

Abstract
Background: Osteopontin (OPN) is a pleiotropic cytokine, which has been shown to a close relationship with cardiac fibrosis. Overexpression of OPN in cardiomyocytes induces dilated cardiomyopathy (DCM). This research is to study whether inhibition of OPN could reduce myocardial remodelling in DCM, and if this process is focal adhesion kinase (FAK) dependent, which is recently found an important signal molecule in fibrosis. Method: Eight-week-old cTnTR141W transgenic mouse of DCM were injected with OPN-shRNA in left ventricular free wall, which could inhibit the OPN expression. Six weeks later, echocardiographic examinations were performed to test left ventricle function and heart tissues were harvested to test the quality of FAK by western blot and severity of fibrosis by masson staining. Human cardiac fibroblast was administrated with OPN, and FAK inhibition by PP2 was treated 2 h before OPN was given. Expression of α-SMA and collagen-I were tested by western blot and real-time PCR assay. Results: OPN-shRNA group has a relatively high ejection fraction (EF), fractional shortening (FS), LV free wall thickness and a less sever cardiac fibrosis. In vitro, OPN could increase collagen-I and α-SMA expression, and this process can be inhibited by FAK inhibitor. Conclusion: Inhibition of OPN could reduce the LV remodeling and dysfunction in DCM mice, which may attribute to the suppression of collagen-I secretion in fibroblast through a FAK/Akt dependent pathway.
Keywords: Osteopontin (OPN), focal adhesion kinase (FAK), cardiac fibrosis, dilated cardiomyopathy (DCM), fibroblast
Introduction
Myocardium fibrosis appears in almost all kinds of heart diseases like ischemic cardiomyopathy [1], dilated cardiomyopathy (DCM) [2] and heart failure [3]. The pathological courses like onset and progress of myocardium fibrosis will lead to a poor prognosis in patients suffered cardiovascular disease since the absent of effective treatments for fibrosis [4].
Osteopontin (OPN) is a large-acid phosphoprotein adhesion molecule secreted by both cardiac interstitial fibroblasts [5], and macrophage [6] which is considered to be closely related to fibrosis process in humans and animal models [7-9]. Previous research shows overexpressing OPN may results in dilated cardiomyopathy [10]. OPN can also acts as an ECM protein and a proinflammatory cytokine [11], containing an arginine-glycine-aspartate-binding (RGD-binding) motif [12]. Recent studies proved that the binding of OPN to cell-surface integrins can increase the synthesis of collagen-I protein [13].
Focal adhesion kinase (FAK) is a 125-kDa non-receptor cytoplasmic tyrosine kinase which plays a pivotal role in regulating cell migration, proliferation and survival in a range of various cell types [14]. FAK is also involved in signal pathways in which OPN participated growth factors signal transduction, previous study demonstrated that integrin beta3-FAK signaling can modulate OPN-induced vascular smooth muscle cells migration during neointimal formation [15].
We have reported that FAK is involved in atrial fibrosis and ischemic cardiomyopathy; inhibition of FAK can suppresses α-SMA expressions in TGFβ1-induced fibroblasts and alleviates post-infarction fibrosis [16,17].
Findings, therefore, we postulate that inhibition of OPN might prevent the cardiac myofibrosis of DCM, and FAK might be a reasonable signal molecular in the process of OPN-induced cardiac fibrosis.
Materials and methods
Cell culture and treatment
Human cardiac fibroblasts (hCF) were purchased from sciencell research laboratories (Cat. No.6300, sciencell research laboratories). Cells were maintained in Fibroblast Medium (FM-2, Cat. No.2331, sciencell research laboratories) supplemented with 5% FBS, 100 U/ml penicillin/streptomycin. Trypsin-EDTA solution (0.05%) was used for subculturing fibroblasts, and the 3rd-7th generation of cells was used for the experiments. All assays in the present study were done at temperatures of 37.8°C, 95% sterile air and 5% CO2 in a saturation humidified incubator. Cells were starved for 12 h then treated with different doses of OPN (0-400 ng/ml; R&D Systems). FAK/Src inhibitor PP2 (5 μM, Calbiochem) was supplied 12 h before treatment of OPN, in order to determine whether FAK pathway is involved in cardiac fibrosis.
Animals and echocardiograph
All procedures involving experimental design were approved by the Ethics Committee of the Chinese Academy of Medical Sciences and Peking Union Medical College (No. 2014-6-24-GZR). Animal care and experimental procedures were conducted in accordance with the European Guidelines on Laboratory Animal Care. The cTnTR141W transgenic male mice were established at the Laboratory of Animal Science of Peking Union Medical College and maintained on a C57BL/6J genetic background. The transgenic mice expressed high levels of the mutant human cTnTR141W protein and showed ventricular chamber enlargement, systolic dysfunction, myocardial hypertrophy, and interstitial fibrosis at 4 months of age [18]. Under open chest surgery with constant volume ventilation, OPN shRNA lentivirus particles were administered intramurally to the left ventricular free wall (10 μl, 1×109 TU/ml), which would inhibit heart tissue expressing OPN. OPN shRNA Lentiviral Particles is a pool of 3 different shRNA plasmids: (1) 5’-GAT CCC CAA GCA ATT CCA ATG AAA TTC AAG AGA TTT CAT TGG AAT TGC TTG GTT TTT-3’; (2) 5’-GAT CCC GTC ACT GCT AGT ACA CAA TTC AAG AGA TTG TGT ACT AGC AGT GAC GTT TTT-3’; (3) 5’-GAT CCC AGA CAC TTT CAC TCC AAT TTC AAG AGA ATT GGA GTG AAA GTG TCT GTT TTT-3’. At the age of 8 weeks, 6 mice were treated with OPN shRNA as OPN-shRNA group, 6 mice were treated with Control shRNA as vehicle group, 6 mice were just under open chest surgery and treated nothing as sham group. Four weeks after administration, animals were sacrificed and hearts were harvested for further analysis. To evaluate cardiac function and morphology, echocardiography was performed before tissues were harvested.
Echocardiography of sedated mice was carried out using a Vevo 770 high resolution imaging system (VisualSonics, Toronto, Canada) equipped with a 40 MHz transducer. Three independent M-mode measurements per animal were performed by an experienced examiner. Echocardiographic data were recorded at heart rates between 450 and 550 BPM. End systolic and end diastolic chamber diameters, and left ventricular fractional shortening were measured in the short axis at the papillary muscle level.
ELISA of collagen I
Concentration of hCF supernatant of collagen-I was determined using Human Pro-Collagen I alpha 1 DuoSet ELISA Development kit (DY6220-05; R&D Systems), using standard procedure according to the manufacturer’s instructions. Briefly, 96-well plates were coated with antibody specific for Pro-Collagen I alpha 1. Biotinylated detection antibody and streptavidin-conjugated horseradish peroxidase were used for detection of captured Pro-Collagen I alpha 1. The plates between steps were aspirated and washed 3 times using microplate washer (Thermo Fisher). Captured Pro-Collagen I alpha 1 was visualized using tetramethylbenzidine/hydrogen peroxide. Absorbance readings were made at 450 nm, using a microtiter plate reader (infinite M200PRO, TECAN). Pro-Collagen I alpha 1 levels in samples were determined by interpolation from a standard curve. Standards and samples were assayed in duplicate.
Western blotting
Cells and mouse myocardial tissue samples were lysed by cOmplete lysis buffer containing protease inhibitor cocktail tablets and phosphatase inhibitor tablets (Roche, Germany) at a working concentration recommended by roche. Protein concentrations were measured using a BCA protein assay. Equal amounts of protein mixtures were subjected to SDS-PAGE; gels were electrophoretically transferred to PVDF membrane (Millipore Inc.) and incubated in TBST with 5% Bovine Serum Albumin for 1 h at room temperature. The blot was incubated with primary antibodies including anti-α-SMA (1:400, Abcam Inc.), anti-phospho-Tyr397 FAK (1:800, Cell Signaling Technology), anti-phospho-Ser473 AKT (1:1000, Cell Signaling Technology), anti-collagen-I (1:400, Abcam Inc.). Anti-GAPDH antibody (1:500, ZSGB-BIO) was used as an internal control. After three washes by TBST, the blot was treated with horseradish-peroxidase-conjugated secondary antibodies. The washes were repeated and membranes were stained with ECL detection reagents (Millipore Inc.). Results were detected using a chemi-doc image analyzer (FluorChemo M FM0488, Protein Simple) and expressed as density values normalized to GAPDH.
Immunofluorescence staining
Cardiac fibroblasts were fixed in 4% PFA for 30 min and permeabilized with 1% Triton X-100 and three-micrometer sections were deparaffinized with xylene and rehydrated with graded alcohol. antigen retrieval method was performed using 10 mM sodium citrate (pH 6.0) in a water bath at 100°C for 2 mins. Preincubation was carried out for 30 min in a PBS solution containing 5% BSA. Samples were then incubated overnight at 4°C in a solution containing primary antibody including anti α-SMA antibody (1:200 Abcam, Inc), anti-vimentin antibody (1:500 Abcam, Inc) and anti-collagen-I antibody (1:200 Abcam, Inc). Coverslips were washed in PBS for three times and treated with secondary antibodies for 1 hour at 37°C After three washings (5 min each) in PBS, the cells were incubated for 1 h at 37°C in secondary antibody 1:300 in PBS. DAPI was used to stain nuclei. Control staining using only secondary antibody was run in parallel. After the slides were mounted in glycerol, we used a Leica TCS-SP5 Confocal Laser Scanning Microscope (Leica, Germany) for observation.
RT-PCR analysis
mRNA expressions in the hCF and LV anterior wall were measured by quantitative real-time polymerase chain reaction (RT-PCR). Total RNA was isolated with Trizol reagent (Invitrogen, USA) according to the manufacturer’s instructions and the concentration was measured by NanoDrop 2000 (Thermo Scientific). RNA samples (2 μg) were further reverse-transcribed with the PrimeScript RT Reagent Kit (Perfect Real Time, TaKaRa Biotechnology, Japan) using the manufacturer’s protocol. The mRNA levels of Collagen I (mouse)(forward 5’-TC TCC TGG TGC TGA TGG AC-3’ and reverse: 5’-GCC TCT TTC TCC TCT CTG ACC-3’), OPN (mouse) (forward 5’-TCC CTC GAT GTC ATC CCT GT-3’ and reverse: 5’-CCC TTT CCG TTG TTG TCC TG-3’) were quantitatively measured using the Applied Biosystems step-one plus 7500 Real-Time PCR System (Life Technologies, USA) using SYBR Select Master Mix (Life Technologies, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH, mouse) (forward 5’-ACA GCA ACA GGG TGG TGG AC-3’ and reverse: 5’-TTT GAG GGT GCA GCG AAC TT-3’) was used as the non-regulated control. The assays were performed three times using triplicate wells, threshold cycle (Ct) was calculated using the second-derivative maximum method. The data were analyzed via the delta-delta method, final values are expressed as the ratio versus the control.
Masson analysis
Masson analysis was used to detect the cardiac fibrosis. Trichrome Stain (Masson) Kit were purchased from Sigma-Aldrich Corporation. According to the instruction of manufacturer, after deparaffinization and rehydration, the section incubated in Scarlet-Acid Fucshin for 5 min, then immersed in Phosphomolybdic Acid Solution for 1 minute. The sections were treated with Aniline Blue Solution for 2 minutes. Then 1% Acetic Acid was used to incubate the slides for 2 min. Rinse slides, dehydrate through alcohol, clear in xylene and mount. The section was analyzed by microscope. To evaluate the fibrosis index of heart tissues, ten random heart fields per tissue section were captured at the 400× magnification.
Statistical analysis
A student’s t test was used to compare continuous variables between two groups and a one-way analysis of variance (ANOVA) was used when three or more experimental conditions were compared. Data for continuous variables were expressed as the mean±SEM (standard error of the mean). A Chi-square test was used to compare categorical variables between two groups. All statistical analyses were performed using SPSS 17.0. Statistical significance was assumed when P<0.05.
Results
Inhibition of OPN reduce the phosphor-FAK and collagen-I expression in myocardial tissue of dilated cardiomyopathy in mouse models
To define whether the shRNA can silence the expression of OPN, immunofluorescence staining were performed in paraffin sections of heart tissue, OPN-shRNA group can hardly express OPN (Figure 1) OPN-shRNA group 0.21%±0.14%, sham group 2.96%±1.02%, vehicle group 3.19%±1.59%, n=3, P<0.001). In contrast, sham group and vehicle group exist a relatively more expression of OPN. And in accordance with immunofluorescence staining the PCR assay also demonstrated control group and vehicle group have a relative high expression of OPN mRNA (Figure 2).
Figure 1.
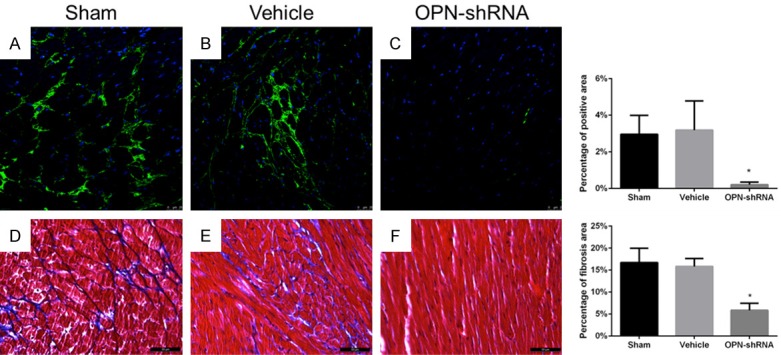
A-C: Paraffin sections were prepared and stained with anti-OPN pAb (green) and DAPI (blue) to stain DNA. OPN expression was inhibited by OPN-shRNA in OPN-shRNA group. Percentage of green stained area: sham group 2.96%±1.02%, vehicle group 3.19%±1.59%, OPN-shRNA group 0.21%±0.14%, n=3, scan bar, 25 μm, *P<0.001. D-F: Interstitial fibrosis of heart assessed by Masson staining, fibrotic area is stained blue, scan bar, 200 μm. Percentage of fibrosis area: sham group 16.72%±3.24%, vehicle group 15.82%±1.80%, OPN-shRNA group 5.86%±1.59%, n=5, *P<0.001.
Figure 2.
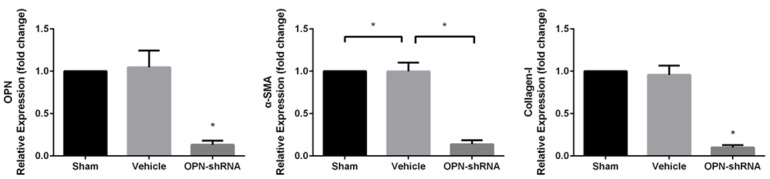
mRNA expression of mice. The expression of OPN, α-SMA and collagen-I mRNA were significant reduced by OPN-shRNA. The mRNA level were determined by RT-PCR and normalized to GAPDH housekeeping gene. Values are mean±SD, *P<0.001.
To test the severity of cardiac fibrosis, Masson staining was applied in paraffin section of heart of mouse. As Figure 1 shows, OPN-shRNA group underwent a less sever cardiac fibrosis than sham group and vehicle group (OPN-shRNA group 5.86%±1.59%, sham group 16.72%±3.24%, vehicle group 15.82%±1.80%, n=5, P<0.001). It demonstrated that OPN might be required during the process of cardiac fibrosis. Western blot shows (Figure 3), p-FAK, p-Akt, α-SMA expressed in the anterior wall of left ventricle of the heart were decreased in shRNA group compared with sham group and vehicle group. This result is in accord with PCR assay, expression levels of OPN and collagen-I were found to be significantly higher in heart tissue of sham group and vehicle group compared with OPN-shRNA group (Figure 2, P<0.001). These results showed that OPN-caused fibrosis might link to its ability to induce the phosphorylation of FAK and affect its downstream pathway.
Figure 3.
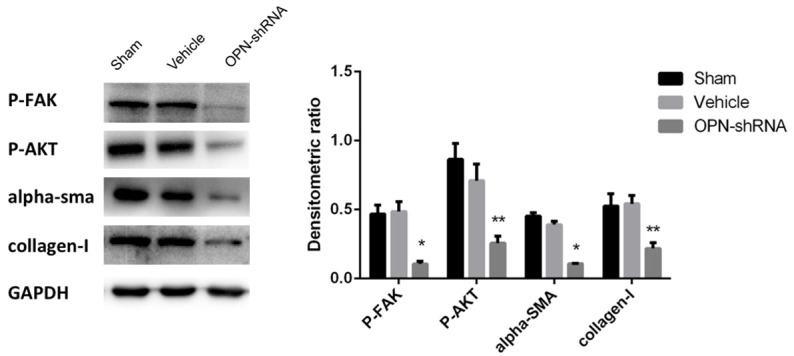
Collagen-I and OPN-related pathway were analyzed by western blot. The results indicated the expression of phosphor-FAK, phosphor-AKT, α-SMA and collagen-I in OPN-shRNA group was down-regulated vs. sham group and vehicle group *P<0.001, **P<0.05.
OPN inhibition can prevent deterioration of cardiac function in DCM
We evaluated the morphological and functional cardiac parameters of all group mice by transthoracic echocardiography. As Table 1 shows, LV Vol;d (Left ventricular volume; diastole) and LV Vol;s (Left ventricular volume; systole) in OPN-shRNA group were both decreased compared with sham group and vehicle group, which suggests that the LV of OPN-shRNA group mice was less severely dilated compared with that of control virus group and sham group. Moreover, LVAW;s (Left ventricular anterior wall; systole) of sham group and vehicle group was significantly lower than in OPN-shRNA group, which suggests the OPN inhibition has a protecting function of a hypokinetic DCM. These structural changes demonstrated the OPN inhibition might attenuate systolic dysfunction in DCM, confirmed by LV fractional shortening (OPN-shRNA group: 33.29%, n=6; sham group: 23.17%, n=6; control virus group: 21.5%; P<0.05) and ejection fractions (OPN-shRNA group: 62.28%, n=6; sham group: 46.17%, n=6; control virus group: 43.37%; P<0.05). Figure 4: typical picture of M-mode echocardiography of three groups, OPN-shRNA group shows a smaller left ventricular internal dimension and thicker ventricular wall.
Table 1.
Echocardiography of mice
| Sham group | Vehicle group | OPN-shRNA group | |
|---|---|---|---|
| BW (g) | 22.43±1.33 | 22.64±1.49 | 22.48±0.87 |
| LVAW;d | 0.81±0.12 | 0.83±0.16 | 0.94±0.17 |
| LVAW;s | 0.92±0.1 | 0.99±0.11 | 1.24±0.19* |
| LVID;d | 4.56±0.25 | 4.75±0.24 | 4.12±0.13* |
| LVID;s | 3.5±0.34 | 3.73±0.29 | 2.75±0.17* |
| LVPW;d | 0.73±0.11 | 0.71±0.09 | 0.69±0.07 |
| LVPW;s | 0.87±0.12 | 0.88±0.16 | 0.97±0.13 |
| EF | 46.17±9.4 | 43.37±7.15 | 62.28±4.47* |
| FS | 23.17±5.6 | 21.5±4.1 | 33.29±3.25* |
| LV Vol;d | 95.56±11.99 | 105.31±12.58 | 75.22±5.78* |
| LV Vol;s | 51.58±12.28 | 59.74±11.72 | 28.4±4.13* |
BW: body weight; d: diastole; s: systole; LVAW: left ventricular anterior wall; LVID: left ventricular internal dimension; LVPW: left ventricular posterior wall; EF ejection fraction; FS: fractional shortening; LV Vol: left ventricular volume. A significant improve of cardiac function was observed in OPN-shRNA group compared with sham group and vehicle group, meanwhile OPN-shRNA group holds a smaller LV volume and relatively thick ventricular wall;
P<0.05.
Figure 4.
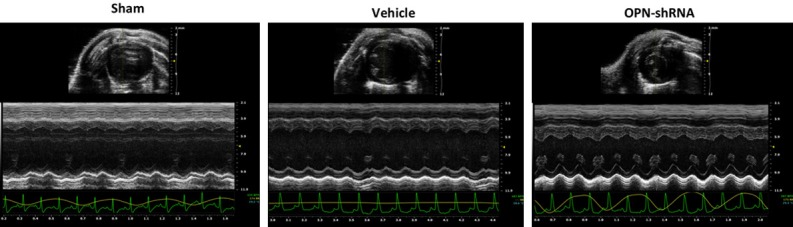
Echocardiography measurement in each group. Representative M-mode echocardiography of all three groups at the 6th week after OPN administration showed increased left ventricular dimension and a thinner anterior wall of LV in sham and vehicle groups vs. OPN-shRNA group, P<0.05.
OPN induces hCF express collagen-I andα-SMA in a dose-dependent manner
To determine how can OPN regulate collagen-I expression in cardiac fibroblasts, human cardiac fibroblasts (hCF) were used and treated with or without recombination human osteopontin (OPN) in different concentrations (0, 25, 50, 100, 200, 400 ng/mL) for 24 h, the expression of α-SMA and collagen-I was detected by western blot while collagen-I alpha 1 in cell supernatant was tested by ELISA. Results showed that the protein expression of collagen-I increased in a dose-dependent manner, the highest level of collagen-I expression induced by OPN was at the concentration of 200 ng/mL and this dose-dependent trend was somehow diminished at the concentration of 400 ng/mL (Figure 5). ELISA assay demonstrates OPN at 200 and 400 ng/mL can induce the highest expression of collagen-I alpha 1 (Figure 6). To get a further validation on the relationship of OPN concentration and fibroblast transformation, α-SMA expression was also tested as a marker of fibroblast differentiation by western blot, results showed α-SMA expression increased in accordance with the increased concentration of OPN, and the most abundant α-SMA expression was also observed at the concentration of 200 and 400 ng/mL (Figure 5). Thus 200 ng/mL seems to be a modest concentration for hCF treatment.
Figure 5.
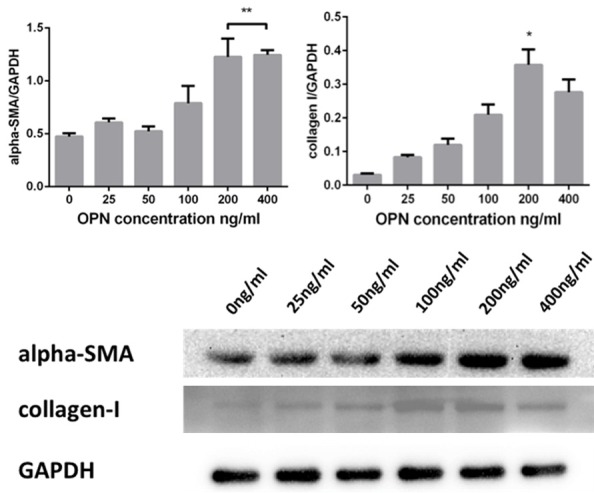
OPN upregulate the expressions of α-SMA and collagen-I, both of which presented as a dose-dependent manner. At the dose of 200 ng/ml, collagen-I reached the highest expression. α-SMA reached highest at 400 ng/ml, no significant increase compared with 200 ng/ml.
Figure 6.
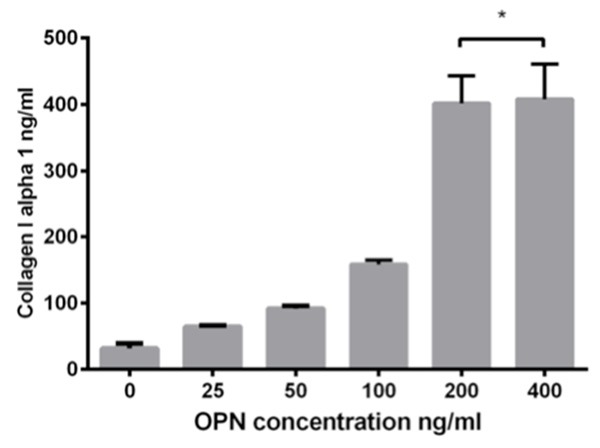
ELISA of supernatant of cardiac fibroblasts treated with OPN of different concentrations. After 24 h, collagen-I alpha 1 concentration increased high at 200 ng/ml, no significant increase exist at 400 ng/ml compared with 200 ng/ml.
OPN induced the transformation from cardiac fibroblast to myofibroblast
To investigate the role of OPN in fibroblast transformation, OPN was used to treat hCF, secretion of α-SMA expression was tested. Optical microscope observation showed that there were no significant appearance difference between hCF cultivated in normal condition or with OPN (200 ng/ml), while western blot and immunofluorescence showed a remarkable increase of α-SMA expression in OPN treated hCF compared with the blank control group (Figure 7). The increasing of α-SMA shows that OPN induced the differentiation of fibroblast to myofibroblast, which has a stronger collagen-I secreting function.
Figure 7.
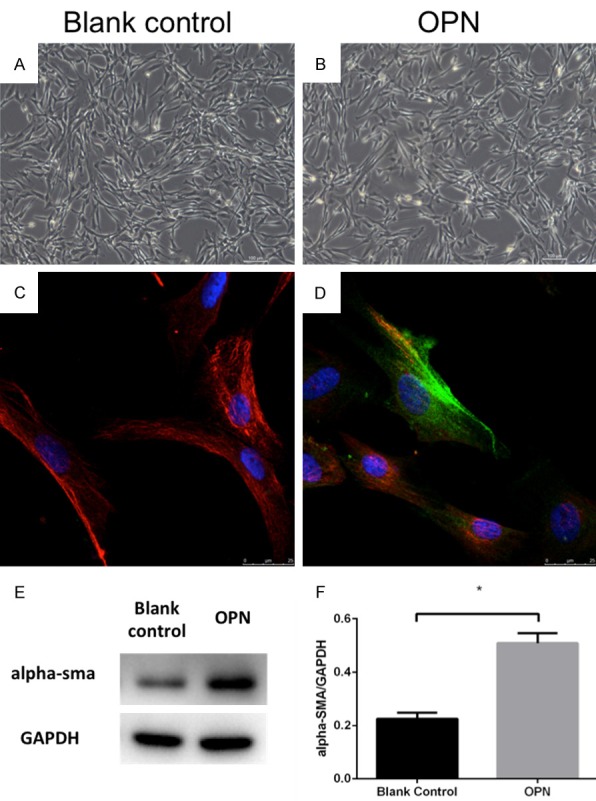
Morphology of human fibroblasts: no significant difference exists between blank control group (A) and OPN (200 ng/ml) group (B) (magnification 100×, scan bar, 100 μm). Both exhibited spindle-shaped morphology. The immunofluorescent staining indicated that the α-SMA expression in fibroblasts of OPN group (D) but not in blank control group (C). Cells were stained with anti-α-SMA mAb (green), anti-vimentin mAb (red) and DAPI (blue) to stain DNA. (E, F) Western blot showed the expression of α-SMA in OPN group was significant higher than in blank control group; *P<0.05.
OPN cause hCF differentiation and collagen-I secretion via FAK/AKT pathway
FAK plays a key role in development of fibrotic disorders [19], we chose FAK/AKT pathway as a potential target for OPN downstream signaling. OPN (200 ng/ml) was preincubated in human cardiac fibroblast, PP2 was used as an FAK inhibitor which was applied 2 h before the OPN treatment. Western blotting demonstrated that expression of fibrosis concerned proteins include α-SMA, collagen-I and phosphor-Tyr397 FAK, phosphor-Ser473 AKT were significantly up-regulated with the present of OPN compared with blank group (Figure 8, P<0.05), this change can be suppressed markedly in OPN treated hCFs preincubated with PP2 (P<0.05 compared with OPN alone group). The PCR assay shows the same results with western bolt, the expression of mRNA of collagen-I and α-SMA is significant increased under the treatment of OPN and attenuated by PP2 (Figure 9, P<0.001). These results indicated that FAK may be necessary in OPN-induced cardiac fibroblast differentiation and collagen-I secretion.
Figure 8.
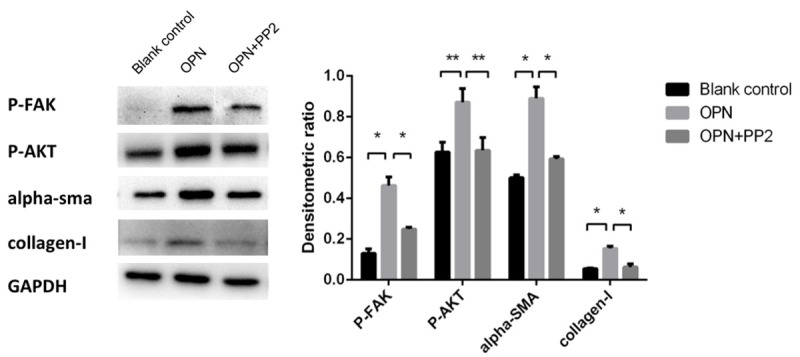
Western blot assay was used to detect whether FAK-dependent pathway is involved in OPN-caused fibrosis. Expression of phosphor-FAK, AKT, α-SMA, collagen-I significant increased in OPN group, and this phenomenon can be attentunated by FAK inhibitor PP2. *P<0.001, **P<0.05.
Figure 9.
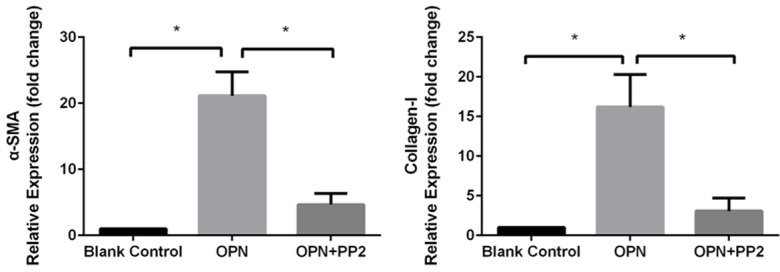
mRNA expression in cardiac fibroblasts, OPN treatment significantly inducedα-SMA and collagen-I expression. FAK inhibitor PP2 can reduce this induction, P<0.001.
Discussion
Several previous findings observed that myocardial OPN expression was abnormally increased in heart failure patients induced by ischaemic heart disease, dilated cardiomyopathy, hypertrophic cardiomyopathy and hypertension [8,20-22], and OPN showed a strong correlation with diseases outcome. Recent studies found that acute congestive heart failure patients with higher OPN level were associated with worse outcomes [23]; moreover, OPN was identified as a strong independent predictor of mortality in patients with chronic heart failure [24]. In human DCM, OPN plays a pivotal role in the development of collagen-I-induced cardiac fibrosis and dysfunction [21], and the viewpoint was supported by Dr. Renault’s research which demonstrated that OPN overexpression in cardiomyocytes could result in DCM in mouse model [10]. So we proposed hypothesis that inhibition of OPN could attenuate the collagen turnover and ameliorate myocardial remodeling in mouse model of DCM.
Focal adhesion kinase (FAK) is a pleiotropic cytokine, which has been shown to be a pivotal factor in cell migration proliferation and survival [14]. Recent studies have defined FAK as an important mediator in the signaling pathway of TGF-β1 and angiotensin II [25,26], and could mediate fibroblast migration and promote fibrosis through integrin β1 [27]. Our previous study demonstrated that FAK and its down-stream signaling pathways Akt/PI3K can regulate the fibrosis development of chronic atrial fibrillation patients with rheumatic mitral valve disease [16]; and also FAK is a critical molecule in mediating the atrial fibrotic process through Akt/S6K signaling [17]. Thus, we designed this experiment to verify our hypothesis above and identify whether FAK was involved in this process.
In our animal experiment, we found EF and FS of OPN-shRNA group were higher than sham group and control virus group, which indicate that the inhibition of OPN can attenuate the left ventricular dysfunction in DCM. In addition, during systole, the LV free wall thickness of control virus group and sham group was significantly thinner than OPN-shRNA group suggesting that inhibition of OPN may be favorable for systolic function reservation during the progression of DCM. Deposition of collagen-I can lead to inferior myocardial compliance [28] and ventricular remodeling in patients with cardiovascular disease like DCM, myocardial infarction and heart failure. Excessive myocardial collagen cross-linking is associated with hospitalization for hypertensive patients result in heart failure [29]. In OPN-shRNA group, a relatively lower ratio of collagen-I was detected, which suggest inhibition of myocardium OPN expression may alleviate the ventricular remodeling progression in DCM mice, this effect may on the account of the reduction of collagen-I synthesis, and the improved left ventricular function may relay on it. Myocardial fibrosis can result in increased tissue stiffness and myocardial viscoelasticity, which finally lead to aggravated LV dysfunction [30], so the results showed in OPN-shRNA group remind us the therapeutic effects of OPN in DCM patients may concerned with reduction of collagen I deposition which attenuate the deterioration of compliance and function of the left ventricle remodeling in DCM.
OPN was found to be participated in the process of fibrosis in different organs like heart, liver, kidney in previous studies [7,31,32], and and also FAK phosphorylation is necessary for the regulation of OPN-mediated cell migration [15]. So we hypothesized that OPN could lead to cardiac fibrosis through FAK-dependent pathway. To verify whether FAK is included in the development of fibrosis caused by OPN, human cardiac fibroblasts were used. Treatment of OPN could lead to the expression of α-SMA in hCF, which means OPN could stimulate fibroblast differentiation into myofibroblasts, which is a dominating source of collagen in myocardium due to its secretory capacity. OPN also upregulated collagen-I expression of CFs accompanied with AKT and FAK phosphorylation, this change can be attenuated with the pre-incubation of FAK inhibitor PP2 in CFs while α-SMA expression, AKT and FAK phosphorylation declined at the same time, which suggest that OPN may activate fibroblasts differentiation in a FAK/Akt dependent manner, result in transformation of human CFs into myofibroblasts and secretion of collagen-I, this result is consistent with our animal experiment. Inhibition of OPN can down-regulate expression of α-SMA and collagen-I in mice in the presence of FAK.
In conclusion, inhibition of OPN could reduce the LV remodeling and dysfunction in DCM mice. The underlying mechanism may attribute to the suppression of cardiac fibroblast differentiation and collagen-I secretion of myofibroblast of OPN in a FAK/Akt dependent pathway. Our results may provide new insights into the pathological development and mechanisms of prevention and treatment of DCM. Since the essential role of OPN in DCM, novel drugs on OPN and FAK is likely to be an effective therapy for DCM.
For all we know, this is the first experiment proved that FAK plays an important role in fibrosis pathological process of DCM, and FAK functions as a key signal molecular in this progress via CFs differentiation and collagen-I secretion. In our future studies, there are still some further issues remain to be solved like if other signal molecular involved in OPN-induced fibrosis and if FAK inhibition would be effective in preventing the progression of DCM.
Acknowledgements
First and foremost, I would like to show my deepest gratitude to my doctoral supervisor, Professor Wei Wang, who has provided me with valuable guidance in every stage of finish this research. We also thank Ms. Qing Xu of Capital Medical University, Beijing, China for providing a sincere echocardiography measurement and Ms. Jue Ye of Fuwai Hospital, for providing technical support of experiment. Contract grant sponsor: the National Natural Science Foundation of China; Contract grant number: 81470423.
Disclosure of conflict of interest
None.
References
- 1.Beltrami CA, Finato N, Rocco M, Feruglio GA, Puricelli C, Cigola E, Quaini F, Sonnenblick EH, Olivetti G, Anversa P. Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circulation. 1994;89:151–163. doi: 10.1161/01.cir.89.1.151. [DOI] [PubMed] [Google Scholar]
- 2.Bortone AS, Hess OM, Chiddo A, Gaglione A, Locuratolo N, Caruso G, Rizzon P. Functional and structural abnormalities in patients with dilated cardiomyopathy. J Am Coll Cardiol. 1989;14:613–623. doi: 10.1016/0735-1097(89)90102-2. [DOI] [PubMed] [Google Scholar]
- 3.Moore-Morris T, Guimaraes-Camboa N, Yutzey KE, Puceat M, Evans SM. Cardiac fibroblasts: from development to heart failure. J Mol Med (Berl) 2015;93:823–830. doi: 10.1007/s00109-015-1314-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mottram PM, Haluska B, Leano R, Cowley D, Stowasser M, Marwick TH. Effect of aldosterone antagonism on myocardial dysfunction in hypertensive patients with diastolic heart failure. Circulation. 2004;110:558–565. doi: 10.1161/01.CIR.0000138680.89536.A9. [DOI] [PubMed] [Google Scholar]
- 5.Ashizawa N, Graf K, Do YS, Nunohiro T, Giachelli CM, Meehan WP, Tuan TL, Hsueh WA. Osteopontin is produced by rat cardiac fibroblasts and mediates A(II)-induced DNA synthesis and collagen gel contraction. J Clin Invest. 1996;98:2218–2227. doi: 10.1172/JCI119031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murry CE, Giachelli CM, Schwartz SM, Vracko R. Macrophages express osteopontin during repair of myocardial necrosis. Am J Pathol. 1994;145:1450–1462. [PMC free article] [PubMed] [Google Scholar]
- 7.Collins AR, Schnee J, Wang W, Kim S, Fishbein MC, Bruemmer D, Law RE, Nicholas S, Ross RS, Hsueh WA. Osteopontin modulates angiotensin II-induced fibrosis in the intact murine heart. J Am Coll Cardiol. 2004;43:1698–1705. doi: 10.1016/j.jacc.2003.11.058. [DOI] [PubMed] [Google Scholar]
- 8.Graf K, Do YS, Ashizawa N, Meehan WP, Giachelli CM, Marboe CC, Fleck E, Hsueh WA. Myocardial osteopontin expression is associated with left ventricular hypertrophy. Circulation. 1997;96:3063–3071. doi: 10.1161/01.cir.96.9.3063. [DOI] [PubMed] [Google Scholar]
- 9.Singh M, Foster CR, Dalal S, Singh K. Osteopontin: role in extracellular matrix deposition and myocardial remodeling post-MI. J Mol Cell Cardiol. 2010;48:538–543. doi: 10.1016/j.yjmcc.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Renault MA, Robbesyn F, Reant P, Douin V, Daret D, Allieres C, Belloc I, Couffinhal T, Arnal JF, Klingel K, Desgranges C, Dos Santos P, Charpentier F, Gadeau AP. Osteopontin expression in cardiomyocytes induces dilated cardiomyopathy. Circ Heart Fail. 2010;3:431–439. doi: 10.1161/CIRCHEARTFAILURE.109.898114. [DOI] [PubMed] [Google Scholar]
- 11.O’Regan A, Berman JS. Osteopontin: a key cytokine in cell-mediated and granulomatous inflammation. Int J Exp Pathol. 2000;81:373–390. doi: 10.1046/j.1365-2613.2000.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oldberg A, Franzen A, Heinegard D. Cloning and sequence analysis of rat bone sialoprotein (osteopontin) cDNA reveals an Arg-Gly-Asp cell-binding sequence. Proc Natl Acad Sci U S A. 1986;83:8819–8823. doi: 10.1073/pnas.83.23.8819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Urtasun R, Lopategi A, George J, Leung TM, Lu Y, Wang X, Ge X, Fiel MI, Nieto N. Osteopontin, an oxidant stress sensitive cytokine, up-regulates collagen-I via integrin alpha(V)beta(3) engagement and PI3K/pAkt/NFkappaB signaling. Hepatology. 2012;55:594–608. doi: 10.1002/hep.24701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Franchini KG. Focal adhesion kinase-the basis of local hypertrophic signaling domains. J Mol Cell Cardiol. 2012;52:485–492. doi: 10.1016/j.yjmcc.2011.06.021. [DOI] [PubMed] [Google Scholar]
- 15.Han M, Wen JK, Zheng B, Liu Z, Chen Y. Blockade of integrin beta3-FAK signaling pathway activated by osteopontin inhibits neointimal formation after balloon injury. Cardiovasc Pathol. 2007;16:283–290. doi: 10.1016/j.carpath.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 16.Zhang P, Wang W, Wang X, Wang X, Song Y, Zhang J, Zhao H. Focal adhesion kinase mediates atrial fibrosis via the AKT/S6K signaling pathway in chronic atrial fibrillation patients with rheumatic mitral valve disease. Int J Cardiol. 2013;168:3200–3207. doi: 10.1016/j.ijcard.2013.04.113. [DOI] [PubMed] [Google Scholar]
- 17.Fan GP, Wang W, Zhao H, Cai L, Zhang PD, Yang ZH, Zhang J, Wang X. Pharmacological Inhibition of Focal Adhesion Kinase Attenuates Cardiac Fibrosis in Mice Cardiac Fibroblast and Post-Myocardial-Infarction Models. Cell Physiol Biochem. 2015;37:515–526. doi: 10.1159/000430373. [DOI] [PubMed] [Google Scholar]
- 18.Juan F, Wei D, Xiongzhi Q, Ran D, Chunmei M, Lan H, Chuan Q, Lianfeng Z. The changes of the cardiac structure and function in cTnTR141W transgenic mice. Int J Cardiol. 2008;128:83–90. doi: 10.1016/j.ijcard.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 19.Lagares D, Kapoor M. Targeting focal adhesion kinase in fibrotic diseases. BioDrugs. 2013;27:15–23. doi: 10.1007/s40259-012-0003-4. [DOI] [PubMed] [Google Scholar]
- 20.Stawowy P, Blaschke F, Pfautsch P, Goetze S, Lippek F, Wollert-Wulf B, Fleck E, Graf K. Increased myocardial expression of osteopontin in patients with advanced heart failure. Eur J Heart Fail. 2002;4:139–146. doi: 10.1016/s1388-9842(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 21.Satoh M, Nakamura M, Akatsu T, Shimoda Y, Segawa I, Hiramori K. Myocardial osteopontin expression is associated with collagen fibrillogenesis in human dilated cardiomyopathy. Eur J Heart Fail. 2005;7:755–762. doi: 10.1016/j.ejheart.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 22.Lopez B, Gonzalez A, Lindner D, Westermann D, Ravassa S, Beaumont J, Gallego I, Zudaire A, Brugnolaro C, Querejeta R, Larman M, Tschope C, Diez J. Osteopontin-mediated myocardial fibrosis in heart failure: a role for lysyl oxidase? Cardiovasc Res. 2013;99:111–120. doi: 10.1093/cvr/cvt100. [DOI] [PubMed] [Google Scholar]
- 23.Behnes M, Brueckmann M, Lang S, Espeter F, Weiss C, Neumaier M, Ahmad-Nejad P, Borggrefe M, Hoffmann U. Diagnostic and prognostic value of osteopontin in patients with acute congestive heart failure. Eur J Heart Fail. 2013;15:1390–1400. doi: 10.1093/eurjhf/hft112. [DOI] [PubMed] [Google Scholar]
- 24.Rosenberg M, Zugck C, Nelles M, Juenger C, Frank D, Remppis A, Giannitsis E, Katus HA, Frey N. Osteopontin, a new prognostic biomarker in patients with chronic heart failure. Circ Heart Fail. 2008;1:43–49. doi: 10.1161/CIRCHEARTFAILURE.107.746172. [DOI] [PubMed] [Google Scholar]
- 25.MacLean J, Pasumarthi KB. Signaling mechanisms regulating fibroblast activation, phenoconversion and fibrosis in the heart. Indian J Biochem Biophys. 2014;51:476–482. [PubMed] [Google Scholar]
- 26.Moraes JA, Frony AC, Dias AM, Renovato-Martins M, Rodrigues G, Marcinkiewicz C, Assreuy J, Barja-Fidalgo C. Alpha1beta1 and integrin-linked kinase interact and modulate angiotensin II effects in vascular smooth muscle cells. Atherosclerosis. 2015;243:477–485. doi: 10.1016/j.atherosclerosis.2015.09.026. [DOI] [PubMed] [Google Scholar]
- 27.Zhao XK, Cheng Y, Liang Cheng M, Yu L, Mu M, Li H, Liu Y, Zhang B, Yao Y, Guo H, Wang R, Zhang Q. Focal Adhesion Kinase Regulates Fibroblast Migration via Integrin beta-1 and Plays a Central Role in Fibrosis. Sci Rep. 2016;6:19276. doi: 10.1038/srep19276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kasner M, Westermann D, Lopez B, Gaub R, Escher F, Kuhl U, Schultheiss HP, Tschope C. Diastolic tissue Doppler indexes correlate with the degree of collagen expression and cross-linking in heart failure and normal ejection fraction. J Am Coll Cardiol. 2011;57:977–985. doi: 10.1016/j.jacc.2010.10.024. [DOI] [PubMed] [Google Scholar]
- 29.Lopez B, Ravassa S, Gonzalez A, Zubillaga E, Bonavila C, Berges M, Echegaray K, Beaumont J, Moreno MU, San Jose G, Larman M, Querejeta R, Diez J. Myocardial Collagen Cross-Linking Is Associated With Heart Failure Hospitalization in Patients With Hypertensive Heart Failure. J Am Coll Cardiol. 2016;67:251–260. doi: 10.1016/j.jacc.2015.10.063. [DOI] [PubMed] [Google Scholar]
- 30.Burlew BS, Weber KT. Connective tissue and the heart. Functional significance and regulatory mechanisms. Cardiol Clin. 2000;18:435–442. doi: 10.1016/s0733-8651(05)70154-5. [DOI] [PubMed] [Google Scholar]
- 31.Dai J, Peng L, Fan K, Wang H, Wei R, Ji G, Cai J, Lu B, Li B, Zhang D, Kang Y, Tan M, Qian W, Guo Y. Osteopontin induces angiogenesis through activation of PI3K/AKT and ERK1/2 in endothelial cells. Oncogene. 2009;28:3412–3422. doi: 10.1038/onc.2009.189. [DOI] [PubMed] [Google Scholar]
- 32.Wolak T, Kim H, Ren Y, Kim J, Vaziri ND, Nicholas SB. Osteopontin modulates angiotensin II-induced inflammation, oxidative stress, and fibrosis of the kidney. Kidney Int. 2009;76:32–43. doi: 10.1038/ki.2009.90. [DOI] [PubMed] [Google Scholar]