, Hemmings AM. Structural and mechanistic basis of immunity toward endonuclease colicins. Nature Struct Biol. 1999;6:243–252. doi: 10.1038/6683. [DOI] [PubMed] [Google Scholar]
- 31.Wallis R, Moore GR, James R, Kleanthous C. Protein-protein interactions in colicin e9 dnase-immunity protein complexes. 1. Diffusion-controlled association and femtomolar binding for the cognate complex. Biochemistry. 1995;34:13743–13750. doi: 10.1021/bi00042a004. [DOI] [PubMed] [Google Scholar]
- 32.Wallis R, Leung KY, Pommer AJ, Videler H, Moore GR, James R, Kleanthous C. Protein-protein interactions in colicin e9 dnase-immunity protein complexes. 2. Cognate and noncognate interactions that span the millimolar to femtomolar affinity range. Biochemistry. 1995;34:13751–13759. doi: 10.1021/bi00042a005. [DOI] [PubMed] [Google Scholar]
- 33.Breuer S, Gerlach H, Kolaric B, Urbanke C, Opitz N, Geyer M. Biochemical indication for myristoylation-dependent conformational changes in hiv-1 nef. Biochemistry. 2006;45:2339–2349. doi: 10.1021/bi052052c. [DOI] [PubMed] [Google Scholar]
- 34.Trott O, Olson AJ. Autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comp Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramezani A, Dubrovsky L, Pushkarsky T, Sviridov D, Karandish S, Raj DS, Fitzgerald ML, Bukrinsky M. Stimulation of liver x receptor has potent anti-hiv effects in a humanized mouse model of hiv infection. J Pharmacol Exp Ther. 2015;354:376–383. doi: 10.1124/jpet.115.224485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morrow MP, Grant A, Mujawar Z, Dubrovsky L, Pushkarsky T, Kiselyeva Y, Jennelle L, Mukhamedova N, Remaley AT, Kashanchi F, Sviridov D, Bukrinsky M. Stimulation of the liver x receptor pathway inhibits hiv-1 replication via induction of atp-binding cassette transporter a1. Mol Pharmacol. 2010;78:215–225. doi: 10.1124/mol.110.065029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Myerson M, Malvestutto C, Aberg JA. Management of lipid disorders in patients living with hiv. J Clin Pharmacol. 2015;55:957–974. doi: 10.1002/jcph.473. [DOI] [PubMed] [Google Scholar]
- 38.Wang T, Yi R, Green LA, Chelvanambi S, Seimetz M, Clauss M. Increased cardiovascular disease risk in the hiv-positive population on art: Potential role of hiv-nef and tat. Cardiovasc Pathol. 2015;24:279–282. doi: 10.1016/j.carpath.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waheed AA, Freed EO. Lipids and membrane microdomains in hiv-1 replication. Virus Res. 2009;143:162–176. doi: 10.1016/j.virusres.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee D, Kraus A, Prins D, Groenendyk J, Aubry I, Liu WX, Li HD, Julien O, Touret N, Sykes BD, Tremblay ML, Michalak M. Ubc9-dependent association between calnexin and protein tyrosine phosphatase 1b (ptp1b) at the endoplasmic reticulum. J Biol Chem. 2015;290:5725–5738. doi: 10.1074/jbc.M114.635474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gerlach H, Laumann V, Martens S, Becker CF, Goody RS, Geyer M. Hiv-1 nef membrane association depends on charge, curvature, composition and sequence. Nature Chem Biol. 2010;6:46–53. doi: 10.1038/nchembio.268. [DOI] [PubMed] [Google Scholar]
- 42.Giese SI, Woerz I, Homann S, Tibroni N, Geyer M, Fackler OT. Specific and distinct determinants mediate membrane binding and lipid raft incorporation of hiv-1(sf2) nef. Virology. 2006;355:175–191. doi: 10.1016/j.virol.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 43.Hussain H, Al-Harrasi A, Al-Rawahi A, Green IR, Csuk R, Ahmed I, Shah A, Abbas G, Rehman NU, Ullah R. A fruitful decade from 2005 to 2014 for anthraquinone patents. Exp Opin Ther Pat. 2015;25:1053–1064. doi: 10.1517/13543776.2015.1050793. [DOI] [PubMed] [Google Scholar]
- 44.Esposito F, Corona A, Zinzula L, Kharlamova T, Tramontano E. New anthraquinone derivatives as inhibitors of the hiv-1 reverse transcriptase-associated ribonuclease h function. Chemotherapy. 2012;58:299–307. doi: 10.1159/000343101. [DOI] [PubMed] [Google Scholar]
- 45.Yuan Y, Li P, Ye J. Lipid homeostasis and the formation of macrophage-derived foam cells in atherosclerosis. Prot Cell. 2012;3:173–181. doi: 10.1007/s13238-012-2025-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Molinari M, Eriksson KK, Calanca V, Galli C, Cresswell P, Michalak M, Helenius A. Contrasting functions of calreticulin and calnexin in glycoprotein folding and er quality control. Mol Cell. 2004;13:125–135. doi: 10.1016/s1097-2765(03)00494-5. [DOI] [PubMed] [Google Scholar]
- 47.Danilczyk UG, Cohen-Doyle MF, Williams DB. Functional relationship between calreticulin, calnexin, and the endoplasmic reticulum luminal domain of calnexin. J Biol Chem. 2000;275:13089–13097. doi: 10.1074/jbc.275.17.13089. [DOI] [PubMed] [Google Scholar]
- 48.Hebert DN, Zhang JX, Chen W, Foellmer B, Helenius A. The number and location of glycans on influenza hemagglutinin determine folding and association with calnexin and calreticulin. J Cell Biol. 1997;139:613–623. doi: 10.1083/jcb.139.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]