

Abstract
Myocyte apoptosis is a key determinant of cardiac recovery and prognosis of patients with acute myocardial infarction (AMI). Tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK), a member of TNF superfamily, is a pro-inflammatory and pro-angiogenic cytokine implicated in physiological tissue regeneration and wound repair and is closely related to cardiac remodeling, dysfunction and fibrosis. However, the role of TWEAK and its receptor Fn14 in the cardiomyocyte apoptosis is still poorly understood. The present study aimed to investigate whether the TWEAK enhanced the cardiomyocyte apoptosis in AMI. The apoptosis of the cardiomyocyte cell line H9C2 was induced by hypoxia/reoxygenation. The apoptosis of H9C2 cells was evaluated by flow cytometry and caspase-3 activity assay under treatment with TWEAK at different concentrations. The phosphorylated signaling molecules and the expression involved in the surprising protection of TWEAK against the apoptosis with a dose-dependent manner (≥50 ng/ml). Furthermore, a rat myocardial ischemia and reperfusion (I/R) model was established by TWEAK preconditioning through injecting the TWEAK into the scar and border after ischemia immediately induced by ligating the left anterior descending coronary artery for 50 min and followed by different reperfusion times. The heart function was significantly improved in TWEAK preconditioning rats compared with controls as well as the infarct size was significantly reduced 21 days after reperfusion. Meanwhile, TWEAK protected the cardiac apoptosis by activation of cardioprotective signaling PI3K/AKT during I/R. Our findings suggest that TWEAK may represent a cardioprotective factor that inhibits the myocyte death of myocardial IRI.
Keywords: TWEAK, cardiac ischemia/reperfusion injury, cardiomyocytes, apoptosis, signaling pathway
Introduction
Ischemic heart disease (IHD) is the main cause of morbidity and mortality worldwide, of which the most severe is myocardial infarction (MI) [1,2]. The therapies for IHD include thrombolysis, primary percutaneous intervention (PPCI) and coronary artery bypass grafting (CABG) [3-7]. These evolving strategies control the etiology and death complicated with MI. However, these strategies produce cardiac reperfusion, which may cause the known “ischemia/reperfusion injury (IRI)” [8]. Evidence indicates that the IRI is more severe than the primary ischemia per se because re-oxygenation may induce the excess production of reactive oxygen species (ROS) resulting in the aggravation of cardiomyocyte death and subsequent heart failure (HF) [8,9]. The loss of cardiomyocytes is primarily caused by autophagy, apoptosis and necrosis, which all determine the progression of IHD [9-11]. It has shown that gene modulation and pharmacological intervention for the inhibition of cardiomyocyte apoptosis are able to reduce the scar area and improve heart function [12,13].
TWEAK is a member of the tumor necrosis factor (TNF) superfamily (TNFSF). It was first reported in 1997 [14] and is expressed in various tissues including the intestine, pancreas, lung, brain, skeletal muscle, heart and vasculature [14,15]. It is a type II transmembrane protein with 249 amino acids (aa) and molecular weight (MW) of 23 kDa. It can be cleaved by furin endoprotease into a soluble TWEAK (sTWEAK) (185 aa, 18 kDa) which then binds to the Fn14 receptor on the cell, the cognate receptor of TWEAK, to induce various biological effects of cells including cell proliferation, migration, differentiation and apoptosis [14-21]. TWEAK has been known as a potent pro-inflammatory [19,20] and pro-angiogenic cytokine [17,21]. Although the expression of TWEAK or Fn14 is usually at very low level under physiological conditions, both can be dramatically induced under pathological conditions including MI [14,16]. Evidence indicates that TWEAK/Fn14 axis is also involved in the pathogenesis of cardiovascular diseases [22-27]. After MI, the synthesis of TWEAK increases significantly in the infiltrated inflammatory cells [15]. The increased serum TWEAK in patients with acute myocardial infarction (AMI) and HF is correlated to their prognosis [26]. TWEAK is also closely related to the proliferation of cardiomyocytes [28], cardiac remodeling [17,22], cardiac fibrosis [22,23] and heart dysfunction [22,25]. The transgenic mice with TWEAK over-expression usually develop dilated cardiomyopathy (DCM) with subsequent severe cardiac dysfunction, which is associated with cardiomyocyte elongation and cardiac fibrosis but not cardiomyocyte apoptosis [28]. More recently, Pachel et al. found that a recombinant variant of TWEAK increased the cardiac rupture without influencing the cardiomyocyte apoptosis after MI [19]. The degree of cardiomyocyte apoptosis is vital for the recovery of heart function after MI. Thus, it is necessary to elucidate the role of TWEAK in the cardiomyocyte apoptosis. Although it has been confirmed that TWEAK is able to bind Fn14 to induce the apoptosis of some types of cancer cells [16,29-31], little is known about the role of TWEAK in the apoptotic cardiomyocyte death following ischemia and reperfusion (I/R). Thus, this study was undertaken to investigate the role of TWEAK in the apoptotic cardiomyocyte death after I/R in rat cardiomyocyte cell line (H9C2 cells) and in rat myocardial I/R model.
Materials and methods
Cell culture
H9C2 cells were obtained from the Cell Bank of Shanxi Medical University. Cells were maintained in Dulbecco’s modified Eagle’s medium-high glucose (GE Healthcare Life Sciences Hyclone Laboratories, South Logan, UT) containing 10% fetal bovine serum (FBS; Life Technologies, NY, USA).
TWEAK treatment in vitro
H9C2 cells were treated with a recombinant human TWEAK (rhTWEAK, Peprotech, Rock Hill, NJ) at different concentrations (0, 5, 10, 50, 100, 500 and 1000 ng/ml) and in a hypoxia environment (0.1% oxygen; New Brunswick Galaxy 48R, Eppendorf Company, Germany) followed by re-oxygenation (19% oxygen).
Pretreatment with inhibitors of signal pathways in vitro
H9C2 cells were pretreated with parthenolide (10 μM; Sigma, Israel) (an inhibitor of NF-κB pathway), cryptotanshinone (5 μM; MCE, NJ, USA) (an inhibitor of STAT3), AG490 (10 μM) (an inhibitor of JAK2), or LY294002 (10 μM; Biyuntian, China) (an inhibitor of PI3K/AKT pathway) for 60 min, and DMSO served as a control. Then, the medium was refreshed, and cells were treated with rhTWEAK (100 ng/ml) under hypoxia/re-oxygenation condition as above mentioned.
RT-PCR
Total RNA was extracted from tissues or cells using Trizol reagent (Life Technologies). RNA (2 μg) was reverse transcribed (Thermo Scientific, Lithuania), and 100 ng of cDNA was amplified (Thermo Scientific). The following primers were used: rat TWEAK, 5’-AGGAGGAGCTGACAGCAGAG-3’ (forward) and 5’-CAGCCCTGATGACCGTAAT-3’ (reverse); rat Fn14, 5’-AGCTCCTCGTGTTG GG ATT-3’ (forward) and 5’-CAGCCTT CTCCACCAGTCTC-3’ (reverse); and rat GAPDH, 5’-ATGGGAAGCTGGTCATCAAC-3’ (forward) and 5’-GGATGCAGGG ATGATGTTCT-3’ (reverse). RT-PCR products were separated by gel electrophoresis, and image analysis was performed using ImageJ software as previously described [30]. The optical density (OD) of each band was determined, and the OD of each target gene was normalized to that of GAPDH as the relative mRNA expression (n=6/group for rat heart, n=4 for H9C2 cells).
Detection of cell apoptosis assay
Apoptosis was tested by flow cytometry (Beckman Coulter) after annexin V and propidium iodide (Life Technologies; n=3) staining. For in vivo assessment of cardiomyocyte apoptosis, terminal deoxynucleotidyl transferase mediated dUTP nick-end labeling (TUNEL) staining (Roche, Basel, Switzerland) was performed following the manufacturer’s instructions in the heart tissues at 1, 2 and 3 days after I/R.
Western blotting
Proteins expression of AKT and phospho-AKT (Ser473) was measured by Western blotting as described previously [32]. The primary antibodies were as follows: AKT (1:1000, #4685), phospho-AKT (Ser473) (1:1000, #9271) (Cell Signaling Technology, USA) and GAPDH (1:1000; Zhongshan Jinqiao, Beijing, China). Experiment was done in 4 times.
Rat ischemia and reperfusion models
Sprague-Dawley rats (weighing 180-200 g) were purchased from the Animal Center of Shanxi Medical University (Taiyuan, Shanxi province, China). Under general anesthesia, ischemia/reperfusion (I/R) was generated by clamping the left anterior descending coronary artery (LAD) with 7-0 suture for 50 min followed by reperfusion, as previously described [33]. The suture was not clamped in sham group. All animal experiments were approved by the Animal Care and Use Committee of Shanxi Medical University.
TWEAK treatment prior reperfusion in rat I/R model
rhTWEAK (1 μg/100 μl/rat) was injected directly into the myocardium in the border and scar immediately after clamping of LAD. After 50 min, reperfusion was induced. In addition, 100 μl of phosphate buffer saline (PBS) was used as vehicle control. The hearts were harvested at 1, 2, 3, 7 and 21 days after reperfusion (n=6 per group).
Caspase-3 activity assay
H9C2 cells (2×106) were treated with rhTWEAK (0, 100 ng/ml) in hypoxia for 24 h and then re-oxygenation for 24 h with pretreatment of inhibitors. Hearts were harvested at 1, 2 and 3 days after myocardial I/R. The caspase-3 activity was measured using a commercially available kit (Abcam, Cambridge, UK; n=6 per group for rat heart; n=3 for H9C2 cells).
Immunohistochemical staining
Immunohistochemistry (IHC) was conducted using a previously described antigen retrieval protocol, followed by primary antibody incubation [34]. Rabbit anti-rat TWEAK antibody (1:1000, NBP1-76695, Novus Biological, USA) and Fn14 antibody (1:1000, #4403, Cell Signal Technology, USA) were used to detect the expression of TWEAK and Fn14. Imaging and data capture were performed under a light microscope. Image analysis was performed using Image J software and optical density (OD)/high power field/cm2 was presented as the relative expression of TWEAK or Fn14.
Detection of inflammatory cells
Hearts were arrested at 1, 3 and 7 days post reperfusion, fixed at physiological pressures via perfusion with 10% formalin, and sectioned. Sections were subjected to H&E staining to assess inflammatory cells. Image analysis was performed with Image J software and the number of inflammatory cells/high power field was presented as the degree of inflammation.
Heart function
Echocardiography was performed in M-mode in the short axis view of the left ventricle to evaluate left ventricular internal end-diastolic and end-systolic diameters (LVIDd and LVIDs) on the day of I/R (prior to surgery), 7 and 21 days after reperfusion (n=6 per group). Ejection fraction (EF%) was calculated as previously [29].
Determining the scar size
Hearts were harvested and cut into 4 slices equally at 21 days after reperfusion. The second slice from the apex of the heart was used to evaluate the scar size. The scar size was presented by the percentage of scar area to total area (×100%).
Statistical analysis
Data are expressed as mean ± standard division (SD). Analyses were performed using GraphPad Prism (v.6) with two tailed unpaired t-test for two-group comparisons. A value of P<0.05 was considered statistically significant.
Results
Both Tweak and Fn14 are expressed in the cardiomyocytes in vivo and H9C2 cells in vitro after I/R
Considering that TWEAK exerts its effects via binding to Fn14 on the cell, the expression of Tweak and Fn14 was detected in the myocardium and H9C2 cells. Results showed the expression of both endogenous Tweak and Fn14 was at a very low level in the myocardium while I/R significantly increased their expression which peaked at 3 days post reperfusion and thereafter reduced to baseline level less than a week after I/R. In addition, the inflammatory cells infiltrated the injured myocardium and the injured myocardium became necrotic (Figure 1A). Meanwhile, the expression of TWEAK and FN14 protein was mainly found in the cardiomyocytes (Figure 1B). Additionally, the H9C2 cells also expressed Tweak and Fn14 after hypoxia/reoxygenation (Figure 1C). These suggest that TWEAK may act directly on cardiomyocytes during I/R.
Figure 1.
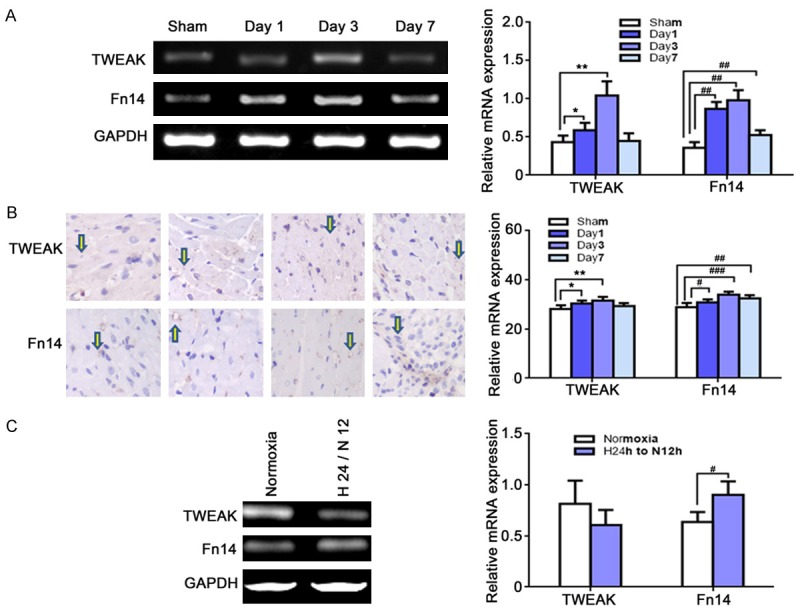
The expression of Tweak and Fn14 in the myocardium and H9C2 cells after I/R. A: mRNA expression of Tweak and Fn14 in the myocardium at 1, 3 and 7 days after I/R (RT-PCR; n=6 per group). Both Tweak and Fn14 were expressed after I/R, which peaked at 3 days after I/R. (TWEAK, *P<0.05, **P<0.01, vs. sham; Fn14, #P<0.05, ##P<0.01 vs. sham). B: Protein expression of TWEAK and Fn14 increased and mainly located in cardiomyocytes (IHC). C: Tweak and Fn14 were expressed in the H9C2 cells under hypoxia (H) for 24 h/re-oxygenation (N) for 12 h. (Fn14, #P<0.05, vs. normoxia).
TWEAK inhibits the apoptosis of H9C2 cells after I/R in vitro
To explore the exact role of TWEAK in the cardiomyocyte apoptosis, a main event in the myocardium following IRI, H9C2 cell apoptosis was detected after hypoxia/reoxygenation. Cells were exposed to hypoxia for 24 h and the reoxygenation for different durations, aiming to identify an optimum condition. Finally, hypoxia for 24 h and subsequent normoxia for 24 h was employed under which the proportion of apoptotic cells was 71.47% as shown by flow cytometry (Figure 2A). Then, H9C2 cells were treated with rhTWEAK at different concentrations (0, 5, 10, 50, 100, 500 and 1000 ng/ml) under hypoxia for 24 h/reoxygenation for 24 h. Unexpected, the apoptosis of H9C2 cells was inhibited by rhTWEAK in a dose-dependent manner (≥50 ng/ml, Figure 2B), and this was also observed in caspase-3 activity (Figure 2C). To further investigate the mechanism involved in the protection of TWEAK against cardiomyocyte apoptosis, rhTWEAK at 100 ng/ml was used in following experiments.
Figure 2.
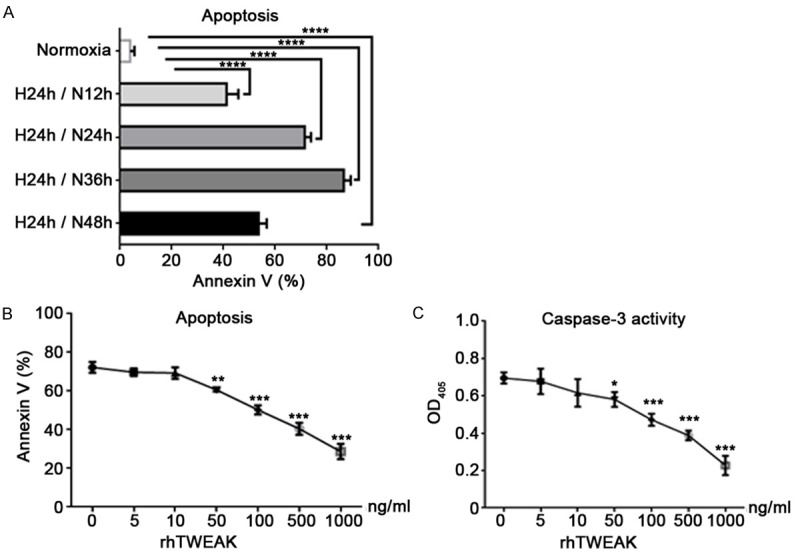
TWEAK inhibited the apoptosis of H9C2 cells under hypoxia (H)/re-oxygenation (N). A: The apoptosis of H9C2 cells was induced by hypoxia for 24 h/reoxygenation for different durations (AnnexinV and PI staining; flow cytometry). H 24 h/N 24 h was employed to treat H9C2 cells, and the apoptosis rate was 71.47%. B: H9C2 cells were treated with rhTWEAK at different concentrations and exposed to H 24 h/N 24 h. TWEAK inhibited the apoptosis of H9C2 cells significantly in a dose-dependent manner when the rhTWEAK concentration was >10 ng/ml. C: The caspase-3 activation was inhibited significantly by rhTWEAK when the rhTWEAK concentration was >50 ng/ml. (*P<0.05, **P<0.01, ***P<0.001 vs. rhTWEWAK 0 ng/ml).
TWEAK protects H9C2 cells against apoptosis mainly through PI3K/AKT signaling pathway
To elicit the mechanism of TWEAK induced inhibition of H9C2 cell apoptosis, some inhibitors of signaling pathways were used: parthenolide, LY294002, cryptotanshinone and AG490, which are inhibitors of NF-κB, PI3K/AKT and JAK/STAT3 pathways, respectively. As shown in Figure 3A, the apoptosis of H9C2 cells was aggravated after above inhibitors pretreatment for 60 min prior to hypoxia, accompanied by increase in caspase-3 activity (Figure 3B). It demonstrates that all three signaling pathways are protective against H9C2 cell apoptosis. Then, H9C2 cells were pretreated with inhibitors and treated with rhTWEAK at 100 ng/ml under hypoxia for 24 h/reoxygenation for 24 h. The apoptosis was evaluated by flow cytometry and caspase-3 activity assay. Results showed the benefit of TWEAK on the H9C2 cell apoptosis was mostly blocked by LY294002 while the parthenolide, cryptotanshinone or AG490 could not abolish the beneficial effect of TWEAK (Figure 3C and 3D). Meanwhile, the phosphorylated AKT at Ser473 (phospho-Ser473 AKT) expression increased after rhTWEAK treatment (Figure 3E, P<0.001, TWEAK vs. control) and the activation of PI3K/AKT pathway was blocked by LY294002 (Figure 3E, P>0.05, LY29400 vs. TWEAK+LY29400). These observations demonstrate that TWEAK protects against H9C2 cell apoptosis through PI3K/AKT signaling pathway.
Figure 3.
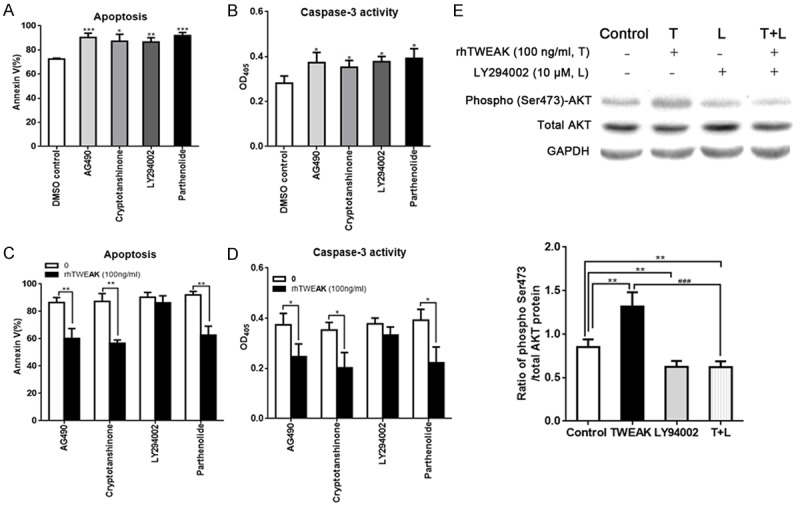
TWEAK inhibited the apoptosis of H9C2 cells under hypoxia/reoxygenation via PI3K/AKT pathway. A: The apoptosis of H9C2 cells was aggravated in the presence of independent pretreatment with inhibitors (AG490 [10 μM], crytotanshinone [5 μM], LY294002 [10 μM] and pathenolide [10 μM]) for 60 min. B: The caspase-3 activation in H9C2 cells was increased in the presence of independent pretreatment with inhibitors (AG490 [10 μM], crytotanshinone [5 μM], LY294002 [10 μM] and pathenolide [10 μM]) for 60 min. C: The TWEAK induced inhibition of H9C2 cell apoptosis under hypoxia for 24 h/reoxygenation for 24 h was blocked by LY294002 pretreatment, not by pathenolide, crytotanshinone and AG490 (flow cytometry: P>0.05, L+rhTWEAK [100 ng/ml] vs. L). D: The TWEAK induced inhibition of caspase-3 activation in H9C2 cell apoptosis under hypoxia for 24 h/reoxygenation for 24 h was blocked by LY294002 pretreatment, not by pathenolide, crytotanshinone and AG490 (flow cytometry: P>0.05, L+rhTWEAK [100 ng/ml] vs. L). E: The TWEAK increased the expression of phosphorylated-AKT (Ser473) and the cardioprotection of TWEAK was abolished by LY294002 pretreatment in H9C2 cells exposed to hypoxia for 24 h/reoxygenation for 24 h (**P<0.01. vs. control: ###P<0.001. vs. TWEAK 100 ng/ml).
TWEAK treatment prior reperfusion reduces cardiomyocyte apoptosis in vivo
To confirm the results from in vitro experiments, TWEAK injection prior reperfusion was performed in rat I/R model. After injection with rhTWEAK (1 μg/rat) into the border and infarct immediately after ischemia, the cardiomyocyte apoptosis at the very early stage of I/R (1, 2 and 3 days after reperfusion) was detected by TUNEL staining. The cardiomyocyte apoptosis decreased by 31.2%, 39.6% and 43.9% at 1, 2 and 3 days after myocardial I/R, respectively (Figure 4A, at day 1 and day 2, P<0.01; day 3, P<0.05). Simultaneously, the caspase-3 activity was inhibited by 57.6%, 63.9% and 52.8%, respectively (Figure 4B, day 1 and day 2, P<0.001; day 3, P<0.01). Similar to the results obtained in vitro, the cardiomyocyte apoptosis significantly decreased in TWEAK group as compared to control group. These indicate that TWEAK preconditioning protect against IRI by inhibiting the cardiomyocyte apoptosis without increasing the inflammation.
Figure 4.
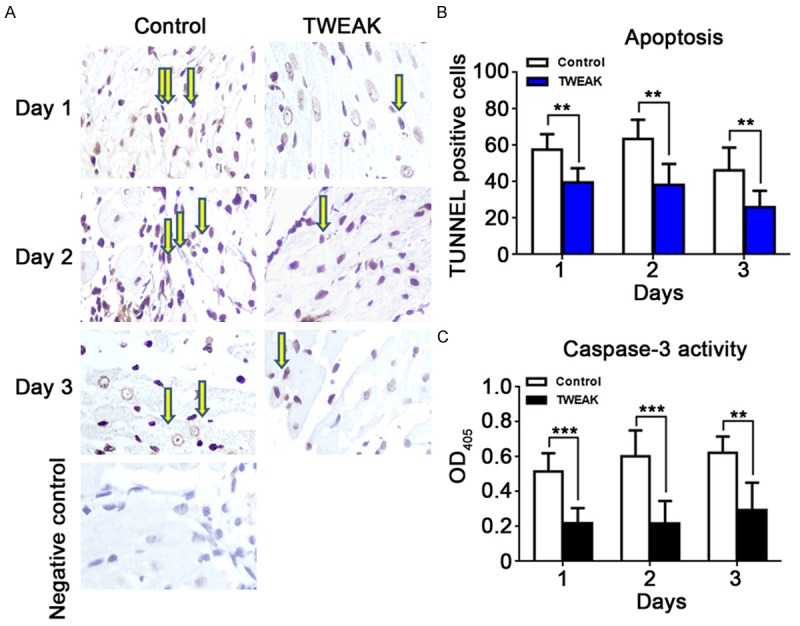
TWEAK treatment decreased cardiomyocyte apoptosis post I/R in vivo. A: The cardiomyocyte apoptosis after I/R was reduced by TWEAK (TUNEL staining). The TUNEL staining of myocardium was done at 1, 2 and 3 days after I/R. B: The proportion of apoptotic cardiomyocytes dramatically decreased by 31.2%, 39.6% and 43.9% at 1, 2 and 3 days after I/R, respectively, in TWEAK group (**P<0.05, vs. control group). C: The caspase-3 activity reduced by 57.6%, 63.9% and 52.8% at 1, 2 and 3 days after I/R, respectively, in TWEAK group (**P<0.01, ***P<0.001, vs. control group).
TWEAK treatment improves the heart function and decreases the scar size post I/R without aggravating the inflammation
To further confirm the benefit of TWEAK, the heart function was detected and scar size measured in I/R rats of TWEAK treatment group and control group. The heart function was comparable between groups prior surgery, but it was dramatically improved in TWEAK group by 24% and 33% at 7 and 21 days after reperfusion, respectively as compared to control group (Figure 5A, P<0.01, TWEAK vs. control). Furthermore, the scar size was decreased by 43.1% at 21 days after reperfusion in TWEAK group (Figure 5B, P<0.01, TWEAK vs. control).
Figure 5.
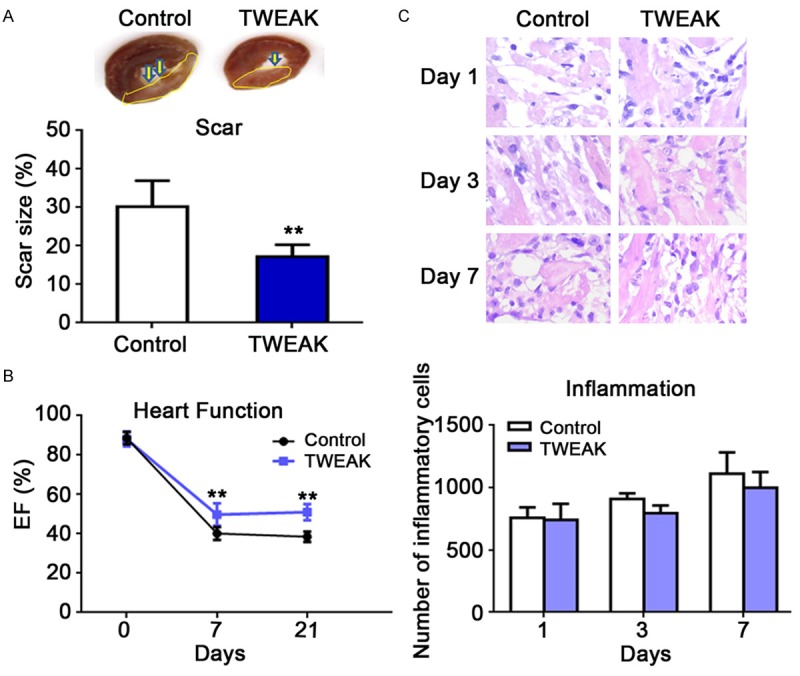
TWEAK treatment improved the heart function and reduced the scar size without aggravating inflammation. A: The heart function was significantly improved by 24% and 33% at 7 and 21 days after I/R, respectively, in TWEAK group as compared to control group (**P<0.01, vs. control). B: The scar size significantly reduced by 43.1% in TWEAK group as compared to control (**P<0.01). C: The degree of inflammation remained unchanged after TWEAK treatment (P>0.05, vs. control group).
As TWEAK is known as a potent pro-inflammatory factor [18-20], and inflammation infiltration and degradation play a role in the cardiac remodeling which occurs at early stage of I/R, the inflammation in the myocardium was also evaluated after HE staining at 1, 3 and 7 days post reperfusion. The inflammatory cells infiltrated into the myocardium, but there was no significant difference between TWEAK group and control group (Figure 5C, P>0.05, TWEAK vs. control).
It is suggested that the TWEAK treatment prior to reperfusion mitigates the IRI by improving the heart function and limiting the scar size without aggravating the inflammation.
Discussion
The loss of cardiomyocytes is crucial in the pathologenesis of HF. The myocardium is comprised of terminally differentiated cardiomyocytes that are responsible for cardiac pump function. Thus, the heart exhibits more lasting pathological effects as a consequence of excessive cardiomyocyte death [35]. The excessive loss of cardiomyocytes has been implicated in many cardiovascular diseases, such as myocardial I/R [8] and congestive HF [36]. Many studies focus on the mechanisms of cardiomyocyte death to improve HF and preserve the cardiac function. Autophagy, apoptosis and necrosis are all responsible to the cardiomyocyte loss, which is the characteristic of heart diseases including I/R, MI and HF [37].
Apoptosis is a highly regulated process of programmed cell death [32]. Classic apoptotic pathways include exogenous death receptor pathway and endogenous mitochondria pathway and are characterized by the activation of caspases, especially the executive caspase-3, and eventually the cell death. After I/R, myocardium suffers from biological and mechanical stress, meanwhile inflammation triggers smaller but more continuous cell death. Currently, researches focus on the pathogenesis of I/R, aiming to develop strategies for the therapy of IHD in clinical practice.
Extracellular ligands such as TNF-α contribute to the apoptosis by binding to their receptors (such as TNF-α receptor, [TNFR]) on cells, which have death receptor domains to induce intracellular signals, eventually leading to apoptosis [38,39]. This binding contributes to of the formation of death-inducing complex (DISC) which includes adaptor as TNF-associated death domain (TRADD), receptor-interacting protein1 (RIP1) and caspase-8. Then, caspase-8 activates the downstream effectors of caspase-3 and -7, resulting in lysis of key proteins and subsequent cell apoptosis [9]. It is a typical exogenous apoptotic pathway. The endogenous apoptotic pathway is activated by intracellular signals in case of hypoxia, oxidative stress and DNA damage, which leads to the increase in the mitochondrial membrane permeability and the release of pro-apoptotic and anti-apoptotic proteins. Then, apoptotic body accumulates and caspase-9 is activated to induce the activation of its effectors caspae-3 and -7 [9]. Studies have shown that reducing the caspas-3 activity by inhibiting the caspase cascade can mitigate IRI and improve cardiac function.
TWEAK, a member of TNFSF, shares some characters with TNF-α. For example, mice with over-expression of TWEAK or TNF-α will develop DCM [28,39]. TWEAK participates in various biological processes, including cell proliferation, migration, apoptosis, angiogenesis, inflammation and cardiac fibrosis depending on the context [14,17,19-21]. TWEAK induces cell apoptosis through a death-signaling complex dependent manner in some cancer cell lines by binding to the receptor Fn14 [14,30,31,40]. However, Jain et al. revealed that mice with TWEAK over-expression did not contribute to the cariomyocyte apoptosis [28]. Pachel et al. found that a recombinant variant of TWEAK increased the cardiac rupture while there was no difference in the apoptosis after MI [20]. It is suggested the unique role of TWEAK in cardiomyocyte apoptosis. There might be some other biological effects of TWEAK that are currently unknown. Therefore, this study focused on whether TWEAK acts to modulate the cardiomyocyte apoptosis and preserve heart function after myocardial I/R.
Unexpectedly, in cardiomyocyte cell line H9C2 cells, our results showed TWEAK (≥50 ng/ml) inhibited the apoptosis of H9C2 cells in dose-dependent manner following hypoxia/re-oxygenation in vitro. Furthermore, the mechanism of TWEAK’s protection against apoptotic cell death was further explored after treatment with various inhibitors for certain signaling pathways.
There are many signaling pathways related to TWEAK’s effects have been reported including NF-κB, JAK/STATs, RhoA ROCK and p38MAPK pathways [40-45]. NF-κB is a oxidative-reduction sensitive transcriptional factor and can regulate the transcription of inflammation related genes in pathological conditions. NF-κB is activated following cardiac I/R, including human cardoplegia and reperfusion during surgery [46]. Nevertheless, NF-κB may also act to inhibit cell death [47]. NF-κB mediated-cardioprotection may be anti-apoptosis through preconditioning. Evidence has shown the close relationship between TWEAK and NF-κB [44,45]. Thus, parthenolide, an inhibitor of NF-κB pathway [48], was used to pretreat H9C2 cells. Although parthenolide pretreatment increased the cardiomyocyte apoptosis after I/R, it failed to abolish the benefit of TWEAK on the apoptosis, which indicates that there are other pathways responsible for the protection of TWEAK.
PI3K/AKT signal pathway is a typical cardioprotection pathway [49]. It is activated by exercise, pressure overload, nutrient and other stimulations. Following PI3K activation, phosphatidylinositol (3,4,5)-trisphosphate (PIP3) accumulates at the membrane and then recruits pleckstrin homology (PH) domain-containing proteins, including phosphoinositide-dependent kinase-1 (PDK1) and AKT. PIP3-activated PDK1 in turn phosphorylates AKT at Thr308 which, together with Ser473 phosphorylation, ensures full AKT activation. This eventually triggers multiple downstream signaling pathways involved in cell proliferation, metabolism and survival [50]. In cardiomyocytes, PI3K is involved the growth and development of cardiomyocyte regulated by insulin and insulin-like growth factor-1 (IGF-1) [50]. PI3K is essential for the cardiac physiology and protection against pathological remodeling and failure. LY294002 is an inhibitor of PI3K and able to inhibit the activation of downstream effectors including AKT [49]. In this study, the phosphorylated AKT at Ser473 increased in H9C2 cells treated with TWEAK, but it was completely blocked by LY294002 pretreatment. The effect of TWEAK on cardiomyocyte apoptosis under I/R is at least partially mediated by the activation of PI3K/AKT signaling pathway.
Moreover, JAK/STATs pathway was tested as well. JAK/STATs pathway has proven to be a cardioprotection signaling pathway during the I/R [51,52]. STATs are latent transcription factors activated by extracellular signaling ligands such as cytokines, growth factors and hormones. STATs may be activated in the cytoplasm by JAK, which is a family of tyrosine kinases. The JAK/STATs signaling pathways have diverse activities and play roles in the cell differentiation, proliferation, development, apoptosis, and inflammation [53,54]. It has reported that TWEAK induces tumor cell apoptosis in some types of tumor cells through JAK/STATs pathway [38]. Thus, the role of JAK/STATs pathway in the TWEAK induced protection on H9C2 cells was further investigated under I/R. Cryptotanshinone was used to blocking the activation of STAT3 [55] but it has no effect on TWEAK. To exclude the effect of other STATs, AG490, a typical inhibitor of JAK, was used [56] to block the JAK/STATs pathways totally. However, AG490 still failed to completely abolish the protection of TWEAK. Therefore, JAK/STATs signaling pathways were not responsible for the protective effects of TWEAK in our study.
Besides PI3K/AKT, JAK/STATs and NF-κB signaling pathways, there are others signaling pathways which can be activated by cardiac I/R [57,58]. For example, p38MAPK pathway is involved in the effect of TWEAK on the inflammatory osteolysis, and the peripheral blood mononuclear cells from patients with lupus nephritis [41,42] are activated by ischemia and their activation sustained during the reperfusion [57-60]. JNK signaling pathway is usually activated by reperfusion and leads to the activation of transcriptional factor AP-1 and subsequent cell apoptosis, which is a special way in the apoptosis induced by I/R [57,58,60,61]. It is necessary to investigate the role of interaction among these pathways in the effect of TWEAK on cardiomyocyte apoptosis under I/R.
To confirm the cardioprotection of TWEAK against cardiomyocyte death after I/R, I/R rats were treated with TWEAK prior to reperfusion. Evidence have proven that ischemic preconditioning has cardioprotective effect against IRI [62]. It is available to decrease IRI through the ischemic preconditioning. The molecular mechanisms underlying the protective effects of ischemic preconditioning include the activation of cellular protective signal pathways and the inhibition of apoptosis, such as the inhibition of mitochondria pathway via MAPK/ERK1/2 and PI3K/AKT pathways [63]. PI3K/AKT pathway mediates the protection against IRI by inhibiting the apoptosis due to increased mitochondrial membrane permeability [64]. For identifying the TWEAK reducing apoptosis in cardiomyocyte occurs after the I/R, TWEAK was directly injected into the infarct and border area immediately after ischemia. Inspiringly, TWEAK treatment significantly decreased the cardiomyocyte apoptosis as compared to control group. Moreover, the heart function was significantly improved after I/R and the scar size reduced in the presence of TWEAK treatment. On the other hand, although TWEAK has the potent ability to aggravate inflammation [15,18,19], there was no significant difference in the inflammation after I/R between TWEAK treatment group and control group. It is suggested that time point of TWEAK treatment might be crucial for preventing the cardiac IRI. These demonstrate that TWEAK can reduce the apoptotic cell death after cardiac I/R without aggravating the inflammation.
Taken together, our study for the first time reports that TWEAK is able to inhibit the apoptosis of cardiomyocytes following I/R in a PI3K/AKT signaling pathway dependent manner. Intra-myocardial treatment with TWEAK protects the myocardium against the apoptotic cell death post I/R without aggravating the inflammation. The study suggests that the time point and the way of therapy are key factors in the therapy of IHD. Nevertheless, the exact mechanism underlying the cardioprotection of TWEAK after cardiac I/R still needs to be further investigated in the future.
Conclusions
TWEAK, as a member of TNFSF, may be a novel target to prevent against IRI by inhibiting the cardiomyocyte apoptosis through PI3K/AKT pathway.
Acknowledgements
This work was funded by a grant from the Natural Science Foundation of China (No. 81500285), Natural Science Foundation of Shanxi Province (No. 2011011041-1) and Doctor Start-up Fund of Shanxi Medical University (No. 03201409).
Disclosure of conflict of interest
None.
References
- 1.Hartley A, Marshall DC, Salciccioli JD, Sikkel MB, Maruthappu M, Shalhoub J. Trends in Mortality From Ischemic Heart Disease and Cerebrovascular Disease in Europe: 1980 to 2009. Circulation. 2016;133:1916–1926. doi: 10.1161/CIRCULATIONAHA.115.018931. [DOI] [PubMed] [Google Scholar]
- 2.Hai JJ, Chan PH, Huang D, Ho MH, Ho CW, Cheung E, Lau CP, Tse HF, Siu CW. Clinical Characteristics, Management, and Outcomes of Hospitalized Heart Failure in a Chinese Population-The Hong Kong Heart Failure Registry. J Card Fail. 2016;22:600–8. doi: 10.1016/j.cardfail.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 3.Indications for fibrinolytic therapy in suspected acute myocardial infarction: collaborative overview of early mortality and major morbidity results from all randomised trials of more than 1000 patients. Fibrinolytic Therapy Trialists’ (FTT) Collaborative Group. Lancet. 1994;343:311–322. [PubMed] [Google Scholar]
- 4.Weintraub WS, Grau-Sepulveda MV, Weiss JM, O’Brien SM, Peterson ED, Kolm P, Zhang Z, Klein LW, Shaw RE, McKay C, Ritzenthaler LL, Popma JJ, Messenger JC, Shahian DM, Grover FL, Mayer JE, Shewan CM, Garratt KN, Moussa ID, Dangas GD, Edwards FH. Comparative effectiveness of revascularization strategies. N Engl J Med. 2012;366:1467–1476. doi: 10.1056/NEJMoa1110717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hannan EL, Racz MJ, Walford G, Jones RH, Ryan TJ, Bennett E, Culliford AT, Isom OW, Gold JP, Rose EA. Long-term outcomes of coronary-artery bypass grafting versus stent implantation. N Engl J Med. 2005;352:2174–2183. doi: 10.1056/NEJMoa040316. [DOI] [PubMed] [Google Scholar]
- 6.Schomig A, Kastrati A, Dirschinger J, Mehilli J, Schricke U, Pache J, Martinoff S, Neumann FJ, Schwaiger M. Coronary stenting plus platelet glycoprotein IIb/IIIa blockade compared with tissue plasminogen activator in acute myocardial infarction. Stent versus Thrombolysis for Occluded Coronary Arteries in Patients with Acute Myocardial Infarction Study Investigators. N Engl J Med. 2000;343:385–391. doi: 10.1056/NEJM200008103430602. [DOI] [PubMed] [Google Scholar]
- 7.Bulluck H, Yellon DM, Hausenloy DJ. Reducing myocardial infarct size: challenges and future opportunities. Heart. 2016;102:341–348. doi: 10.1136/heartjnl-2015-307855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest. 1994;94:1621–1628. doi: 10.1172/JCI117504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orogo AM, Gustafsson AB. Cell death in the myocardium: my heart won’t go on. IUBMB Life. 2013;65:651–656. doi: 10.1002/iub.1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thapalia BA, Zhou Z, Lin X. Autophagy, a process within reperfusion injury: an update. Int J Clin Exp Pathol. 2014;7:8322–8341. [PMC free article] [PubMed] [Google Scholar]
- 11.Linkermann A, Hackl MJ, Kunzendorf U, Walczak H, Krautwald S, Jevnikar AM. Necroptosis in immunity and ischemia-reperfusion injury. Am J Transplant. 2013;13:2797–2804. doi: 10.1111/ajt.12448. [DOI] [PubMed] [Google Scholar]
- 12.Barile L, Lionetti V, Cervio E, Matteucci M, Gherghiceanu M, Popescu LM, Torre T, Siclari F, Moccetti T, Vassalli G. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc Res. 2014;103:530–541. doi: 10.1093/cvr/cvu167. [DOI] [PubMed] [Google Scholar]
- 13.Bergmann MW, Rechner C, Freund C, Baurand A, El Jamali A, Dietz R. Statins inhibit reoxygenation-induced cardiomyocyte apoptosis: role for glycogen synthase kinase 3beta and transcription factor beta-catenin. J Mol Cell Cardiol. 2004;37:681–690. doi: 10.1016/j.yjmcc.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 14.Chicheportiche Y, Bourdon PR, Xu H, Hsu YM, Scott H, Hession C, Garcia I, Browning JL. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem. 1997;272:32401–32410. doi: 10.1074/jbc.272.51.32401. [DOI] [PubMed] [Google Scholar]
- 15.Blanco-Colio LM. TWEAK/Fn14 Axis: A Promising Target for the Treatment of Cardiovascular Diseases. Front Immunol. 2014;5:3. doi: 10.3389/fimmu.2014.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Novoyatleva T, Sajjad A, Engel FB. TWEAK-Fn14 Cytokine-Receptor Axis: A New Player of Myocardial Remodeling and Cardiac Failure. Front Immunol. 2014;5:50. doi: 10.3389/fimmu.2014.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wiley SR, Cassiano L, Lofton T, Davis-Smith T, Winkles JA, Lindner V, Liu H, Daniel TO, Smith CA, Fanslow WC. A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity. 2001;15:837–846. doi: 10.1016/s1074-7613(01)00232-1. [DOI] [PubMed] [Google Scholar]
- 18.Winkles JA. The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat Rev Drug Discov. 2008;7:411–425. doi: 10.1038/nrd2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rayego-Mateos S, Morgado-Pascual JL, Sanz AB, Ramos AM, Eguchi S, Batlle D, Pato J, Keri G, Egido J, Ortiz A, Ruiz-Ortega M. TWEAK transactivation of the epidermal growth factor receptor mediates renal inflammation. J Pathol. 2013;231:480–494. doi: 10.1002/path.4250. [DOI] [PubMed] [Google Scholar]
- 20.Pachel C, Mathes D, Bayer B, Dienesch C, Wangorsch G, Heitzmann W, Lang I, Ardehali H, Ertl G, Dandekar T, Wajant H, Frantz S. Exogenous administration of a recombinant variant of TWEAK impairs healing after myocardial infarction by aggravation of inflammation. PLoS One. 2013;8:e78938. doi: 10.1371/journal.pone.0078938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lynch CN, Wang YC, Lund JK, Chen YW, Leal JA, Wiley SR. TWEAK induces angiogenesis and proliferation of endothelial cells. J Biol Chem. 1999;274:8455–8459. doi: 10.1074/jbc.274.13.8455. [DOI] [PubMed] [Google Scholar]
- 22.Ren MY, Sui SJ. The role of TWEAK/Fn14 in cardiac remodeling. Mol Biol Rep. 2012;39:9971–9977. doi: 10.1007/s11033-012-1867-6. [DOI] [PubMed] [Google Scholar]
- 23.Novoyatleva T, Schymura Y, Janssen W, Strobl F, Swiercz JM, Patra C, Posern G, Wietelmann A, Zheng TS, Schermuly RT, Engel FB. Deletion of Fn14 receptor protects from right heart fibrosis and dysfunction. Basic Res Cardiol. 2013;108:325. doi: 10.1007/s00395-012-0325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turkmen K, Tonbul HZ, Erdur FM, Toker A, Biyik Z, Ozbiner H, Gaipov A, Gul EE, Kayrak M, Solak Y, Ozbek O, Turk S, Covic A. Soluble TWEAK independently predicts atherosclerosis in renal transplant patients. BMC Nephrol. 2013;14:144. doi: 10.1186/1471-2369-14-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi J, Jiang B, Qiu Y, Guan J, Jain M, Cao X, Bauer M, Su L, Burkly LC, Leone TC, Kelly DP, Liao R. PGC1alpha plays a critical role in TWEAK-induced cardiac dysfunction. PLoS One. 2013;8:e54054. doi: 10.1371/journal.pone.0054054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richter B, Rychli K, Hohensinner PJ, Berger R, Mortl D, Neuhold S, Zorn G, Huber K, Maurer G, Wojta J, Pacher R, Hulsmann M, Niessner A. Differences in the predictive value of tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in advanced ischemic and non-ischemic heart failure. Atherosclerosis. 2010;213:545–548. doi: 10.1016/j.atherosclerosis.2010.08.061. [DOI] [PubMed] [Google Scholar]
- 27.Mustonen E, Sakkinen H, Tokola H, Isopoussu E, Aro J, Leskinen H, Ruskoaho H, Rysa J. Tumour necrosis factor-like weak inducer of apoptosis (TWEAK) and its receptor Fn14 during cardiac remodelling in rats. Acta Physiol (Oxf) 2010;199:11–22. doi: 10.1111/j.1748-1716.2010.02080.x. [DOI] [PubMed] [Google Scholar]
- 28.Jain M, Jakubowski A, Cui L, Shi J, Su L, Bauer M, Guan J, Lim CC, Naito Y, Thompson JS, Sam F, Ambrose C, Parr M, Crowell T, Lincecum JM, Wang MZ, Hsu YM, Zheng TS, Michaelson JS, Liao R, Burkly LC. A novel role for tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in the development of cardiac dysfunction and failure. Circulation. 2009;119:2058–2068. doi: 10.1161/CIRCULATIONAHA.108.837286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meighan-Mantha RL, Hsu DK, Guo Y, Brown SA, Feng SL, Peifley KA, Alberts GF, Copeland NG, Gilbert DJ, Jenkins NA, Richards CM, Winkles JA. The mitogen-inducible Fn14 gene encodes a type I transmembrane protein that modulates fibroblast adhesion and migration. J Biol Chem. 1999;274:33166–33176. doi: 10.1074/jbc.274.46.33166. [DOI] [PubMed] [Google Scholar]
- 30.Ikner A, Ashkenazi A. TWEAK induces apoptosis through a death-signaling complex comprising receptor-interacting protein 1 (RIP1), Fas-associated death domain (FADD), and caspase-8. J Biol Chem. 2011;286:21546–21554. doi: 10.1074/jbc.M110.203745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakayama M, Ishidoh K, Kojima Y, Harada N, Kominami E, Okumura K, Yagita H. Fibroblast growth factor-inducible 14 mediates multiple pathways of TWEAK-induced cell death. J Immunol. 2003;170:341–348. doi: 10.4049/jimmunol.170.1.341. [DOI] [PubMed] [Google Scholar]
- 32.Krijnen PA, Nijmeijer R, Meijer CJ, Visser CA, Hack CE, Niessen HW. Apoptosis in myocardial ischaemia and infarction. J Clin Pathol. 2002;55:801–811. doi: 10.1136/jcp.55.11.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan P, Chen KJ, Wu J, Sun L, Sung HW, Weisel RD, Xie J, Li RK. The use of MMP2 antibody-conjugated cationic microbubble to target the ischemic myocardium, enhance Timp3 gene transfection and improve cardiac function. Biomaterials. 2014;35:1063–1073. doi: 10.1016/j.biomaterials.2013.10.043. [DOI] [PubMed] [Google Scholar]
- 34.Yan P, Gong H, Zhai X, Feng Y, Wu J, He S, Guo J, Wang X, Guo R, Xie J, Li RK. Decreasing CNPY2 Expression Diminishes Colorectal Tumor Growth and Development through Activation of p53 Pathway. Am J Pathol. 2016;186:1015–1024. doi: 10.1016/j.ajpath.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 35.Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol. 2010;72:19–44. doi: 10.1146/annurev.physiol.010908.163111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–1141. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 37.Gottlieb RA. Cell death pathways in acute ischemia/reperfusion injury. J Cardiovasc Pharmacol Ther. 2011;16:233–238. doi: 10.1177/1074248411409581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dorge H, Schulz R, Belosjorow S, Post H, van de Sand A, Konietzka I, Frede S, Hartung T, Vinten-Johansen J, Youker KA, Entman ML, Erbel R, Heusch G. Coronary microembolization: the role of TNF-alpha in contractile dysfunction. J Mol Cell Cardiol. 2002;34:51–62. doi: 10.1006/jmcc.2001.1489. [DOI] [PubMed] [Google Scholar]
- 39.Kubota T, McTiernan CF, Frye CS, Slawson SE, Lemster BH, Koretsky AP, Demetris AJ, Feldman AM. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ Res. 1997;81:627–635. doi: 10.1161/01.res.81.4.627. [DOI] [PubMed] [Google Scholar]
- 40.Chapman MS, Wu L, Amatucci A, Ho SN, Michaelson JS. TWEAK signals through JAK-STAT to induce tumor cell apoptosis. Cytokine. 2013;61:210–217. doi: 10.1016/j.cyto.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 41.Chorianopoulos E, Heger T, Lutz M, Frank D, Bea F, Katus HA, Frey N. FGF-inducible 14-kDa protein (Fn14) is regulated via the RhoA/ROCK kinase pathway in cardiomyocytes and mediates nuclear factor-kappaB activation by TWEAK. Basic Res Cardiol. 2010;105:301–313. doi: 10.1007/s00395-009-0046-y. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Z, Fang Y, Wang Q, Sun Y, Xiong C, Cao L, Wang B, Bao N, Zhao J. Tumor necrosis factor-like weak inducer of apoptosis regulates particle-induced inflammatory osteolysis via the p38 mitogen-activated protein kinase signaling pathway. Mol Med Rep. 2015;12:1499–1505. doi: 10.3892/mmr.2015.3529. [DOI] [PubMed] [Google Scholar]
- 43.Zhi-Chun L, Qiao-Ling Z, Zhi-Qin L, Xiao-Zhao L, Xiao-xia Z, Rong T. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) mediates p38 mitogen-activated protein kinase activation and signal transduction in peripheral blood mononuclear cells from patients with lupus nephritis. Inflammation. 2012;35:935–943. doi: 10.1007/s10753-011-9396-3. [DOI] [PubMed] [Google Scholar]
- 44.Burkly LC. Regulation of Tissue Responses: The TWEAK/Fn14 Pathway and Other TNF/TNFR Superfamily Members That Activate Non-Canonical NFkappaB Signaling. Front Immunol. 2015;6:92. doi: 10.3389/fimmu.2015.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tran NL, McDonough WS, Savitch BA, Sawyer TF, Winkles JA, Berens ME. The tumor necrosis factor-like weak inducer of apoptosis (TWEAK)-fibroblast growth factor-inducible 14 (Fn14) signaling system regulates glioma cell survival via NFkappaB pathway activation and BCL-XL/BCL-W expression. J Biol Chem. 2005;280:3483–3492. doi: 10.1074/jbc.M409906200. [DOI] [PubMed] [Google Scholar]
- 46.Valen G, Yan ZQ, Hansson GK. Nuclear factor kappa-B and the heart. J Am Coll Cardiol. 2001;38:307–314. doi: 10.1016/s0735-1097(01)01377-8. [DOI] [PubMed] [Google Scholar]
- 47.Wilhide ME, Tranter M, Ren X, Chen J, Sartor MA, Medvedovic M, Jones WK. Identification of a NF-kappaB cardioprotective gene program: NF-kappaB regulation of Hsp70.1 contributes to cardioprotection after permanent coronary occlusion. J Mol Cell Cardiol. 2011;51:82–89. doi: 10.1016/j.yjmcc.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riggins RB, Zwart A, Nehra R, Clarke R. The nuclear factor kappa B inhibitor parthenolide restores ICI 182,780 (Faslodex; fulvestrant)-induced apoptosis in antiestrogen-resistant breast cancer cells. Mol Cancer Ther. 2005;4:33–41. [PubMed] [Google Scholar]
- 49.Tong Y, Zhu W, Huang X, You L, Han X, Yang C, Qian W. PI3K inhibitor LY294002 inhibits activation of the Akt/mTOR pathway induced by an oncolytic adenovirus expressing TRAIL and sensitizes multiple myeloma cells to the oncolytic virus. Oncol Rep. 2014;31:1581–1588. doi: 10.3892/or.2014.3020. [DOI] [PubMed] [Google Scholar]
- 50.Ghigo A, Li M. Phosphoinositide 3-kinase: friend and foe in cardiovascular disease. Front Pharmacol. 2015;6:169. doi: 10.3389/fphar.2015.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G, Schulz R. The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther. 2008;120:172–185. doi: 10.1016/j.pharmthera.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 52.Mascareno E, El-Shafei M, Maulik N, Sato M, Guo Y, Das DK, Siddiqui MA. JAK/STAT signaling is associated with cardiac dysfunction during ischemia and reperfusion. Circulation. 2001;104:325–329. doi: 10.1161/01.cir.104.3.325. [DOI] [PubMed] [Google Scholar]
- 53.Abroun S, Saki N, Ahmadvand M, Asghari F, Salari F, Rahim F. STATs: An Old Story, Yet Mesmerizing. Cell J. 2015;17:395–411. doi: 10.22074/cellj.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Villarino AV, Kanno Y, Ferdinand JR, O’Shea JJ. Mechanisms of Jak/STAT signaling in immunity and disease. J Immunol. 2015;194:21–27. doi: 10.4049/jimmunol.1401867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li W, Saud SM, Young MR, Colburn NH, Hua B. Cryptotanshinone, a Stat3 inhibitor, suppresses colorectal cancer proliferation and growth in vitro. Mol Cell Biochem. 2015;406:63–73. doi: 10.1007/s11010-015-2424-0. [DOI] [PubMed] [Google Scholar]
- 56.Kobayashi A, Tanizaki Y, Kimura A, Ishida Y, Nosaka M, Toujima S, Kuninaka Y, Minami S, Ino K, Kondo T. AG490, a Jak2 inhibitor, suppressed the progression of murine ovarian cancer. Eur J Pharmacol. 2015;766:63–75. doi: 10.1016/j.ejphar.2015.09.039. [DOI] [PubMed] [Google Scholar]
- 57.Brown DI, Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res. 2015;116:531–549. doi: 10.1161/CIRCRESAHA.116.303584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bogoyevitch MA, Gillespie-Brown J, Ketterman AJ, Fuller SJ, Ben-Levy R, Ashworth A, Marshall CJ, Sugden PH. Stimulation of the stress-activated mitogen-activated protein kinase subfamilies in perfused heart. p38/RK mitogen-activated protein kinases and c-Jun N-terminal kinases are activated by ischemia/reperfusion. Circ Res. 1996;79:162–173. doi: 10.1161/01.res.79.2.162. [DOI] [PubMed] [Google Scholar]
- 59.Yokota T, Wang Y. p38 MAP kinases in the heart. Gene. 2016;575:369–376. doi: 10.1016/j.gene.2015.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin KH, Kuo WW, Jiang AZ, Pai P, Lin JY, Chen WK, Day CH, Shen CY, Padma VV, Huang CY. Tetramethylpyrazine Ameliorated Hypoxia-Induced Myocardial Cell Apoptosis via HIF-1alpha/JNK/p38 and IGFBP3/BNIP3 Inhibition to Upregulate PI3K/Akt Survival Signaling. Cell Physiol Biochem. 2015;36:334–344. doi: 10.1159/000374076. [DOI] [PubMed] [Google Scholar]
- 61.Hreniuk D, Garay M, Gaarde W, Monia BP, McKay RA, Cioffi CL. Inhibition of c-Jun N-terminal kinase 1, but not c-Jun N-terminal kinase 2, suppresses apoptosis induced by ischemia/reoxygenation in rat cardiac myocytes. Mol Pharmacol. 2001;59:867–874. doi: 10.1124/mol.59.4.867. [DOI] [PubMed] [Google Scholar]
- 62.Pac-Soo CK, Mathew H, Ma D. Ischaemic conditioning strategies reduce ischaemia/reperfusion-induced organ injury. Br J Anaesth. 2015;114:204–216. doi: 10.1093/bja/aeu302. [DOI] [PubMed] [Google Scholar]
- 63.Xue Y, Xie N, Lin Y, Xu J, Han Y, Wang S, Jiang H, Chi Z. Role of PI3K/Akt in diazoxide preconditioning against rat hippocampal neuronal death in pilocarpine-induced seizures. Brain Res. 2011;1383:135–140. doi: 10.1016/j.brainres.2011.01.037. [DOI] [PubMed] [Google Scholar]
- 64.Jang HS, Kim J, Kim KY, Kim JI, Cho MH, Park KM. Previous ischemia and reperfusion injury results in resistance of the kidney against subsequent ischemia and reperfusion insult in mice; a role for the Akt signal pathway. Nephrol Dial Transplant. 2012;27:3762–3770. doi: 10.1093/ndt/gfs097. [DOI] [PubMed] [Google Scholar]