

Abstract
Background
Survival after sudden cardiac arrest is limited by post-arrest myocardial dysfunction but understanding of this phenomenon is constrained by lack of data from a physiological model of disease. In this study, we established an in vivo model of cardiac arrest and resuscitation, characterized the biology of the associated myocardial dysfunction, and tested novel therapeutic strategies.
Methods
We developed rodent models of in vivo post-arrest myocardial dysfunction using extra-corporeal membrane oxygenation (ECMO) resuscitation followed by invasive hemodynamics measurement. In post-arrest isolated cardiomyocytes, we assessed mechanical load and Ca2+ induced Ca2+ release (CICR) simultaneously using the micro-carbon-fiber technique and observed reduced function and myofilament calcium sensitivity. We used a novel-designed fiber optic catheter imaging system, and a genetically encoded calcium sensor GCaMP6f, to image CICR in vivo.
Results
We found potentiation of CICR in isolated cells from this ECMO model and also in cells isolated from an ischemia-reperfusion Langendorff model perfused with oxygenated blood from an arrested animal, but not when reperfused in saline. We established that CICR potentiation begins in vivo. The augmented CICR observed post-arrest was mediated by the activation of Ca2+/calmodulin kinase II (CaMKII). Increased phosphorylation of CaMKII, phospholamban and ryanodine receptor 2 (RyR2) was detected in the post-arrest period. Exogenous adrenergic activation in vivo recapitulated Ca2+ potentiation but was associated with lesser CaMKII activation. Since oxidative stress and aldehydic adduct formation were high post arrest, we tested a small molecule activator of aldehyde dehydrogenase type 2, Alda-1, which reduced oxidative stress, restored calcium and CaMKII homeostasis, and improved cardiac function and post-arrest outcome in vivo.
Conclusions
Cardiac arrest and reperfusion lead to CaMKII activation and calcium long-term potentiation which support cardiomyocyte contractility in the face of impaired post-ischemic myofilament calcium sensitivity. Alda-1 mitigates these effects, normalizes calcium cycling and improves outcome.
Keywords: Post-cardiac arrest myocardial dysfunction, ECMO resuscitation, calcium-induced calcium release (CICR), CaMKII, aldehyde dehydrogenase type 2 (ALDH2), oxidative stress
Introduction
Sudden cardiac arrest is a major cause of death in the Western World,1 and mortality after resuscitation remains greater than 50%.2,3 This excess mortality is reflected in the post-cardiac arrest syndrome (PCAS),2 a condition that comprises: 1) post-arrest central nervous system dysfunction, 2) post-arrest ischemia-reperfusion (I-R), 3) post-myocardial arrest dysfunction (PMAD), and 4) continuation of the factors precipitating sudden cardiac arrest. PMAD is believed to be a form of myocardial stunning4–6 and has been observed for hypoxic arrest and following ventricular fibrillation.7 Recognizing that mortality remains high after resumption of circulation, the International Liaison Committee on Resuscitation has recommended more detailed resuscitation research including the phenomenon of PMAD.1
PMAD may result from oxidative stress induced cytosolic calcium overload. Calcium overload has been traditionally explained by two processes: i) reverse mode operation of the sodium-calcium exchanger (NCX) related to intracellular sodium overload during ischemia and ii) sarcoplasmic reticulum ATPase (SERCA) dysfunction related to ATP depletion.8 These processes have been suggested to lead to both reversible myofilament dysfunction and, subsequently, to irreversible cell death partly through calcium/calmodulin-dependent protein kinase II (CaMKII) signaling pathways.8–10 The relationship between acute calcium overload and long-term augmentation of calcium-induced calcium release (CICR) is critical but understudied. In particular, the biochemical pathways identified would be predicted to potentiate CICR. In addition, since normal CICR generates, on a beat-to-beat basis, systolic calcium levels in localized domains tenfold higher than predicted to occur during post-arrest calcium overload,11 any CICR augmentation might be positioned to play a dominant signaling role. Moreover, as it relates to excitation-contraction coupling, potentiated CICR transients would be expected to result in more forceful contractions, not the opposite. For example, in one post-cardiac arrest study, ryanodine receptor (RyR) dependent calcium release was shown to support reversible injury.12 However, in spite of data in support of such a mechanism, no long-lasting changes in CICR have been demonstrated following reperfusion13 except for one early report.14 Alternatively, the increased CICR transients could be a consequence of a mis-match between cell contractility and external mechanical demand, whereby reduced shortening, or diastolic stretch, of cells may raise intracellular Ca2+ levels 15or Ca2+-releasability from the sarcoplasmic reticulum.16 Two limitations of previous experimental models are that i) there is an absence of in vivo data, and ii) much of the in vitro mechanistic data have come from the buffered saline-perfused Langendorff preparations that suffer from significant oedema.17 In addition, CICR and CaMKII signaling have been linked to beta-adrenergic signaling under pathological conditions,15 and cardiac arrest has been associated with dramatic elevations in catecholaminergic signaling which is not present in the isolated heart model.18 We believed a model preserving the neurohumoral axis might better reflect the in vivo environment and developed an in vivo model of cardiac arrest with resuscitation using extracorporeal membrane oxygenation (ECMO).19 Here, we report findings from this in vivo cardiac arrest-ECMO model relating to post-myocardial arrest dysfunction. Two novel approaches were introduced in this study. First, we examined cellular force in vitro using micro-carbon fiber assessment simultaneously with CICR measurements. Second, we developed an approach to measuring calcium dynamics in vivo using a genetically engineered calcium indicator (GECI). Our data from this clinically relevant in vivo model support the contribution of calcium potentiation, CaMKII signaling and oxidative stress to post arrest myocardial dysfunction and suggested a potential new therapeutic candidate, Alda-1.
Methods
Studies were approved by the Stanford Administrative Panel on Laboratory Animal Care and conform to the Guide for the Care and Use of Laboratory Animals published by the NIH.
Invasive hemodynamics and development of extracorporeal membrane oxygenation
Male Sprague-Dawley rats (250-350 g) were anesthetized and oxygenated/ventilated with 1-2% isoflurane, maintained at 37°C, and monitored throughout for depth of anesthesia. An extracorporeal membrane oxygenation of blood circuit was built to support re-animation following cardiac arrest (see Figure 1A).20 A conductance catheter (Millar Instruments, Houston, TX) was introduced into the LV for Pressure-Volume (PV) loop measurements as described previously.21,22
FIGURE 1. In vivo cardiac arrest models.

A. ECMO circuit consisting of a venous reservoir (VR), roller pump (RP), miniature membrane oxygenator (MO) between arterial (femoral artery, FA) and venous (right atrium via external jugular (EJ) systems perfused in a retrograde fashion. B. Timeline of experiment (CA=cardiac arrest) followed by 15 mins downtime and then ECMO initiation. C. An example arrhythmic sudden cardiac arrest protocol. D. Ordinate for D-F is the same, and shown in 1D only. Zoomed in view of the beginning of Rapid Ventricular Pacing (RVP) at 12 Hz pacing over the LV apex leading to hypotension. BP (black) is shown on top with the scale bar on the left, and heart rate (far field EKG; gray) is shown underneath. E. Both ventricular fibrillation (VF) followed by pulseless electrical activity (PEA) later in time in a zoomed in view. F. Zoomed in view of the heart off ECMO after full resuscitation. G, H. Representative PV loops from pre (G) and post-arrest (H) with ECMO resuscitation in the same animal. The ESPVR (solid line) and EDPVR (dotted line) are superimposed. The ESPVR slope was 1.38 and 0.6 mmHg/μl.
Animal Models of Disease
After institution of the ECMO circuit (Figure 1A), sudden cardiac arrest was initiated either by cessation of ventilation or by rapid ventricular pacing to induce ventricular fibrillation (Figure 1B-E).19 Both processes led to a rapid hypotension, followed by pulseless electrical activity and/or asystole (Figure 1C). The arrest period was 15 minutes after which ECMO was initiated and the heart resumed spontaneous beating (Figure 1F). We saw no differences between these two modes of cardiac arrest. Data are presented from the hypoxic cardiac arrest model because of the similarity to existing descriptions of isolated Langendorff heart ischemia-reperfusion. For selected experiments, rat hearts were explanted and retrogradely perfused on a Langendorff system. After stabilization (pre-ischemia) for 10 minutes, hearts were subjected to 15 minutes of no-flow ischemia, and coronary perfusion was restored for 30 minutes.13 During explanted IR experiments, HEPES-buffered saline (see below) was bubbled with 98% oxygen/2% isoflurane. In select experiments, heparinized blood was used to perfuse Langendorff hearts.
Isolation of left ventricular cardiac myocytes
Within 5 minutes of heart explantation from arrested animals weaned from ECMO (typically within 20 minutes after initiation of ECMO) or Langendorff hearts where specified, left ventricular cardiac myocytes were isolated based on our published protocols.23 Experiments were performed with HEPES-buffered solution containing (in mM): 1 CaCl2, 137 NaCl, 5.4 KCl, 15 dextrose, 1.3 MgSO4, 1.2 NaH2PO4, and 20 HEPES (pH 7.4) maintained at 37.7°C with a feedback control system (IonOptix, Milton, MA).
Myocyte unloaded shortening and relaxation
Myocyte contraction properties including sarcomeric length were evaluated with a sarcomeric imaging acquisition system (SarcLen; IonOptix) as previously described.23
Two-Carbon-Fiber Technique
Carbon fibers (CF) (12 μm diameter, kindly provided by Prof Jean-Yves LeGuennec)22 affixed to miniature hydraulic manipulators (SM-28; Narishige, Tokyo, Japan),computer-controlled with a piezoelectric translator (P-621.1CL; Physik Instruments, Karlsruhe/Palmbach, Germany) and mounted on a custom-made railing system (IonOptix) were attached to single-isolated ventricular myocytes. CFs were stretched axially by the piezoelectric translator controlled with custom software (Matlab; MathWorks, Natick, MA). CF bending and force were measured and analyzed with IonWizard.24 Individual systolic and diastolic cell results were plotted and fitted using a linear regression (Origin; OriginLab, Northampton, MA) to give both end-systolic and end-diastolic length-tension relationships (ESLTR and EDLTR, respectively). Data were normalized to the starting systolic force or as the Frank-Starling gain where specified.25
Calcium transient measurements
In vivo CICR measurement was performed using a GCaMP6f genetically encoded calcium indicator (GECI) delivered by an AAV9 vector which was directly injected into the LV apex of anesthetized rats (described below). After three weeks, fluorescence transients were imaged using a custom built fiber optic excitation imaging system (AUST Development, LLC) attached to a camera system described previously26,27 and analyzed using ImageJ (NIH). For cell experiments, cardiomyocytes were loaded with 0.2 μM Fluo-5f-AM (Molecular Probes, now part of Thermo Fisher, Waltham, MA) for 30 min, and then allowed to incubate in dye-free HEPES-buffered saline for additional 30 min to allow for de-esterification of the calcium dye. Spatially averaged electrically evoked calcium transients were measured using a standard FITC cube (Chroma, Bella Falls, VT) using the HyperSwitch system (IonOptix). Fluorescence transients were normalized to ΔF/F units.26Myocyte calcium transient properties including ΔF/F, rise time, and decay time were evaluated with a caclium imaging acquisition system (IonOptix) as previously described.23
Delivery of AAV9-GCaMP6f by direct intramyocardial injection
Adult male Sprague-Dawley rats (275-300 g) were anesthetized with 2% isoflurane. A left thoracotomy was performed after intubation and ventilation. 150 ul of normal saline containing AAV9-GCaMP6f (total titer 2e12, Penn Vector Core) was injected into 3 spots at equal volume at the apex of the left ventricle wall. After the successful injection, the chest was closed and the animal was extubated and allowed to recover. GCaMP6f signal was detected at 3-4 weeks post delivery.
Pharmacological reagents
Epinephrine infusion was performed with 23 μg/kg/min for 30 minutes through venous access. In select experiments, control cardiomyocytes were exposed to 100 nM epinephrine in the extracellular solution. For Ryanodine experiments, 1 mg dissolved ryanodine (Sigma, St. Louis, MO) was injected into the femoral vein empirically to stop the heart. To inhibit CaMKII, 1 mg KN92 or KN93 (Sigma, St. Louis, MO) or 100 μg myristoylated-AIP (Autocamtide-2-related Inhibitory Peptide) (Calbiochem or Anaspec) was used. A myristoylated AC3-C peptide (KKALHAQERVDCL, synthesized by Anaspec), an inactive peptide inhibitor of CaMKII, was used as a control.28 To inhibit protein kinase A (PKA), 1 mg H89 (Sigma) was raised in 1 mL 0.9% normal saline and infused over 10 minutes. Experimental protocols were initiated 10 minutes after infusion. Alda-1, 0.0324 g Alda-1 dissolved in 2 ml 50%PEG/50%PBS, was injected intraperitoneally 30-60 minutes before prior to experimental protocol.
Sham experiments
Sham experiments with animals on ECMO for 30 minutes yielded normal values in isolated cells (data not shown). Control animals refer to no ECMO unless otherwise stated.
Lucigenin-enhanced chemiluminescence
Total O2•– was measured by lucigenin-enhanced chemiluminescence as described previously.29 Chemiluminescence was recorded by luminometer (Bio-Orbit, Turku, Finland) using 5 μmol/l lucigenin. In parallel experiments, ventricular tissue was incubated in l-NMMA (1 mmol/L), diphenylene iodonium (DPI)(10 μmol/L), or Alda-1 (20 μmol/L). After measuring baseline readings for 4 minutes, samples were equilibrated, dark adapted for 5 minutes, and chemiluminescence was recorded for 10 minutes. Recordings were performed blinded to the sample identity. Results were expressed as counts per second per milligram of tissue dry weight.
Oxidative fluorescent microtopography
O2•– was detected in the ventricle using the fluorescent probe dihydroethidium (DHE; Molecular Probes) as described previously.29 Cryosections (30 μm) were incubated in physiological buffer for 30 minutes at 37°C, with either Alda-1 (20 μmol/l) or mito-TEMPO (10 μmol/l) followed by 5 minutes dark incubation with 2 μmol/l dihydroethidium. Images were obtained on a confocal microscope at 60x (Bio-Rad MRC-1024 laser; filter settings, excitation filter at 488 nm; emission filter at 550 nm) and quantified (red intensity x area) using Image-Pro Plus software (Media Cybernetics, Rockville, MD). Analysis was performed blind to the sample identity. Mean fluorescence was calculated from 4 separate, high-power fields from each quadrant to produce n = 1.
Protein extracts and Western blot
Heart tissues were homogenized in lysis buffer containing 50mM Tris-HCl (pH7.5), 150mM NaCl, 1% NP-40, 0.5% Sodium deoxycholate, 1% SDS and Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Heart tissue homogenates were separated on 4-20% TGX gel (Bio-Rad) and transferred to Immobilon-FL PVDF membrane (Millipore). Membranes were blocked in Odyssey TBS blocking buffer and incubated with respective primary antibodies: CaM Kinase II (G1, Santa Cruz Biotechnology), Phospholamban (2D12), Ryanodine Receptor (34C) (Thermo Fisher Scientific) (under non-reducing conditions), phospho-CaMKII Thr286/287 (D21E4, Cell Signaling), Phospholamban pThr-17, Phospholamban pSer-16, RyR2 pSer-2814 (Badrilla), and GAPDH (Sigma). IRDye 800CW Goat anti-Mouse IgG and IRDye 680LT Goat anti-Rabbit IgG were used as secondary antibodies (LI-COR). Quantitative Western images were acquired using Odyssey Fc Imager and images were analyzed with Image Studio Software (LI-COR).
Statistics
For comparisons between independent samples, the t-test using the Welch-Satterthwaite correction for unequal variances was used. One-way Kruskal-Wallis tests were performed for multiple group comparisons. For significant differences, post-hoc testing was performed. P-values from post-hoc testing were corrected for multiple comparisons using the Bonferroni correction. Exact P values are stated in most cases, and a p-value of <0.05 was taken to indicate a statistically significant difference between means. All data are presented as mean ± standard error of the mean (SEM).
For comparisons with nested designs (multiple cells from different number of rats), a mixed effects model was used to account for the correlation between cells from the same rat. The treatment (PMAD vs. control) was modeled as a fixed effect while the animal was modeled as a random effect. We fit the mixed models using maximum likelihood, and tested the significance of the treatment fixed effect.
RESULTS
Myocardial dysfunction is found in a rodent model of cardiac arrest and resuscitation
Pressure-volume hemodynamic measurements were made before (Figure 1G) and after (Figure 1H) cardiac arrest. Analysis showed significant decrement in contraction and relaxation following cardiac arrest (ESPVR −44%; CI-48%; PRSW −31%; τ +500%; Table 1). These data establish post-cardiac arrest myocardial dysfunction in our model.
TABLE 1.
In vivo and isolated cell load
| control | PMAD | |
|---|---|---|
| ESPVR (mmHg/μl) | 1.0±0.10 | 0.5±0.1 (p=0.02) |
| PRSW (mmHg) | 58±8 | 30±6 (p=0.02) |
| CI (mmHg/μl*s) | 93±6 | 64±6 (p=0.005) |
| τ (ms) | 12±3 | 45±6 (p=7e-11) |
| ESLTR (au) | 84±11 | 34±6 (p=0.04) |
| EDLTR (au) | 44±6 | 28±6 (p=0.02) |
| Peak PRSWcell (au) | 8±2 | 3.0±1 (p=0.006) |
ESPVR: end-systolic pressure volume relationship; PRSW: Preload Recruitable Stroke Work; CI: cardiac input; τ: isovolumic relaxation constant; ESLTR: end-systolic length-tension relationship; EDLTR: end-diastolic length-tension relationship.
From the same hearts for above, we next examined unloaded sarcomere shortening (%SL), peak contraction (vc) and relaxation (vr) velocity in isolated adult cells from control and PMAD hearts. For control vs PMAD cells, respectively, the %SL was 13±0.3 vs. 9±0.3 (p=1.61 x 10−6), a 23% decrease in the %SL in unloaded cells (data not shown; n=143 cells from 25 animals and 187 cells from 26 animals for control and PMAD, respectively for above data), while vc was 4.5±0.2 μ/ms vs. 2.6±0.1 μ/ms (p=6.78e-7) and vr was 2.7±0.2 μ/ms vs. 2.0±0.1 1 μ/ms (p= 0.0078 )(data not shown;; n=136 cells from 11 animals and 164 cells from 12 animals for control and PMAD, respectively for above data)).
We further characterized this contractile dysfunction in PMAD by measuring single-cell force and contractility using the two-carbon-fiber technique.30 Figure 2A showed an adult left ventricular myocyte before and during contraction in response to electrical stimulation. Figure 2B and C showed the stepwise stretch protocol used to assess myocyte function. Figure 2C showed carbon fiber bending for the same stretched cell.30 Corresponding to the derivation by regression of load independent values in vivo, the ESLTR and EDLTR are defined in single cells as the slopes of the linear regressions of systolic and diastolic force (proportional to CF bending; see Methods) and plotted vs. sarcomere length (Figure 2D, E). In Table 1, pooled analysis of the slopes of the ESTLR and EDTLR for both control and PMAD cells was shown, demonstrating both to be depressed (60% and 36% of control, respectively) in PMAD cells (p<0.05). In addition, the Frank-Starling gain was used to compare the change in linear slopes as well as at each stretched length by dividing the active force by the passive force (Figure 2F).25 These cellular data recapitulated load varying curves obtained with PV loops, and demonstrated impaired force development. We also explored the force-frequency relationship (FFR) for both control and PMAD cells (Figure 2G). This demonstrated that FFR was depressed in PMAD cells at all pacing frequencies investigated (0.5 - 4.0 Hz).
FIGURE 2. In vivo and isolated cell load dependent parameters in control and cardiac arrest.

A. Isolated twitching myocyte before (top) and during twitch in response to electrical activation demonstrating carbon fiber bending. B The sarcomere length (μm) in response to 1 Hz pacing vs time as CF stretch was applied to three different endpoint lengths demonstrates the cellular Frank-Starling effect. Between each step, the CFs were returned to baseline. Each stretch produced increased sarcomere length change. C. Carbon fiber bending (CFB) (proportional to force) vs time demonstrating the increase in both diastolic (passive) and systolic (active) single cell force, respectively. D. Stretch induced peak systolic (open squares) and baseline diastolic (filled squares) tension from a control cell. The linear fits to the data represented the EDLTR (black line) and ESLTR (gray line). In this case, the ESLTR was 22 μN/μm (R2=0.96), and the EDLTR was 6.6 μN/μm (R2=0.95). E. Stretch induced peak systolic (open squares) and baseline diastolic (filled squares) tension from a PMAD cell. In this case, the ESLTR was 11 μN/μm(R2=0.92), and the EDLTR was 6 μN/μm (R2=0.98). F. PRSWcell vs afterloadcell force for control (open squares) and PMAD (filled squares), n= 8 cells from 3 hearts and 8 cells from 2 hearts for control and PMAD, respectively. Exponential fits to the data were done using the equation FSG= y0+A1e(−x/t1). For control and PMAD, respectively, y0, A1, t1 were: 0.94 and 1.47, 9.5 and 2.7, 0.78 s and 0.54 s. R2 were 0.96 and 0.92. N= 8 cells from 3 hearts and 8 cells from 2 hearts for control and PMAD, respectively. G. Force frequency relationship (FFR) for pooled BSF (μm) vs pacing frequency for control (black squares) and PMAD (open squares) plotted with SE, n= 8 cells from 3 hearts and 8 cells from 2 hearts for control and PMAD, respectively. H. Example traces for simultaneous CICR (bottom, gray) and cellular force (top, black) measurements, shown for an isolated cell electrically paced at 1 Hz. The SL was (in μm) ~1.87,~1.94 and ~2.0 (at slack length, first, and second stretch, respectively).
Cardiac myocytes from post arrest animals exhibit markedly enhanced peak calcium transients
Force development is critically regulated by CICR. Previous reports examining post-arrest CICR have found little change, but did so using high-affinity calcium indicators,13, 31 while rapid calcium transients were subject to dye kinetic filtering that could both mask important phenotypic differences and alter cellular contractility.26 Our findings were consistent with this effect for Fluo-4 but not for the lower affinity dye Fluo-5f (Supplemental Figure 1). As such, we combined measurements of mechanical load and CICR simultaneously in dynamically contracting cardiomyocytes (Figure 2H). It has been reported that CICR does not change with acute stretch in either amphibian cells29 or in mammalian heart cells. We observed these to be the case (Figure 2H).
We next explored CICR after cardiac arrest using the approach outlined above in Figure 2H. In Figure 3A, a single sweep of a steady-state pacing train was shown for a PMAD and a control cell. Consistent with one report,14 we found post-arrest cells to have markedly enhanced peak calcium transients. In pooled analysis (Figure 3B), a significant 290% increase in CICR amplitude was present (ΔF/Fpeak from control (n=97 cells from 11 animals) and PMAD (n=72 cells from 12 animals) were 99±8 and 272±13, respectively; p=1.53 x 10−6). We found no difference in the time-to-peak (tp) of the calcium transient (52±4 ms and 46±10 ms, p=0.11, same n as above). The time to 90% decay (td) of the CICR in PMAD was prolonged (340±9 ms vs. 456±9 ms, p=4e-15), as was the time constant of decay (τ) (152±5 ms vs. 216±7 ms). We cannot exclude that this effect is from dye kinetic filtering as the dye nears saturation inthe PMAD group, rather than a true finding, so we did evaluate this data for significance. The augmented CICR was present in isolated cells long after in vivo cardiac arrest (at least 8 hours which was the longest time point investigated), hence, we labeled this phenomenon calcium “long-term potentiation”. In contrast, consistent with predicted effects of high affinity indicators,25 we found that Fluo-4 minimized this difference in CICR (Supplemental Figure 1). At times, the calcium potentiation led to spontaneous delayed calcium increases, a cellular surrogate candidate for delayed after depolarizations. In Figure 3C, the top panel shows sarcomere shortening under load, and the bottom panel displays CICR for the same fiber. After stretch (arrow), a delayed calcium increase occurred and induced chaotic contraction without coordinated force generation. In this case, with rapid pacing, the “fibrillatory” behavior converted to organized contraction. All cells investigated from all PMAD hearts exposed to load displayed this phenomenon (n=12 from 4 rat hearts). Rarely, load-independent spontaneous delayed calcium increases were seen as shown in Figure 3D. Such behavior was never seen in control cells.
FIGURE 3. Cardiac calcium memory after cardiac arrest.

A. Example CICR transients measured by Fluo-5f in control compared against PMAD, and also PMAD in which arrest was generated in the presence of KN93 (PMAD-KN93) or ryanodine (PMAD-Rya), as well as a control animal exposed to IV infusion of epinephrine (Epi) for 30 minutes. Cells were paced steadily at 1Hz. B. Pooled analysis of ΔF/Fpeak for all conditions tested. The ΔF/Fpeak was significantly higher in animals exposed to epinephrine infusion and cardiac arrest. Ryanodine and KN93 reduced ΔF/Fpeak to control levels in PMAD ventricular myocytes. WT was no different than sham. n=114, 135, 89, 16, 17, 9, 11, 15, 11, 31, 20, 51, 32, 119, 19, 11 cells for control, PMAD, PMADAIP, PMAD-AIP-IN, PMAD-KN93, PMAD-KN92 PMAD-Rya, Epi infusion, control-KN93 (Cntr-KN93), control ryanodine (Cntr-Rya), epi infusion with ryanodine (Epi-Rya), PMAD-Langendorff-Saline (PMAD-Lan-Sal), PMAD-Langendorff-PMAD blood (PMAD-Lan-Blood), Brainstem herniation (BSH), BSH-ryanodine (BSH-Rya), and PMAD-H89. Cntr-Sham experiments demonstrated no difference compared with control. P values compared to control for PMAD, Epi infusion, BSH, PMAD-Lan-Blood were 6.4e-24, 2.2e-18, 2e-25, and 2e-14 (corrected for multiple comparisons). C. Example CICR transients (red) and sarcomere shortening (black) prior to and after carbon fiber stretch. Top set showed stretch followed by a delayed calcium release (black arrow) initiating fibrillatory behavior in both traces. Lower panel showed 30 seconds later termination of “arrhythmia” through rapid pacing. D. Example CICR transients from two different PMAD cells. Top panel showed CICR in response to electrical stimulation followed delayed calcium transients. Bottom panel showed spontaneous calcium transients competing for paced rhythm. E. Pooled analysis of velocity of contraction (Vc) vs sarcomere length for control and PMAD. Baseline (filled) and stretched (open) for pairs of similar stretches from ~1.8 μm ~ 2.0 μm. PMAD (red) than control (black); n= 8, 9 cells from 3 and 4 hearts for PMAD and control hearts respectively. F. Pooled data comparing unloaded %SL vs ΔF/Fpeak of CICR under control, PMAD, epinephrine infusion in animal, but not cells (EPI inf), and epinephrine in isolated cells (EPI cell), but not in whole animal), as well as PMAD in the presence of Alda-1 (PMAD-Alda, n=25 cells from 4 animals). N for control, PMAD, and Epi infusion are the same as above. G. Baseline Force (BSF) in example cells paced steadily at 1 Hz in control, PMAD, PMADKN93 and PMAD-Rya cells. H. Pooled data for BSF measurements at 1 Hz steady-state pacing in control, PMAD in the presence and absence of KN93 or ryanodine. n= 8, 8, 5, 4 cells for control, PMAD, PMAD-KN93, PMAD-Rya.
Simultaneous measurements of CICR and force generation allowed us to test whether length-dependent activation, a putative mechanism for the Frank-Starling effect,30 was impaired in PMAD by comparing contraction velocity immediately before and after stretch (Figure 3E). Care was taken to equalize baseline and stretched sarcomere lengths. The post-stretch change in velocity was similar between control and PMAD, but the absolute velocity was also lower in PMAD (ratio post to pre-stretch vc: 1.7±0.2 vs. 1.8±0.2 for control vs PMAD, p=0.5). This is important because while PMAD is associated with reduced baseline myofilament calcium sensitivity, the response to stretch (length-dependent activation) is preserved.
To examine the multidimensional relationship between force, frequency, and CICR we plotted these together in three dimensional space. This demonstrates the reduced myofilament sensitivity seen at all frequencies as well as the extent to which the potentiation of the calcium transient abrogates the reduced contractile force (Figure 4A and 4B).
FIGURE 4. Calcium Force Frequency Relationships after cardiac arrest.

A. Plot of force (z-axis), pacing frequency (x-axis) and calcium expressed as ΔF/Fpeak (y-axis) for control cells at slack length loaded with Fluo-5f and simultaneously stretched with CF. n=4 cells from 2 control hearts. The curve showed a steep curve shifted to lower ΔF/Fpeak. The color represents force and is coded from low to high as from blue to red, B. Similar plot in post arrest cells. n=4 cells from 2 control hearts. By comparison to panel A, the curve is shifted to a flatter curve despite also being shifted to higher ΔF/Fpeaks (rightward) at all pacing frequencies. Force was color coded with the same palette as in panel A, and all scale bars were identical.
Since previous studies in vitro have not found an augmented CICR post-arrest, we compared our data with that from cells isolated from a saline perfused bubble-oxygenated Langendorff “resuscitated” hearts. Cells from these hearts showed a more minor depression of %SL and no difference in CICR as compared to controls (Figure 3B), in agreement with most reported results.6,13 However, when the control Langendorff heart was perfused with oxygenated and pH-controlled blood from a cardiac arrest animal, augmented CICR was demonstrated (Figure 3B, PMAD-Lan-Blood), suggesting a dependency of effect not on neural innervation of the heart but on an intact circulation. Since cardiac arrest is known to be associated with marked increases in both catecholamines18,32 and reactive oxygen species, we hypothesized these as key mechanisms.
Potentiation of CICR begins in vivo
Although responses in isolated cells are often used as a surrogate for in vivo effects, and the control animals gave us confidence that this effect related to the cardiac arrest, there remained the possibility that this was an artifact of that isolation process for those cells. In order to assess whether that CICR was augmented in vivo, we transduced rat hearts with AAV9-GCaMP6f vectors and measured in vivo CICR using a custom built fiber optic imaging system (AUST Development, LLC) tunneled under the skin to the heart (Figure 5A, B). Using this system, we were able to make sequential measurements in the same rat (for example a weeks apart in Figure 5C). An example fluorescent image through fiber optic on heart from baseline and peak calcium transient was shown (Figure 5D). We then performed a cardiac arrest experiment as described above with additional measurement of CICR by way of GECI fluorescence. CICR post-arrest was potentiated in this in vivo measurement, similarly to our findings at the cellular level from this same model, and the blood perfused preparation described above, but in contrast to the traditional saline perfused Langendorff preparation. Furthermore, the proarrhythmic potential of delayed after calcium depolarizations in this post-arrest setting was seen as well, in agreement with our in vitro data (Figure 5E). We found the peak ΔF/F to be ~300% increased post-cardiac arrest compared to internal baseline pre-arrest control (n=2 hearts). This increase, when corrected for heart rate by atrial pacing to similar heart rates is less dramatic and approximates ~200%.
FIGURE 5. In vivo CICR by AAV9-GCaMP6f fluorescence.
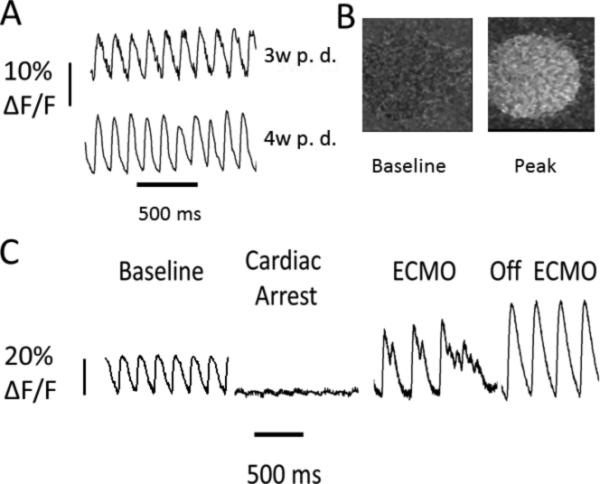
A. Example fluorescence measurements of CICR (y-axis F/F vs x-axis time). In vivo fluorescence imaging was performed 3 or 4 weeks after GCaMP6f delivery in rat (3w P.D., 4w P.D.). B. Example image of raw fluorescence seen through fiber optic on heart by camera at baseline and peak fluorescence during a calcium transient. C. Example cardiac arrest experiment from a different GECI-rat while measuring CICR fluorescence continuously through experiment (y-axis F/F vs x-axis time). Shown were measurements at baseline, after cardiac arrest, on ECMO demonstrating delayed calcium release initiating fibrillatory behavior similar to what was seen in isolated cells, and finally off ECMO.
Catecholamine excess reproduces calcium potentiation
It has been demonstrated that catecholamines are dramatically elevated with in vivo cardiac arrest.18 To test the hypothesis directly in vivo that catecholamine signaling was responsible for the augmented CICR, we blunted catecholamine signaling with beta- and alpha-blockade (labetalol). However, elevated catecholamines are also required for survival and the presence of signaling antagonists in vivo prevented adequate resuscitation, as might be expected. Therefore, to examine the role of catecholaminergic activation in the promotion of cardiac myocyte calcium potentiation in vivo, a stable, ventilated animal was infused with epinephrine for 30 minutes based on our data demonstrating its profound elevation post-arrest. Infusion induced a typical adrenergic response, characterized by an increase in heart rate (HR) and Mean Arterial Pressure (MAP). Upon termination of epinephrine infusion prior to cell isolation, blood pressure and heart rate normalized (blood pH was also normal) within 10 min. In isolated cardiomyocytes from this model, a pronounced increase in CICR was seen similar to post cardiac arrest (Figures 3A and B, Epi). We further explored whether cardiac myocyte calcium potentiation was sustained after brainstem herniation (BSH), a condition known to induce a physiologic endogenous catecholamine surge.33 This led to a similar dramatic increase in heart rate and blood pressure. In cells isolated from these hearts, a similar increase in the ΔF/Fpeak was seen (271±21, n=110 cells from n=9 animals (p=1e-14 against control; Figure 3B, BSH, Supplemental Figure 2).
We also added epinephrine directly to the cell bath of control cells which as expected acutely manifested a markedly increased contractile performance (%SL 20±0.5) in association with a more modest increase CICR (ΔF/Fpeak 141±0.53%). This was a marked contrast to the prolonged administration in vivo as described above.
Cardiac myocyte calcium potentiation is dependent on CaMKII and ryanodine but not on PKA
Since a link between sub-acute and chronic beta-adrenergic signaling and Ca2+/Calmodulin kinase II (CaMKII), rather than the acute PKA, dependent pathways has been observed,34–36 we tested whether CaMKII inhibition could block the induction of CICR potentiation in our in vivo cardiac arrest model. As shown in Figure 3B, AIP, a CaMKII inhibitor, given prior to and during cardiac arrest, abolished the CICR increase in both PMAD and epinephrine-infusion models. The lower affinity inhibitor KN93 blunted cardiac myocyte calcium potentiation in PMAD (Figure 3A, B) but not in epinephrine infusion. In the presence of AIP or KN93, no delayed after depolarizations or stretch-induced cellular fibrillatory behavior was seen. Because CaMKII activation is partly mediated by calcium-dependent pathways and CaMKII is known to be localized to the dyad, we tested whether inhibiting CICR during cardiac arrest would blunt cardiac myocyte calcium potentiation. We found that both for epinephrine infusion and PMAD, simultaneous ryanodine application blocked the augmentation in CICR (Figure 3B), suggesting that calcium release by ryanodine receptor (RyR) was a critical contributor (in the presence of ryanodine, animals were supported with ECMO). In contrast, the PKA inhibitor H89 did not prevent the increase in CICR either prior to and during cardiac arrest (Figure 3B) suggesting CaMKII activation was a critical component. The sham inhibitor KN92 had no effect on CICR in PMAD (Figure 3B).
Cardiac myocyte calcium potentiation supports impaired cardiac function
We measured both %SL (Figure 3F) and force (Figure 3G, H) in cells isolated from PMAD hearts that were exposed to CaMKII inhibitors, KN93, AIP or ryanodine, during the initial cardiac arrest to inhibit CICR augmentation. We found that both %SL and baseline force were lower under these conditions, suggesting that the augmented CICR is necessary to support cardiac contractility after cardiac arrest.
CaMKII-dependent phosphorylation in PMAD
CaMKII is a mediator of long-term potentiation in the brain, a form of synaptic plasticity and is associated with enhanced calcium signaling.37,38 CaMKII activity is important for Ca regulated/mediated activities such as excitation-contraction coupling (ECC) and excitation-transcription coupling (ETC). CaMKII activity may also become Ca/CaM autonomous and such activity is increased in myocardial disease contributing to apoptosis, arrhythmia, disrupted ECC and ETC leading to pathological hypertrophy.39 CaMKII and its targets, RyR2 and phospholamban (PLN), have also been shown to be critical in cardiac ischemia/reperfusion injury.40,41 To assess CaMKII activation/autophosphorylation as a potential mediator of cardiomyocyte calcium potentiation in our in vivo cardiac arrest model, we examined the phosphorylation status of CaMKII Thr 287 (PT287) and total CaMKII protein in ventricular tissue lysates after cardiac arrest (Figure 6A). After 30 minutes of cardiac arrest and 30 minutes of ECMO reperfusion, the ratio of pCaMKII PT287 to total CaMKII nearly doubled in PMAD group compared with the control group (Figure 6C) while total CaMKII protein remained unchanged (Figure 6D), indicating an activation of CaMKII. This activation was reduced by 30% in the presence of the CaMKII inhibitor AIP, given 30-40 minutes prior to the onset of cardiac arrest (Figure 6C). We also examined the phosphorylation state of RyR2 and PLN, the targets of both CaMKII and PKA at the sarcoplasmic reticulum (SR). CaMKII dependent phosphorylation of PLN at Thr 17 (PT17) showed similar patterns (Figure 6E, F). The ratio of PT17/total PLN increased nearly two-fold in the cardiac arrest group than in the control group and PT17 phosphorylation was abolished by AIP treatment prior to cardiac arrest. On the other hand PKA-dependent phosphorylation of PLN at serine 16 is indifferent between the cardiac arrest group and the control group (Figure 6J).
FIGURE 6. CaMKII-dependent phosphorylation is increased in PMAD and can be inhibited by AIP and Alda-1.

A and B. Example Western blots showing CaMKII-dependent phosphorylation and protein level of CaMKII, phospholamban and RyR2 in Control group, PMAD model (30 minutes of cardiac arrest and 30 minutes of ECMO reperfusion) and PMAD in the presence of AIP (A) and Alda-1 (B), both were given 30 minutes before the onset of cardiac arrest. C-I. Quantitative summary of Western blot analysis. CaMKII-dependent phosphorylation of CaMKII at T287 (C) and phosphorylation of PLN at T17 (E) increased significantly in PMAD and inhibited by AIP while the protein level of CaMKII (D) and PLN (F) did not change significantly. CaMKII autophosphorylation (PT287) in PMAD was also inhibited by Alda-1(C). CaMKII-dependent RyR2 phosphorylation at S2814 did not vary considerably in PMAD or with AIP or Alda-1 treatment, illustrated as PS2814/RyR2 in G. However, RyR2 protein level surged 5-fold in PMAD compared with Control group (RyR2/GAPDH in H) therefore overall pRyR2 PS2814 was also shown to increase dramatically (normalized to GAPDH in I). Neither AIP nor Alda-1 treatment reduced the protein level or total RyR2 phosphorylation at S2814 (H, I). J. Western blot and quantitative assessment showed that PKA phosphorylation of PLN at S16 and PLN protein level were indifferent between PMAD and Control group. K. Representative Western blot and quantitative summary showed that CaMKII was activated after PMAD but not epinephrine infusion for 30 minutes. Graph data represent the Mean ± SEM of each group. *, p<0.05; **, p<0.005; ns, not-significant. p-values were reported by ordinary one-way ANOVA with multiple comparisons (C-I and K) or t-test (J).
The most striking change during PMAD was found on RyR2, both on the detection of total protein and phosphorylation at Ser 2814 (PS2814)(Figure 6A). However the ratio of PS2814 to total RyR2 did not vary significantly (Figure 6G). Signals for RyR2 protein and PS2814 surged approximately 7-fold in PMAD (Figure 6H, I) and were not significantly altered by AIP or Alda-1 treatment. In addition, a single band of RyR2 protein of 565 kD was detected in hearts from the control group while a doublet of bands became prominent after cardiac arrest, suggesting some form of oligomerization or protein modification (see discussion). As similar calcium potentiation was observed in cardiomyocyte isolated from rats treated with epinephrine as in PMAD (Figure 3B), the phosphorylation status of CaMKII, phospholamban and RyR2 were also examined in heart tissues collected after 30 minutes of epinephrine infusion. CaMKII phosphorylation was also increased in this model but to a lesser extent than that seen during cardiac arrest (Figure 6K) leading us to consider the contributory role of oxidative stress (reactive oxygen species) in the effects seen in vivo.
Cardiac arrest increased myocardial O2•– generation predominantly from mitochondria
We measured myocardial superoxide (O2•–) production using lucigenin-enhanced chemiluminescence. We found it to be elevated after epinephrine infusion but significantly more elevated after PMAD (+70% increase in total chemiluminescence compared with control; P < 0.001; Figure 7A). When ventricular tissue was incubated with the NOS inhibitor L-NMMA or the NADPH oxidase inhibitor DPI, total O2•– levels were not significantly different, suggesting that the major contribution of O2•– was mitochondrial. To confirm the chemiluminescence findings and investigate further the cellular source of O2•–, we measured its production using dihydroethidium oxidative fluorescence microtopography (Figure 7B) finding again significant O2•– production in PMAD. Finally, to confirm the source of O2•– we used the mitochondria-targeted antioxidant with O2•– and alkyl scavenging properties Mito-TEMPO. Mito-TEMPO led to a significant reduction in O2•– generation (P < 0.01, Figure 7B), confirming the dominant mitochondrial source.
FIGURE 7. Cardiac arrest hearts demonstrated increased myocardial O2•– generation, which can be inhibited by Alda-1.

A. Myocardial O2•– production, measured by lucigenin, was mildly increased in hearts treated with epinephrine, but markedly elevated after cardiac arrest (†P < 0.01). The NADPH oxidase inhibitor DPI and the NOS inhibitor L-NMMA did not change O2•– generation compared to uninhibited controls, however the ALDH2 inhibitor Alda-1 did inhibit O2•– (*P < 0.01). B. Dihydroethidium (DHE) fluorescence microscopy allowed topographic assessment of O2•– production in the ventricular wall, and showed mild increase in O2•– production with epinephrine infusion, compared with marked increase after cardiac arrest (†P < 0.01). Alda-1 inhibited O2•– generation. Mito-TEMPO, used as the mitochondria-targeted antioxidant with superoxide and alkyl scavenging properties, confirmed that the majority of O2•– being generated was from mitochondria (compared to uninhibited *P < 0.01). Myocytes exhibited green autofluorescence. White arrows denoted endothelial O2•– producing cells. Scale bar: 5 μm.
Alda-1 reduces calcium potentiation and improves cardiac performance in PMAD
Since the augmented CICR triggered by catecholamines and maintained by CaMKII was supportive of cardiac function and survival in the early stages, we looked towards downstream effects of oxygen radicals as a potential therapeutic intervention. Recently, activation of aldehyde dehydrogenase type 2 (ALDH2) by Alda-1 has been shown to play a pivotal role in cardioprotection in the setting of myocardial infarction through detoxification of O2•– induced reactive aldehydes (such as 4-HNE and acetaldehyde) to less reactive acids (such as 4-hydroxynon-2-enoic acid and acetic acid).42 We hypothesized the reactive aldehydes were playing an important role in PMAD. In keeping with this hypothesis, we found 4-HNE adducts to be significantly increased after cardiac arrest, and administration of Alda-1 in vivo during cardiac arrest significantly decreased both 4-HNE adducts (Supplemental Figure 3) and O2•– to levels seen after Mito-TEMPO inhibition (P < 0.01 compared with control, Figure 7A,B). Alda-1 also had a dramatic beneficial impact on PMAD both in vitro and in vivo. At the cellular level, Alda-1 treatment blunted the increase in CICR seen in PMAD by 30% similarly to CaMKII inhibition (Figure 3B). However, while myofilament calcium sensitivity was unchanged with CaMKII inhibition, it was enhanced with Alda-1 (Figure 3F). In addition, immunoblotting of CaMKII and autophosphorylated CaMKII revealed that in hearts exposed to Alda-1 during cardiac arrest, T287 autophosphorylation was reduced by 30%, similarly to the effect of AIP (Figure 6B, C). As expected, CaMKII inhibition had no impact on 4-HNE adduct formation while Alda-1 reduced 4-HNE adduct formation after cardiac arrest (Supplemental Figure 3). We also found that Alda-1 reduced malondialdehyde fluorescence after myocardial infarction in a separate pig model (data not shown).
Alda-1 improved outcome after cardiac arrest
We found weaning from resuscitation (defined as reaching 20% of the pre-arrest MAP) was successfully achieved in 5 out of 8 animals (62.5%) treated with Alda-1 compared to only 4 out of 30 animals (13%) in the absence of Alda-1. Moreover, with Alda-1 administration, we found that whole animal cardiac contractility, as indexed by ESPVR, PRSW, CI, and τ returned to near control levels (Figure 8A-D, Table 1). Alda-1 had no effect on these same parameters pre-arrest (data not shown).
FIGURE 8.

Alda-1 improved in vivo contractility and recovery rate to spontaneous circulation after cardiac arrest. A. Summary data for ESPVR for control, PMAD, PMAD-Alda (Alda-1) p=0.006. B. Summary data for PRWV for control, PMAD, PMAD-Alda (Alda-1), p=0.04. C. Summary data for CI for control, PMAD, PMAD-Alda (Alda-1)D. Summary data for tau for control, PMAD, PMAD-Alda (Alda-1). P-values in graphs are with respect to control using post-hoc analysis. Comparisons between groups performed using Kruskall-Wallis nonparamateric test. E. Proposed mechanism for post-arrest myocardial dysfunction, mediated by calcium overload, CaMKII, and oxidative stress (ROS). Catecholamines released during cardiac arrest and the ischemic/reperfusion process triggered cytosolic Ca2+ influx and SR Ca2+ release/uptake. The activation of RyR2 (by oxidative stress or other unknown mechanism**) also increased CICR. Elevated cytosolic Ca2+ -activated CaMKII further enhanced CICR and lead to mitochondrial Ca2+ overload. This Ca2+ overload contributed to excess ROS (O2•–) and reactive aldehyde (4-HNE) generation, more CaMKII activation, and other cellular damages. AIP inhibited CaMKII autophosphorylation but did not improve cardiac function after arrest. Alda-1 reduced oxidative stress, inhibited CaMKII auto-phosphorylation and improved cardiac function.
DISCUSSION
For more than three decades, post myocardial arrest dysfunction has been recognized as the critical factor in recovery and survival after cardiac arrest.5, 6 To date, most research focused on ischemia-reperfusion has used variants of a Langendorff isolated-heart model. Here, we present an in vivo model of post-myocardial arrest dysfunction, which, in leaving the neurohumoral axis intact, uncovered a significant increase in CICR in vivo that continued in isolated cardiac myocytes. A similar increase in CICR was reproduced when blood from an arrested animal was used as perfusate in the Langendorff system. Notably, we did not see this potentiation of CICR in the saline perfused Langendorff model. This cardiac myocyte calcium ‘memory’ seems to be triggered by a combination of catecholamines and reactive oxygen species activation and maintained by autophosphorylation of CaMKII with subsequent phosphorylation of RyR2 and PLN.
The role of CAMKII in cardiomyopathy has been described in seminal investigations.34,43–45 Its activation is important in the generation of electrical storm,46,47 and in reperfusion-related arrhythmias,48 while its inhibition can prevent the occurrence of ventricular arrhythmias.49 In our study, we found that inhibition of CaMKII also reduced cellular stretch-induced arrhythmias. We propose that oxidative stress and aldehydic adduct formation contribute to acute myofilament dysfunction and impaired myofilament calcium sensitivity which is partially compensated by this increase in CICR. Consistent with this hypothesis, the small molecule activator of the aldehyde dehydrogenase type 2, Alda-1, dramatically improved cardiac myofilament calcium sensitivity at the whole heart and cellular level by acting upstream of calcium signaling to reduce both mitochondrial O2•– production and toxic aldehydic-adduct formation. In so doing, myofilament calcium sensitivity after arrest was improved, and simultaneously autophosphorylation of CaMKII and CaMKII augmentation of CICR was reduced. This proposed mechanism (Figure 8E) has similarities to neuronal plasticity where CaMKII senses phasic changes in and then augments the same post-synaptic signals.37 We speculate that just as in the brain, CaMKII might induce long-term potentiation38 in the heart. The unique features of CaMKII autophosphorylation and its ability to follow calcium changes phasically37 allow integration of calcium signal in the “post-synaptic” dyadic cleft.34,35,41,43,50–52
In an attempt to track the timing of signaling events that accompanied the calcium potentiation during cardiac arrest and ECMO reperfusion, we traced the dynamics of CaMKII signaling in our in vivo PMAD model. We varied the time of cardiac arrest and reperfusion/resuscitation and indeed observed variations in the phosphorylation status of CaMKII (PT287) and PLN (PT17) (Supplemental Figure 4). Notably, the surge of total RyR2 protein and PS2814 detection was prominent in almost all the heart samples collected after cardiac arrest/reperfusion despite variation in CaMKII activation. It is likely that in addition to CaMKII, RyR2 is responsive to other signaling pathways involved in oxygen homeostasis and return of spontaneous contraction in this cardiac arrest rat model. The overall elevation of RyR2 in its active form (PS2814) would independently increase SR calcium release and contribute to augmented CICR. The elevation of RyR2 signal seen in the Western blots could indicate a rapid increase of RyR2 proteins after cardiac arrest and reperfusion, although it was unclear which signals may lead to such induction. Alternatively, RyR2 proteins may be post-translationally modified in response to cardiac arrest/reperfusion and become stabilized against degradation. It is also possible that cardiac arrest and oxidative stress resulted in RyR2 conformational change that leads to increased antigenicity. The Western blots of RyR2 and PS2814 were performed under non-reducing conditions, in which the RyR2 protein could retain some folded structure. It has been reported that oxidative stress (ROS) could induce conformational change in RyR2.53 Such conformational changes could possibly make the epitope more accessible to the antibodies and resulted in higher detectable signals.
The SR Ca2+ -ATPase regulator phospholamban (PLN) is a common target of both CaMKII (phosphorylation at Thr 17, PT17) and PKA (phosphorylation at Ser 16, PS16). Examination of these phosphorylation sites after in vivo cardiac arrest-reperfusion demonstrated clearly that CaMKII but not PKA pathway was activated after cardiac arrest, as PT17 /PLN ratio surged while PS16/PLN ratio remained unchanged. Inhibition of CaMKII with AIP prior to cardiac arrest reduced CaMKII activation and abolished PLN PT17 phosphorylation and calcium potentiation, however, in vivo and in vitro contractility was not improved. It is possible that CaMKII-mediated PLN PT17 phosphorylation is not contributing to the post-arrest myocardial dysfunction. A previous study on the PLN S16A/T17A (non-phosphorylatable PLN) mutant mice showed that CaMKII-dependent increase of PLN PT17 opposes rather than contributes to ischemia/reperfusion damage.40 Ideally genetic models such as PLN S16A/S17A mutant mice, CaMKII mutant mice (overexpression and knockouts) and RyR2 S2814D, S2814A mutant mice would be studied in this in vivo cardiac arrest model to define the molecular pathway and clarify the roles of individual contributors. However the technical challenge of reproducing cardiac arrest and ECMO resuscitation in the mouse has not yet been achieved.
In conclusion, using new approaches for simultaneous force and CICR measurement in vitro and GECI enabled calcium fluorescence measurements in vivo, we present a novel mechanism of post myocardial arrest dysfunction (Figure 8E). We propose that hypoxia triggers a surge in both catecholamines and reactive oxygen species that triggers an increase in calcium induced calcium release that is further maintained by auto-phosphorylation of CaMKII. In addition, stretch of cardiac cells may increase calcium uptake and sarcoplasmic reticulum releasability, aiding CICR.16,54 This increased CICR supports contraction in the face of reduced myofilament sensitivity. We show that inhibiting aldehydic adduct formation during arrest/reperfusion increases myofilament sensitivity, reduces CaMKII activation, and improves survival. These mechanisms should be explored further as potential targets for human therapeutic trials.
Major limitations to our work remain. First, the rat is obviously not a clinical model system, so whether the results translate well to clinical studies is not known. Second, the role of the action potential has not been investigated in this work because of technical limitations to twitching cells and photostability from fluorescence voltage dyes. In addition, the interplay of CaMKII activation by calcium versus oxidation has not been fully elucidated.
Supplementary Material
Clinical Perspective.
What is new?
We developed a rodent model of cardiac arrest using ECMO resuscitation. We observed calcium-induced calcium release (CICR) in vivo before and after resuscitation using a genetically encoded calcium sensor and a novel fiber-optic catheter imaging system. In cardiomyocytes isolated from this model, we assessed mechanical load and CICR simultaneously using the micro-carbon-fiber technique and observed reduced myocardial performance but potentiation of CICR.
This “memory” phenomenon was CaMKII dependent and mechanistically similar to Long-Term-Potentiation, a form of hippocampal learning. Since aldehydic adduct formation and oxidative stress were high post arrest, we used a small molecule activator of aldehyde dehydrogenase type 2 which restored calcium signaling and improved cardiac performance.
What are the clinical perspectives?
Despite return of circulation, survival after sudden cardiac arrest is low in part due to post-myocardial arrest dysfunction. Our data demonstrate that this myopathy is associated with a CaMKII dependent increase in calcium induced calcium release which seems to “support” the function of the heart short term but carries the potential for long term negative consequences. In our model, a small molecular activator of aldehyde dehydrogenase restored heart function and improved survival.
This may represent a novel therapeutic approach to a condition with a high mortality.
Acknowledgements
We thank Dr. Peter Lee for his construction of the Matlab control device for carbon fiber stretch. We also thank Dr. Kathia Zaleta for assisting in experiments.
Funding Sources
Papworth Hospital NHS Foundation Trust: Gledhill Fellowship Reference 175-2009-MD398 (F.T.), NIH New Investigator DP2 award OD004613 (E.A.), and NIH T32 award HL094274-01A2 (Stanford) and NIH T32 award HL 007731 (UCSF), and NIH R43 HL116017 (C.W.), NIH R37 AA011147 MERIT award (D.M-R), the British Heart Foundation (C.B., P.K.), and the European Research Council Advanced Grant CardioNect (P.K.).
Footnotes
Disclosures
DM-R is a founder of ALDEA Pharmaceuticals. However, she has no active role in the company nor any consulting arrangements and none of the work in her lab is supported by or in collaboration with the company. C.W. is a senior scientist at AUST Development, LLC and inventor of percutaneous optical mapping catheter system.
References
- 1.Nolan JP, Neumar RW, Adrie C, Aibiki M, Berg RA, Böttiger BW, Callaway C, Clark RSB, Geocadin RG, Jauch EC, Kern KB, Laurent I, Longstreth WT, Merchant RM, Morley P, Morrison LJ, Nadkarni V, Peberdy MA, Rivers EP, Rodriguez-Nunez A, Sellke FW, Spaulding C, Sunde K, Hoek TV. Post-cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A Scientific Statement from the International Liaison Committee on Resuscitation; the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; the Council on Stroke. Resuscitation. 2008;79:350–379. doi: 10.1016/j.resuscitation.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 2.Stub D, Bernard S, Duffy SJ, Kaye DM. Post cardiac arrest syndrome: a review of therapeutic strategies. Circulation. 2011;123:1428–1435. doi: 10.1161/CIRCULATIONAHA.110.988725. [DOI] [PubMed] [Google Scholar]
- 3.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Després J-P, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER, III, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB. Executive Summary: Heart Disease and Stroke Statistics—2015 Update: A Report From the American Heart Association. Circulation. 2015;131:434–441. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 4.Zia A, Kern KB. Management of postcardiac arrest myocardial dysfunction. Curr Opin Crit Care. 2011;17:241–246. doi: 10.1097/MCC.0b013e3283447759. [DOI] [PubMed] [Google Scholar]
- 5.Kern KB, Hilwig RW, Rhee KH, Berg RA. Myocardial dysfunction after resuscitation from cardiac arrest: an example of global myocardial stunning. J Am Coll Cardiol. 1996;28:232–240. doi: 10.1016/0735-1097(96)00130-1. [DOI] [PubMed] [Google Scholar]
- 6.Bolli R, Marbán E. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev. 1999;79:609–634. doi: 10.1152/physrev.1999.79.2.609. [DOI] [PubMed] [Google Scholar]
- 7.Zaugg CE, Ziegler A, Lee RJ, Barbosa V, Buser PT. Postresuscitation stunning: postfibrillatory myocardial dysfunction caused by reduced myofilament Ca2+ responsiveness after ventricular fibrillation-induced myocyte Ca2+ overload. J Cardiovasc Electrophysiol. 2002;13:1017–1024. doi: 10.1046/j.1540-8167.2002.01017.x. [DOI] [PubMed] [Google Scholar]
- 8.Vittone L, Mundina-Weilenmann C, Mattiazzi A. Phospholamban phosphorylation by CaMKII under pathophysiological conditions. Front Biosci. 2008;13:5988–6005. doi: 10.2741/3131. [DOI] [PubMed] [Google Scholar]
- 9.Kim S-J, Depre C, Vatner SF. Novel mechanisms mediating stunned myocardium. Heart Fail Rev. 2003;8:143–153. doi: 10.1023/a:1023040718319. [DOI] [PubMed] [Google Scholar]
- 10.Solaro RJ, Arteaga GM. Heart failure, ischemia/reperfusion injury and cardiac troponin. Adv Exp Med Biol. 2007;592:191–200. doi: 10.1007/978-4-431-38453-3_17. [DOI] [PubMed] [Google Scholar]
- 11.Bers DM. Cardiac excitation–contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 12.Valverde CA, Mundiña-Weilenmann C, Reyes M, Kranias EG, Escobar AL, Mattiazzi A. Phospholamban phosphorylation sites enhance the recovery of intracellular Ca2+ after perfusion arrest in isolated, perfused mouse heart. Cardiovasc Res. 2006;70:335–345. doi: 10.1016/j.cardiores.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 13.Valverde CA, Kornyeyev D, Ferreiro M, Petrosky AD, Mattiazzi A, Escobar AL. Transient Ca2+ depletion of the sarcoplasmic reticulum at the onset of reperfusion. Cardiovasc Res. 2010;85:671–680. doi: 10.1093/cvr/cvp371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kusuoka H, Koretsune Y, Chacko VP, Weisfeldt ML, Marban E. Excitation-contraction coupling in postischemic myocardium. Does failure of activator Ca2+ transients underlie stunning? Circ Res. 1990;66:1268–1276. doi: 10.1161/01.res.66.5.1268. [DOI] [PubMed] [Google Scholar]
- 15.Calaghan SC, Belus A, White E. Do stretch-induced changes in intracellular calcium modify the electrical activity of cardiac muscle? Prog Biophys Mol Biol. 2003;82:81–95. doi: 10.1016/s0079-6107(03)00007-5. [DOI] [PubMed] [Google Scholar]
- 16.Iribe G, Ward CW, Camelliti P, Bollensdorff C, Mason F, Burton RAB, Garny A, Morphew MK, Hoenger A, Lederer WJ, Kohl P. Axial stretch of rat single ventricular cardiomyocytes causes an acute and transient increase in Ca2+ spark rate. Circ Res. 2009;104:787–795. doi: 10.1161/CIRCRESAHA.108.193334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bub G, Camelliti P, Bollensdorff C, Stuckey DJ, Picton G, Burton RAB, Clarke K, Kohl P. Measurement and analysis of sarcomere length in rat cardiomyocytes in situ and in vitro. Am J Physiol Heart Circ Physiol. 2010;298:H1616–25. doi: 10.1152/ajpheart.00481.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ali AA, White P, Xiang B, Lin H-Y, Tsui SS, Ashley E, Lee TW, Klein JRH, Kumar K, Arora RC, Large SR, Tian G, Freed DH. Hearts from DCD donors display acceptable biventricular function after heart transplantation in pigs. Am J Transplant. 2011;11:1621–1632. doi: 10.1111/j.1600-6143.2011.03622.x. [DOI] [PubMed] [Google Scholar]
- 19.Ali AA, Downey P, Singh G, Qi W, George I, Takayama H, Kirtane A, Krishnan P, Zalewski A, Freed D, Large SR, Ashley EA, Leon MB, Bacchetta M, Ali ZA. Rat model of veno-arterial extracorporeal membrane oxygenation. J Transl Med. 2014;12:37. doi: 10.1186/1479-5876-12-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Günzinger R, Wildhirt SM, Schad H, Heimisch W, Gurdan M, Mendler N, Grammer J, Lange R, Bauernschmitt R. A rat model of cardiopulmonary bypass with cardioplegic arrest and hemodynamic assessment by conductance catheter technique. Basic Res Cardiol. 2007;102:508–517. doi: 10.1007/s00395-007-0668-x. [DOI] [PubMed] [Google Scholar]
- 21.Ashley EA, Powers J, Chen M, Kundu R, Finsterbach T, Caffarelli A, Deng A, Eichhorn J, Mahajan R, Agrawal R, Greve J, Robbins R, Patterson AJ, Bernstein D, Quertermous T. The endogenous peptide apelin potently improves cardiac contractility and reduces cardiac loading in vivo. Cardiovasc Res. 2005;65:73–82. doi: 10.1016/j.cardiores.2004.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suga H. Ventricular energetics. Physiol Rev. 1990;70:247–277. doi: 10.1152/physrev.1990.70.2.247. [DOI] [PubMed] [Google Scholar]
- 23.Charo DN, Ho M, Fajardo G, Kawana M, Kundu RK, Sheikh AY, Finsterbach TP, Leeper NJ, Ernst KV, Chen MM, Ho YD, Chun HJ, Bernstein D, Ashley EA, Quertermous T. Endogenous regulation of cardiovascular function by apelin-APJ. Am J Physiol Heart Circ Physiol. 2009;297:H1904–13. doi: 10.1152/ajpheart.00686.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iribe G, Helmes M, Kohl P. Force-length relations in isolated intact cardiomyocytes subjected to dynamic changes in mechanical load. Am J Physiol Heart Circ Physiol. 2007;292:H1487–97. doi: 10.1152/ajpheart.00909.2006. [DOI] [PubMed] [Google Scholar]
- 25.Bollensdorff C, Lookin O, Kohl P. Assessment of contractility in intact ventricular cardiomyocytes using the dimensionless “Frank-Starling Gain” index. Pflugers Arch. 2011;462:39–48. doi: 10.1007/s00424-011-0964-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woods CE, Novo D, DiFranco M, Vergara JL. The action potential-evoked sarcoplasmic reticulum calcium release is impaired in mdx mouse muscle fibres. J Physiol. 2004;557:59–75. doi: 10.1113/jphysiol.2004.061291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee P, Wang K, Woods CE, Yan P, Kohl P, Ewart P, Loew LM, Terrar DA, Bollensdorff C. Cardiac electrophysiological imaging systems scalable for high-throughput drug testing. Pflugers Arch. 2012;464:645–656. doi: 10.1007/s00424-012-1149-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu Y, Gao Z, Chen B, Koval OM, Singh MV, Guan X, Hund TJ, Kutschke W, Sarma S, Grumbach IM, Wehrens XHT, Mohler PJ, Song L-S, Anderson ME. Calmodulin kinase II is required for fight or flight sinoatrial node physiology. Proc Natl Acad Sci U S A. 2009;106:5972–5977. doi: 10.1073/pnas.0806422106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ali ZA, Bursill CA, Douglas G, McNeill E, Papaspyridonos M, Tatham AL, Bendall JK, Akhtar AM, Alp NJ, Greaves DR, Channon KM. CCR2-mediated antiinflammatory effects of endothelial tetrahydrobiopterin inhibit vascular injury-induced accelerated atherosclerosis. Circulation. 2008;118:S71–7. doi: 10.1161/CIRCULATIONAHA.107.753558. [DOI] [PubMed] [Google Scholar]
- 30.Le Guennec JY, Peineau N, Argibay JA, Mongo KG, Garnier D. A new method of attachment of isolated mammalian ventricular myocytes for tension recording: length dependence of passive and active tension. J Mol Cell Cardiol. 1990;22:1083–1093. doi: 10.1016/0022-2828(90)90072-a. [DOI] [PubMed] [Google Scholar]
- 31.Carrozza JP, Jr, Bentivegna LA, Williams CP, Kuntz RE, Grossman W, Morgan JP. Decreased myofilament responsiveness in myocardial stunning follows transient calcium overload during ischemia and reperfusion. Circ Res. 1992;71:1334–1340. doi: 10.1161/01.res.71.6.1334. [DOI] [PubMed] [Google Scholar]
- 32.Laing GS, Kumar PS, Frayn KN, Little RA. Cardiac arrest and plasma catecholamines. J R Soc Med. 1983;76:1080–1081. [PMC free article] [PubMed] [Google Scholar]
- 33.Berman M, Ali A, Ashley E, Freed D, Clarke K, Tsui S, Parameshwar J, Large S. Is stress cardiomyopathy the underlying cause of ventricular dysfunction associated with brain death? J Heart Lung Transplant. 2010;29:957–965. doi: 10.1016/j.healun.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 34.Zhang R, Khoo MSC, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr, Thiel W, Guatimosim S, Song L-S, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 35.Grimm M, Brown JH. Beta-adrenergic receptor signaling in the heart: role of CaMKII. J Mol Cell Cardiol. 2010;48:322–330. doi: 10.1016/j.yjmcc.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang W, Zhu W, Wang S, Yang D, Crow MT, Xiao R-P, Cheng H. Sustained beta1-adrenergic stimulation modulates cardiac contractility by Ca2+/calmodulin kinase signaling pathway. Circ Res. 2004;95:798–806. doi: 10.1161/01.RES.0000145361.50017.aa. [DOI] [PubMed] [Google Scholar]
- 37.Amici M, Doherty A, Jo J, Jane D, Cho K, Collingridge G, Dargan S. Neuronal calcium sensors and synaptic plasticity. Biochem Soc Trans. 2009;37:1359–1363. doi: 10.1042/BST0371359. [DOI] [PubMed] [Google Scholar]
- 38.Okamoto K, Bosch M, Hayashi Y. The roles of CaMKII and F-actin in the structural plasticity of dendritic spines: a potential molecular identity of a synaptic tag? Physiology. 2009;24:357–366. doi: 10.1152/physiol.00029.2009. [DOI] [PubMed] [Google Scholar]
- 39.Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51:468–473. doi: 10.1016/j.yjmcc.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Di Carlo MN, Said M, Ling H, Valverde CA, De Giusti VC, Sommese L, Palomeque J, Aiello EA, Skapura DG, Rinaldi G, Respress JL, Brown JH, Wehrens XHT, Salas MA, Mattiazzi A. CaMKII-dependent phosphorylation of cardiac ryanodine receptors regulates cell death in cardiac ischemia/reperfusion injury. J Mol Cell Cardiol. 2014;74:274–283. doi: 10.1016/j.yjmcc.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salas MA, Valverde CA, Sánchez G, Said M, Rodriguez JS, Portiansky EL, Kaetzel MA, Dedman JR, Donoso P, Kranias EG, Mattiazzi A. The signalling pathway of CaMKII-mediated apoptosis and necrosis in the ischemia/reperfusion injury. J Mol Cell Cardiol. 2010;48:1298–1306. doi: 10.1016/j.yjmcc.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen C-H, Budas GR, Churchill EN, Disatnik M-H, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Passier R, Zeng H, Frey N, Naya FJ, Nicol RL, McKinsey TA, Overbeek P, Richardson JA, Grant SR, Olson EN. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Invest. 2000;105:1395–1406. doi: 10.1172/JCI8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA, Katus HA, Bassel-Duby R, Maier LS, Olson EN. The δ isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proceedings of the National Academy of Sciences. 2009;106:2342–2347. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D, Brown JH, Heller Brown J. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119:1230–1240. doi: 10.1172/JCI38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsuji Y. Electrical storm and calcium signaling: a review. J Electrocardiol. 2011;44:725–729. doi: 10.1016/j.jelectrocard.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 47.Tsuji Y, Hojo M, Voigt N, El-Armouche A, Inden Y, Murohara T, Dobrev D, Nattel S, Kodama I, Kamiya K. Ca2+-Related Signaling and Protein Phosphorylation Abnormalities Play Central Roles in a New Experimental Model of Electrical Storm. Circulation. 2011;123:2192–2203. doi: 10.1161/CIRCULATIONAHA.110.016683. [DOI] [PubMed] [Google Scholar]
- 48.Said M, Becerra R, Valverde CA, Kaetzel MA, Dedman JR, Mundiña-Weilenmann C, Wehrens XH, Vittone L, Mattiazzi A. Calcium-calmodulin dependent protein kinase II (CaMKII): a main signal responsible for early reperfusion arrhythmias. J Mol Cell Cardiol. 2011;51:936–944. doi: 10.1016/j.yjmcc.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bell JR, Curl CL, Ip WTK, Delbridge LMD. Ca2+/calmodulin-dependent protein kinase inhibition suppresses post-ischemic arrhythmogenesis and mediates sinus bradycardic recovery in reperfusion. Int J Cardiol. 2012;159:112–118. doi: 10.1016/j.ijcard.2011.02.038. [DOI] [PubMed] [Google Scholar]
- 50.Saucerman JJ, Bers DM. Calmodulin binding proteins provide domains of local Ca2+ signaling in cardiac myocytes. J Mol Cell Cardiol. 2012;52:312–316. doi: 10.1016/j.yjmcc.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vila-Petroff M, Salas MA, Said M, Valverde CA, Sapia L, Portiansky E, Hajjar RJ, Kranias EG, Mundiña-Weilenmann C, Mattiazzi A. CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury. Cardiovasc Res. 2007;73:689–698. doi: 10.1016/j.cardiores.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 52.Schott P, Jacobshagen C, Köhler J, Seidler T, Asif AR, Dihazi H, Hasenfuss G, Maier LS. Proteome changes in CaMKIIδC-overexpressing cardiac myocytes. Cardiovasc Pathol. 2010;19:e241–50. doi: 10.1016/j.carpath.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 53.Oda T, Yang Y, Uchinoumi H, Thomas DD, Chen-Izu Y, Kato T, Yamamoto T, Yano M, Cornea RL, Bers DM. Oxidation of ryanodine receptor (RyR) and calmodulin enhance Ca release and pathologically alter, RyR structure and calmodulin affinity. J Mol Cell Cardiol. 2015;85:240–248. doi: 10.1016/j.yjmcc.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peyronnet R, Nerbonne JM, Kohl P. Cardiac Mechano-Gated Ion Channels and Arrhythmias. Circ Res. 2016;118:311–329. doi: 10.1161/CIRCRESAHA.115.305043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.