

Abstract
Cure rates for acute myeloid leukemia (AML) remain suboptimal; thus new treatment strategies are needed for this deadly disease. Poor clinical outcomes have been associated with overexpression of the anti-apoptotic Bcl-2 family proteins Bcl-2, Bcl-xL, and Mcl-1, which have garnered great interest as therapeutic targets. While the Bcl-2-selective inhibitor ABT-199 has demonstrated promising preclinical anti-leukemic activities, intrinsic drug resistance remains a problem. In our most recent study, we identified Mcl-1 sequestration of Bim as a mechanism of intrinsic resistance to ABT-199 in AML cells, thus upregulating Bim could overcome such resistance. Histone deacetylase (HDAC) inhibitors (HDACI) are a class of agents that have been confirmed to upregulate Bim. This prompted our hypothesis that combining an HDACI with ABT-199 would overcome intrinsic resistance to ABT-199 and result in synergistic anti-leukemic activity against AML. In this study, we investigated the anti-leukemic activity of panobinostat, a pan-HDACI, in combination with ABT-199 in AML cell lines and primary patient samples. We found that the combined drug treatment resulted in synergistic induction of cell death in both AML cell lines and primary patient samples. Panobinostat treatment resulted in upregulation of Bim, which remained elevated in the presence of ABT-199. In addition, shRNA knockdown of Bim in AML cell lines significantly attenuated apoptosis induced by combined panobinostat and ABT-199. Our results provide compelling evidence that Bim plays a key role in the combined anti-leukemic activity of panobinostat and ABT-199 against AML, and support clinical evaluation of combined panobinostat and ABT-199 in the treatment of AML.
Keywords: ABT-199, Bcl-2, Bim, panobinostat, acute myeloid leukemia
Introduction
The five year survival rate for adults with acute myeloid leukemia (AML) is a dismal 25% [1]. Although the survival rate for the pediatric population is 65%, this significantly lags behind the overall survival rate for pediatric acute lymphoblastic leukemia (approximately 90%) [1]. Resistance to chemotherapy in leukemic cell line models and poor clinical outcomes of adult patients with AML have been associated with overexpression of the anti-apoptotic Bcl-2 family proteins Bcl-2, Bcl-xL, and Mcl-1, which have garnered great interest as potential therapeutic targets [2]. Inhibitors targeting multiple anti-apoptotic Bcl-2 family proteins have shown great potential in preclinical models but have limited clinical applicability due to increased incidence of thrombocytopenia; which was associated with inhibition of Bcl-xL [2]. Thus, attention has shifted to the Bcl-2-selective inhibitor ABT-199 (Venetoclax), which has shown excellent anti-leukemic activity against chronic lymphoblastic leukemia [3] and was approved by the US FDA in April 2016. Although ABT-199 has been shown to have promising preclinical effects on AML, intrinsic drug resistance remains a concern [4-6].
Mcl-1 has been identified as playing a key role in resistance to ABT-199 in AML and lymphoid malignancies [5,7,8]. In our most recent study, ABT-199 treatment was found to result in dissociation of pro-apoptotic Bim from Bcl-2, demonstrating its canonical role in inducing apoptosis in AML cells [5]. However, ABT-199 treatment also resulted in increased sequestration of Bim by Mcl-1, preventing Bim from inducing apoptosis, while also resulting in increased Mcl-1 protein levels in ABT-199-resistant AML cells. These results suggest that Mcl-1 is a key player in the intrinsic resistance to ABT-199 in AML cells. CRISPR knockdown of Mcl-1 significantly enhanced ABT-199-induced apoptosis in AML cells, confirming its role in this aspect [5]. While there have been a number of reports focused on downregulating Mcl-1 [6,8,9], another approach to overcome this mechanism of intrinsic resistance to ABT-199 would be to increase Bim levels, leading to increased Bim/Mcl-1 ratio favoring apoptosis. Histone deacetylase (HDAC) inhibitors (HDACI) are a class of agents that have been demonstrated to upregulate Bim [10-12]. In particular, panobinostat, a highly potent pan-HDACI [13], has recently been approved by the US FDA for the treatment of multiple myeloma [14] and has been demonstrated to upregulate Bim in cancer cells [10,12]. Therefore, panobinostat may overcome intrinsic ABT-199 resistance by increasing Bim protein levels. In this study, we investigated the combination of panobinostat and ABT-199 in AML cell lines and primary patient samples. We found synergistic induction of cell death by the combined drug treatment. In addition, we provided evidence that Bim played a key role in the combined anti-leukemic activity against AML. These findings support clinical evaluation of combined panobinostat and ABT-199 in the treatment of AML.
Materials and methods
Drugs
ABT-199 and panobinostat were purchased from Selleck Chemicals (Houston, TX, USA).
Cell culture
THP-1 and U937 cell lines were purchased from the American Type Culture Collection (Manassas, VA, USA). The cell lines were cultured in RPMI 1640 media with 10% fetal bovine serum (Life Technologies, Carlsbad, CA, USA) and 2 mM L-glutamine, plus 100 U/ml penicillin and 100 µg/ml streptomycin, in a 37°C humidified atmosphere containing 5% CO2/95% air. The cell lines were tested for the presence of mycoplasma on a monthly basis.
Diagnostic blast samples were purified by standard Ficoll-Hypaque density centrifugation, then cultured in RPMI 1640 with 20% fetal bovine serum (Life Technologies) supplemented with ITS solution (Sigma-Aldrich, St. Louis, MO, USA) and 20% supernatant of the 5637 bladder cancer cell line (as a source of granulocyte-macrophage colony-stimulating factor) [5-7,15].
Clinical samples
Diagnostic AML blast samples were obtained from the First Hospital of Jilin University, Changchun, China. Written informed consent was provided according to the Declaration of Helsinki. Clinical samples were screened for gene mutations and fusion genes as previously described [5-7,16,17]. Patient characteristics are presented in Table 1.
Table 1.
Patient characteristics for the primary acute myeloid leukemia patient samples
| Patient | Gender | Age (year) | Disease status | FAB subtype | Cytogenetics | Blast purity (%) | Fusion gene/Gene mutation |
|---|---|---|---|---|---|---|---|
| AML#18 | Female | 19 | Relapsed | M2 | 46, XX, t(8;21)(q22;q22) | 54 | AML1-ETO |
| AML#19 | Male | 4 | Newly diagnosed | M4 | 46, XY | 40.5 | |
| AML#21 | Male | 42 | Newly diagnosed | M4 | 46, XY/44, XY, -17, -19, (11q-?) | 51 | MLL-ELL |
| AML#23 | Male | 60 | Newly diagnosed | M5 | 48, XY, +2,+8, i(12)(q10) | 81 | MLL-AF10 |
| AML#25 | Female | 12 | Newly diagnosed | M3 | 46, XX, t(15;17)(q22;q21) | 88 | |
| AML#29 | Male | 9 | Newly diagnosed | M4 | 46, XY, t(6;9)(p22;q34) | 31 | DEK-CAN |
| AML#30 | NA | NA | NA | NA | NA | NA | NA |
| AML#31 | Male | 17 | Newly diagnosed | M2 | 46, XY | 68.5 | CEBPA |
| AML#33 | Female | 76 | Newly diagnosed | M5 | 46, XX | 84.5 | dupMLL, CEBPA |
| AML#34 | Male | 52 | Newly diagnosed | M4 | 46, XY | 96 | DEK-CAN |
| AML#35 | Male | 65 | Newly diagnosed | M5 | 47, XY, add(7q), -16, -17, +marx3 | 76 | |
| AML#38 | Male | 27 | Newly diagnosed | M3 | 46, XY, t(15;17)(q22;q21) | 90.5 | PML-RARα |
Abbreviation: NA, not available.
In vitro cytotoxicity assays
In vitro cytotoxicities of the diagnostic AML blast samples were measured by using MTT (3-[4,5-dimethyl-thiazol-2-yl]-2,5-diphenyltetrazoliumbromide, Sigma-Aldrich) assays, as previously described [18,19]. Briefly, the cells were either treated with variable concentrations of ABT-199 or panobinostat alone, or in combination, for 72 hours. The cells were lysed using 10% SDS in 10 mM HCL. IC50 values were calculated as drug concentrations necessary to inhibit 50% proliferation compared to vehicle control treated cells. The IC50 values are means of duplicates from one experiment due to limited sample. Standard isobologram analysis was performed to determine the extent and direction of anti-leukemic interactions [20]. The IC50 values of each drug are plotted on the axes; the solid line represents the additive effect, whereas the points represent the concentrations of each drug resulting in 50% inhibition of proliferation. Points falling below the line indicate synergistic effect, whereas those above the line indicate antagonistic effect. Patient sample selection was merely based on sample availability.
Western blot analysis
Cells were lysed in Tris buffer (10 mM, pH 8.0) containing protease inhibitors (Roche Diagnostics, Indianapolis, IN, USA). Whole cell lysates were subjected to SDS-polyacrylamide gel electrophoresis, electrophoretically transferred onto polyvinylidene difluoride (PVDF) membranes (Thermo Fisher Inc., Rockford, IL, USA) and immunoblotted with anti-Bim, -Bcl-2, -Mcl-1, -PARP, -Cleaved Caspase 3 (Cell Signaling Technology, Danvers, MA, USA), or -β-actin (A2228, Sigma-Aldrich) antibody, as previously described [18,21,22]. Immunoreactive proteins were visualized using the Odyssey Infrared Imaging System (Li-Cor, Lincoln, NE, USA), as described by the manufacturer. Western blots were repeated at least 4 times and one representative blot is shown. Densitometry measurements were made using Odyssey V3.0 (Li-Cor), normalized to β-actin first and then to the no drug treatment control (set as 1).
Apoptosis
AML cells were treated with variable concentrations of ABT-199 or panobinostat, alone or in combination, for 24 h, and then subjected to flow cytometry analysis to determine drug-induced apoptosis using the Annexin V-fluorescein Isothiocyanate (FITC)/Propidium Iodide (PI) Apoptosis Kit (Beckman Coulter; Brea, CA, USA), as previously described [18,23]. Results were expressed as percent of Annexin V+ cells. AML cell line experiments were performed 3 independent times in triplicate and data presented are from one representative experiment, while the patient sample experiments were performed once in triplicate due to limited sample availability. The combination index (CI) values were determined using CompuSyn software. CI<0.9 indicates synergistic, 0.9<CI<1.1 indicates additive, and CI>1.1 indicates antagonistic anti-leukemic interactions [24].
Production of lentivirus particles and transduction of AML cells
The pMD-VSV-G and delta 8.2 plasmids were gifts from Dr. Dong at Tulane University. Bim and non-target control (NTC) shRNA lentiviral vectors were purchased from Sigma-Aldrich. Lentivirus production and transduction were carried out as previously described [25,26]. Briefly, TLA-HEK293T cells were transfected with pMD-VSV-G, delta 8.2, and lentiviral shRNA constructs using Lipofectamine and Plus reagents (Life Technologies) according to the manufacturer’s instructions. Virus containing culture medium was harvested 48 h post-transfection. Cells were transduced overnight using 1 mL of virus supernatant and 4 µg of polybrene and then cultured for an additional 48 h prior to selection with puromycin.
Statistical analysis
Differences in cell apoptosis between treated (individually or combined) and untreated cells were compared using the pair-wise two-sample t-test. Statistical analyses were performed with GraphPad Prism 5.0. Error bars represent ± s.e.m.
Results
ABT-199 and panobinostat synergize in inducing apoptosis in AML cell lines and primary patient samples
To determine if panobinostat enhances ABT-199-induced apoptosis in AML cells, we treated two ABT-199-resistant AML cell lines, THP-1 and U937, (ABT-199 IC50s of 2.4 mM for THP-1 and 13.5 mM for U937 cells, as determined previously by MTT assays [7]), with both drugs individually or simultaneously for 24 h. Annexin V/PI staining and flow cytometry analyses revealed significantly increased annexin V+ cells for the combined drug treatments compared to that for the individual drug treatments (Figure 1A, 1B). This was accompanied by increased cleavage of caspase-3 and PARP in the combined treatments as compared to the individual drug treatments (Figure 1C, 1D). These results demonstrate that the combined drug treatments caused significantly increased apoptosis in the two AML cell lines as compared to the individual drug treatments. Calculation of CI values by using the CompuSyn software showed that the anti-leukemic interactions between the two drugs were synergistic, as indicated by CI<0.9 (Figure 1A, 1B).
Figure 1.
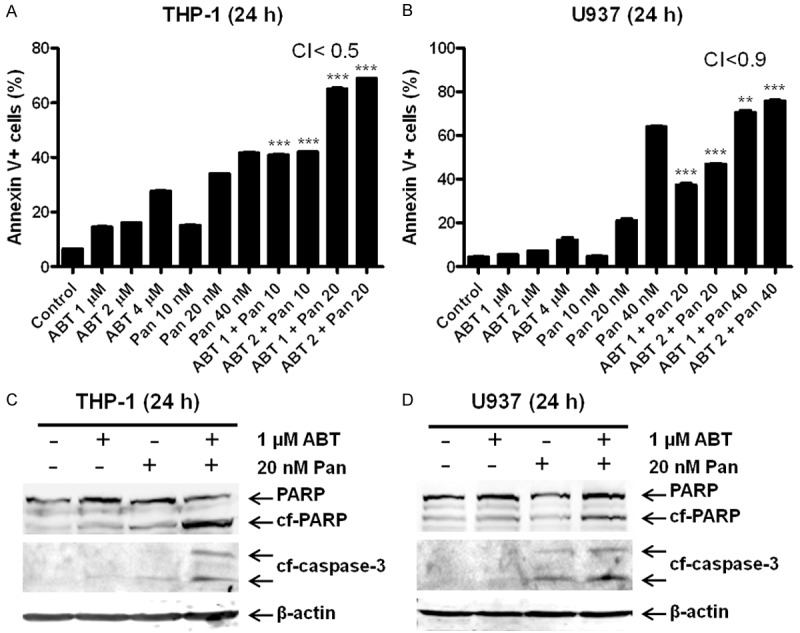
Synergistic induction of apoptosis by ABT-199 and panobinostat (Pan) in AML cell lines determined by annexin V/PI staining and flow cytometry and CompuSyn software analyses. (A and B) THP-1 (A) and U937 (B) cells were treated with ABT-199 and panobinostat, alone or in combination, for 24 h and then subjected to annexin V/PI staining and flow cytometry analysis. CI values were calculated using CompuSyn software. **indicates P<0.01 and ***indicates P<0.001. (C and D) Whole cell lysates were subjected to Western blotting and probed with the indicated antibodies.
To confirm that ABT-199 and panobinostat also synergize in primary AML patient samples, we tested the drugs in two freshly isolated AML blast samples (AML#31 and AML#38; samples were chosen based on availability of adequate number of cells for the assay) ex vivo. Interestingly, simultaneous combination of the two drugs caused strong synergistic induction of apoptosis in the two primary AML patient samples, as indicated by CI<0.25 (Figure 2).
Figure 2.
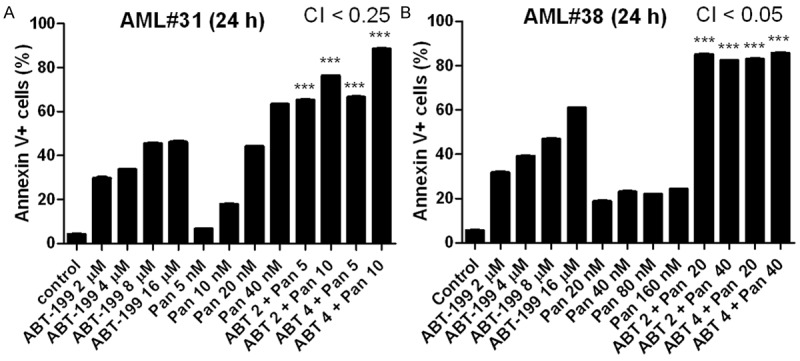
Synergistic induction of apoptosis by ABT-199 and panobinostat in primary AML patient samples determined by annexin V/PI staining and flow cytometry and CompuSyn software analyses. Primary AML patient samples, AML#31 (A) and AML#38 (B) were treated as indicated for 24 h and then subjected to annexin V/PI staining and flow cytometry analyses. CI values were calculated using CompuSyn software. ***indicates P<0.001.
ABT-199 and panobinostat synergize in inhibiting cell proliferation in primary AML patient samples
To confirm the synergistic anti-leukemic interactions between ABT-199 and panobinostat in AML cells detected by annexin V/PI staining and flow cytometry analyses, MTT assays and standard isobologram analyses were performed in the AML#31 and AML#38 primary patient samples. As shown in Figure 3, simultaneous combination of the two drugs resulted in synergistic proliferation inhibition in the two primary patient samples.
Figure 3.

Panobinostat synergizes with ABT-199 in primary patient samples determined by MTT assays and standard isobologram analyses. Primary AML patient samples were treated with ABT-199 and panobinostat, alone or in combination, for 72 h and then viable cells were determined using MTT reagent. The IC50 values are means of duplicates from one experiment due to limited sample. Standard isobologram analysis was performed to determine the extent and direction of the anti-leukemic interaction between the two agents. The IC50 values of each drug are plotted on the axes; the solid line represents the additive effect, while the points represent the concentrations of each drug resulting in 50% inhibition of proliferation. Points falling below the line indicate synergistic effect, whereas those above the line indicate antagonistic effect.
To further enhance the clinical relevance of our study, we included 10 additional primary AML patient samples in our drug interaction experiments. Due to limited sample availability, these additional 10 primary AML patient samples were tested by MTT assays and standard isobologram analyses, but not by annexin V/PI staining and flow cytometry analyses, which require large number of cells. Essentially the same results were obtained in the 10 additional samples as those in the AML#31 and AML#38 samples (Figure 3). These results further confirm the synergistic anti-leukemic interactions between ABT-199 and panobinostat in AML cells and the clinical relevance of our study.
Panobinostat upregulates Bim expression in AML cell lines
To determine the effects of panobinostat on Bim expression in the absence or presence of ABT-199 in AML cells, the THP-1 and U937 AML cell lines were treated with panobinostat and ABT-199, alone or in combination, for 24 h and then whole cell lysates were subjected to western blotting. Bim protein levels were increased after treatment with panobinostat alone or in combination with ABT-199 (~30-40%, Figure 4), which is consistent with other studies reporting Bim upregulation following HDACI treatment [11,27]. In contrast, Bcl-2 levels remained unchanged. As we found most recently [5], ABT-199 treatment resulted in increased Mcl-1 protein levels in relatively resistant AML cells, which remained elevated in the presence of panobinostat (Figure 4). These results indicate that even though panobinostat treatment does not block Mcl-1 upregulation induced by ABT-199, it upregulates Bim expression, ultimately resulting in increased ratio of Bim/Mcl-1 favoring apoptosis.
Figure 4.
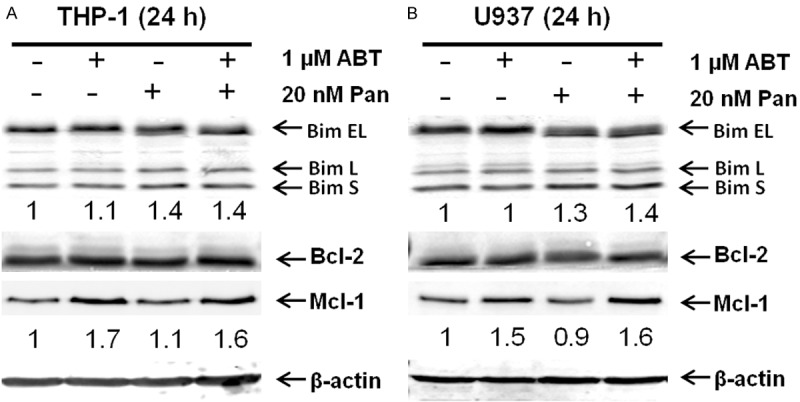
The effect of panobinostat on Bim expression in AML cell lines. THP-1 (A) and U937 (B) cells were treated with ABT-199 and panobinostat, alone or in combination, for 24 h. Whole cell lysates were subjected to Western blotting and probed with the indicated antibodies.
Bim plays a critical role in apoptosis induced by ABT-199 and panobinostat, alone or in combination
To confirm the functional role Bim plays in apoptosis induced by ABT-199 and panobinostat, alone or in combination, we used shRNA to knock down Bim in both THP-1 and U937 cells (Figure 5A, 5B). Cells infected with non-target control shRNA (NTC) showed a similar induction of Bim by panobinostat, and the combination of ABT-199 and panobinostat resulted in similar synergistic induction of apoptosis. As expected, Bim knockdown (40% compared to the NTC) significantly attenuated apoptosis induced by combined panobinostat and ABT-199 (Figure 5C, 5D). These results provide compelling evidence that panobinostat enhances ABT-199-induced apoptosis in AML cells through upregulation of Bim.
Figure 5.
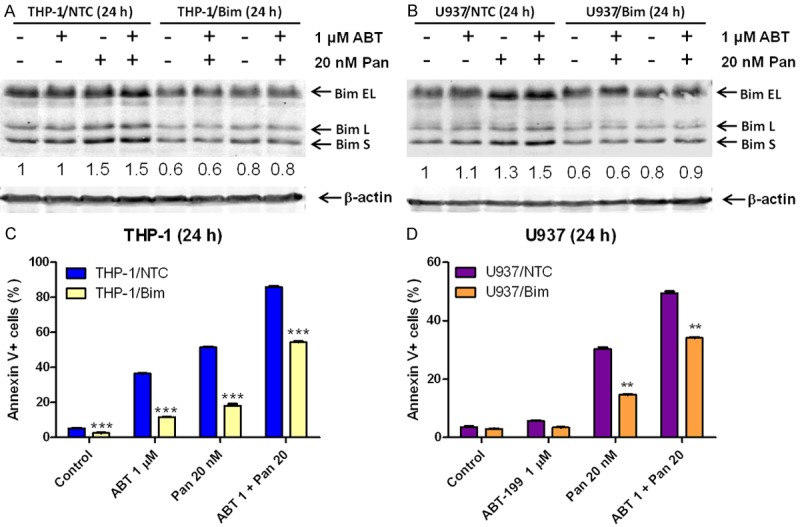
The role of Bim in apoptosis induced by ABT-199 and panobinostat, alone or in combination, in AML cell lines. (A and B) THP-1 (A) and U937 (B) cells were infected with non-target control (NTC) or Bim shRNA lentivirus particles overnight, then washed and incubated for 48 h prior to adding puromycin to the culture medium. The cells were then treated with ABT-199 and panobinostat, alone or in combination, for 24 h. Whole cell lysates were subjected to Western blotting. The fold changes for the Bim densitometry measurements, normalized to β-actin and then compared to no drug treatment control, are indicated. (C and D) THP-1/NTC, THP-1/Bim (C), U937/NTC and U937/Bim (D) cells were treated with ABT-199 and panobinostat, alone or in combination, for 24 h and then subjected to annexin V/PI staining and flow cytometry analyses. **indicates P<0.01 and ***indicates P<0.001.
Discussion
In this study, we investigated the ability of panobinostat to overcome ABT-199 resistance in AML cells. We found that panobinostat synergistically enhanced ABT-199-induced apoptosis in two AML cell lines and two primary patient samples, which were relatively resistant to ABT-199. These results were further confirmed in 10 additional primary AML patient samples, as determined by MTT assays and standard isobologram analyses. Our results were similar to those of Chen et al. who used ABT-737, a Bcl-2 and Bcl-xL inhibitor, in combination with the HDACI suberoyl bis-hydroxamic acid (SBHA) and found that upregulation of Bim by SBHA contributed to the synergistic induction of cell death [11]. Another study, in mantle cell lymphoma, also found synergistic anti-cancer activity for ABT-263, which inhibits both Bcl-2 and Bcl-xL, in combination with suberoylanilide hydroxamic acid (SAHA) [27].
Consistent with other studies reporting Bim upregulation following HDACI treatment [11,27] we confirmed that panobinostat treatment caused upregulation of Bim (30-40%) in AML cell lines. Our shRNA knockdown experiments convincingly revealed that Bim was a critical mediator of panobinostat enhancement on ABT-199-induced apoptosis. Although we did not investigate how panobinostat induces Bim expression, SAHA has been demonstrated to induce acetylation of the Bim, Bmf and NOXA promoters [27], and E2F1 has been shown to the increase of Bim expression following HDACI treatment [28], suggesting that increased transcription may be responsible for the upregulation of Bim by panobinostat.
However, it is important to note that although expression of the Bim shRNA in AML cell lines completely abolished Bim induction by panobinostat, it did not completely abolish drug-induced apoptosis suggesting that other factors contributed. Panobinostat has been demonstrated to affect numerous proteins which could also contribute to the enhanced cell death induced by ABT-199 [29].
In conclusion, we provide compelling evidence to support the development of panobinostat in combination with ABT-199 in the treatment of AML. Our cell line data demonstrates that ABT-199 synergizes with panobinostat in ABT-199 resistant cells. Furthermore, our MTT data using primary patient samples demonstrates that the combination also synergized in ABT-199 sensitive samples, suggesting that screening for ABT-199 resistance may not be necessary.
Acknowledgements
This study was supported by the Barbara Ann Karmanos Cancer Institute, Wayne State University School of Medicine and Jilin University, Changchun, China, grants from the Children’s Hospital of Michigan Foundation, Hyundai Hope On Wheels, the Natural Science Foundation of China (NSFC 31271477 and NSFC 31471295), the Graduate Innovation Fund of Jilin University (NX), the Christoph A.L.L. Star Foundation, and the Ring Screw Textron Endowed Chair for Pediatric Cancer Research. The funders had no role in study design, data collection, analysis and interpretation of data, decision to publish, or preparation of the manuscript.
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Davids MS, Brown JR. Targeting the B cell receptor pathway in chronic lymphocytic leukemia. Leuk Lymphoma. 2012;53:2362–2370. doi: 10.3109/10428194.2012.695781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, Wong S, Dunbar M, Zhu M, Desai MB, Cerri E, Heitner Enschede S, Humerickhouse RA, Wierda WG, Seymour JF. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2016;374:311–322. doi: 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 5.Niu X, Zhao J, Ma J, Xie C, Edwards H, Wang G, Caldwell JT, Xiang S, Zhang X, Chu R, Wang J, Lin H, Taub JW, Ge Y. Binding of released Bim to Mcl-1 is a mechanism of intrinsic resistance to ABT-199 which can be overcome by combination with daunorubicin or cytarabine in AML cells. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-15-3057. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao J, Niu X, Li X, Edwards H, Wang G, Wang Y, Taub JW, Lin H, Ge Y. Inhibition of CHK1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Oncotarget. 2016 doi: 10.18632/oncotarget.9185. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Niu X, Wang G, Wang Y, Caldwell JT, Edwards H, Xie C, Taub JW, Li C, Lin H, Ge Y. Acute myeloid leukemia cells harboring MLL fusion genes or with the acute promyelocytic leukemia phenotype are sensitive to the Bcl-2-selective inhibitor ABT-199. Leukemia. 2014;28:1557–1560. doi: 10.1038/leu.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choudhary GS, Al-Harbi S, Mazumder S, Hill BT, Smith MR, Bodo J, Hsi ED, Almasan A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015;6:e1593. doi: 10.1038/cddis.2014.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vaillant F, Merino D, Lee L, Breslin K, Pal B, Ritchie ME, Smyth GK, Christie M, Phillipson LJ, Burns CJ, Mann GB, Visvader JE, Lindeman GJ. Targeting BCL-2 with the BH3 mimetic ABT-199 in estrogen receptor-positive breast cancer. Cancer Cell. 2013;24:120–129. doi: 10.1016/j.ccr.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Inoue S, Riley J, Gant TW, Dyer MJ, Cohen GM. Apoptosis induced by histone deacetylase inhibitors in leukemic cells is mediated by Bim and Noxa. Leukemia. 2007;21:1773–1782. doi: 10.1038/sj.leu.2404760. [DOI] [PubMed] [Google Scholar]
- 11.Chen S, Dai Y, Pei XY, Grant S. Bim upregulation by histone deacetylase inhibitors mediates interactions with the Bcl-2 antagonist ABT-737: evidence for distinct roles for Bcl-2, Bcl-xL, and Mcl-1. Mol Cell Biol. 2009;29:6149–6169. doi: 10.1128/MCB.01481-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fiskus W, Sharma S, Saha S, Shah B, Devaraj SG, Sun B, Horrigan S, Leveque C, Zu Y, Iyer S, Bhalla KN. Pre-clinical efficacy of combined therapy with novel beta-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia. 2015;29:1267–1278. doi: 10.1038/leu.2014.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett. 2009;280:233–241. doi: 10.1016/j.canlet.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 14.Garnock-Jones KP. Panobinostat: first global approval. Drugs. 2015;75:695–704. doi: 10.1007/s40265-015-0388-8. [DOI] [PubMed] [Google Scholar]
- 15.Quentmeier H, Zaborski M, Drexler HG. The human bladder carcinoma cell line 5637 constitutively secretes functional cytokines. Leuk Res. 1997;21:343–350. doi: 10.1016/s0145-2126(96)00132-4. [DOI] [PubMed] [Google Scholar]
- 16.Qi W, Zhang W, Edwards H, Chu R, Madlambayan GJ, Taub JW, Wang Z, Wang Y, Li C, Lin H, Ge Y. Synergistic anti-leukemic interactions between panobinostat and MK-1775 in acute myeloid leukemia ex vivo. Cancer Biol Ther. 2015;16:1784–1793. doi: 10.1080/15384047.2015.1095406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qi W, Xie C, Li C, Caldwell JT, Edwards H, Taub JW, Wang Y, Lin H, Ge Y. CHK1 plays a critical role in the anti-leukemic activity of the wee1 inhibitor MK-1775 in acute myeloid leukemia cells. J Hematol Oncol. 2014;7:53. doi: 10.1186/s13045-014-0053-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie C, Edwards H, Xu X, Zhou H, Buck SA, Stout ML, Yu Q, Rubnitz JE, Matherly LH, Taub JW, Ge Y. Mechanisms of synergistic antileukemic interactions between valproic acid and cytarabine in pediatric acute myeloid leukemia. Clin Cancer Res. 2010;16:5499–5510. doi: 10.1158/1078-0432.CCR-10-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu X, Xie C, Edwards H, Zhou H, Buck SA, Ge Y. Inhibition of histone deacetylases 1 and 6 enhances cytarabine-induced apoptosis in pediatric acute myeloid leukemia cells. PLoS One. 2011;6:e17138. doi: 10.1371/journal.pone.0017138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tallarida RJ. Drug synergism: its detection and applications. J Pharmacol Exp Ther. 2001;298:865–872. [PubMed] [Google Scholar]
- 21.Ge Y, Stout ML, Tatman DA, Jensen TL, Buck S, Thomas RL, Ravindranath Y, Matherly LH, Taub JW. GATA1, cytidine deaminase, and the high cure rate of Down syndrome children with acute megakaryocytic leukemia. J Natl Cancer Inst. 2005;97:226–231. doi: 10.1093/jnci/dji026. [DOI] [PubMed] [Google Scholar]
- 22.Ge Y, Dombkowski AA, LaFiura KM, Tatman D, Yedidi RS, Stout ML, Buck SA, Massey G, Becton DL, Weinstein HJ, Ravindranath Y, Matherly LH, Taub JW. Differential gene expression, GATA1 target genes, and the chemotherapy sensitivity of Down syndrome megakaryocytic leukemia. Blood. 2006;107:1570–1581. doi: 10.1182/blood-2005-06-2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edwards H, Xie C, LaFiura KM, Dombkowski AA, Buck SA, Boerner JL, Taub JW, Matherly LH, Ge Y. RUNX1 regulates phosphoinositide 3-kinase/AKT pathway: role in chemotherapy sensitivity in acute megakaryocytic leukemia. Blood. 2009;114:2744–2752. doi: 10.1182/blood-2008-09-179812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 25.Xie C, Drenberg C, Edwards H, Caldwell JT, Chen W, Inaba H, Xu X, Buck SA, Taub JW, Baker SD, Ge Y. Panobinostat enhances cytarabine and daunorubicin sensitivities in AML cells through suppressing the expression of BRCA1, CHK1, and Rad51. PLoS One. 2013;8:e79106. doi: 10.1371/journal.pone.0079106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caldwell JT, Edwards H, Buck SA, Ge Y, Taub JW. Targeting the wee1 kinase for treatment of pediatric Down syndrome acute myeloid leukemia. Pediatr Blood Cancer. 2014;61:1767–1773. doi: 10.1002/pbc.25081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xargay-Torrent S, Lopez-Guerra M, Saborit-Villarroya I, Rosich L, Campo E, Roue G, Colomer D. Vorinostat-induced apoptosis in mantle cell lymphoma is mediated by acetylation of proapoptotic BH3-only gene promoters. Clin Cancer Res. 2011;17:3956–3968. doi: 10.1158/1078-0432.CCR-10-3412. [DOI] [PubMed] [Google Scholar]
- 28.Zhao Y, Tan J, Zhuang L, Jiang X, Liu ET, Yu Q. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc Natl Acad Sci U S A. 2005;102:16090–16095. doi: 10.1073/pnas.0505585102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]