

Abstract
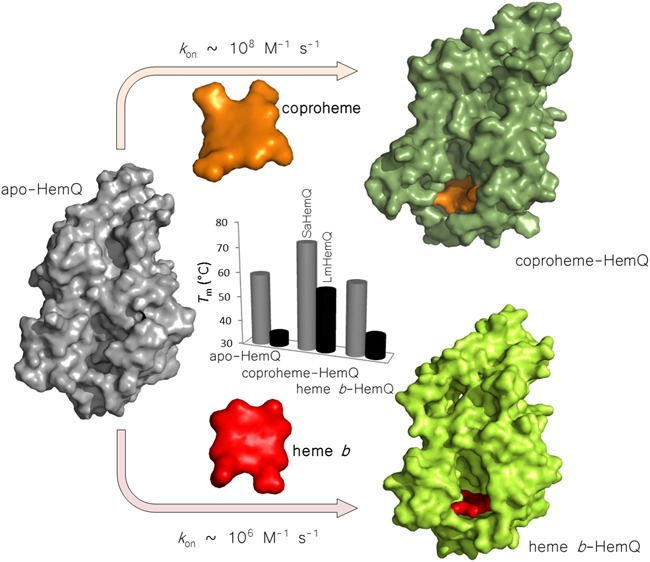
Recently, a novel pathway for heme b biosynthesis in Gram-positive bacteria has been proposed. The final poorly understood step is catalyzed by an enzyme called HemQ and includes two decarboxylation reactions leading from coproheme to heme b. Coproheme has been suggested to act as both substrate and redox active cofactor in this reaction. In the study presented here, we focus on HemQs from Listeria monocytogenes (LmHemQ) and Staphylococcus aureus (SaHemQ) recombinantly produced as apoproteins in Escherichia coli. We demonstrate the rapid and two-phase uptake of coproheme by both apo forms and the significant differences in thermal stability of the apo forms, coproheme-HemQ and heme b-HemQ. Reduction of ferric high-spin coproheme-HemQ to the ferrous form is shown to be enthalpically favored but entropically disfavored with standard reduction potentials of −205 ± 3 mV for LmHemQ and −207 ± 3 mV for SaHemQ versus the standard hydrogen electrode at pH 7.0. Redox thermodynamics suggests the presence of a pronounced H-bonding network and restricted solvent mobility in the heme cavity. Binding of cyanide to the sixth coproheme position is monophasic but relatively slow (∼1 × 104 M–1 s–1). On the basis of the available structures of apo-HemQ and modeling of both loaded forms, molecular dynamics simulation allowed analysis of the interaction of coproheme and heme b with the protein as well as the role of the flexibility at the proximal heme cavity and the substrate access channel for coproheme binding and heme b release. Obtained data are discussed with respect to the proposed function of HemQ in monoderm bacteria.
HemQ is an enzyme involved in the late stages of the heme biosynthetic pathway of monoderm (Gram-positive) bacteria.1−3 It catalyzes the decarboxylation of iron coproporphyrin III (coproheme) to yield heme b via an unusual peroxide-dependent reaction that is poorly understood.4 HemQs are structurally and phylogenetically related to two families of heme enzymes, i.e., chlorite dismutases (Cld) and dye-decolorizing peroxidases (DyP), and were originally designated in the literature as Cld-like proteins (Figure 1A,B).5−7 However, the functions of Cld, DyP, and HemQ are very diverse and specialized.
Figure 1.

Sequence analysis and phylogenetics of HemQs and chlorite dismutases. (A) Sequence alignment of selected chlorite dismutase (Cld) and HemQ sequences. Completely conserved residues are highlighted in blue background (proximal histidine in dark blue); relevant conserved residues are highlighted in yellow for Cld, red for HemQs, green for HemQs from Firmicutes, and orange for Actinobacteria. (B) Phylogenetic tree (maximum likelihood) of Clds and HemQs based on the sequence alignment presented in panel A. Proteins from proteobacteria, Firmicutes, Actinobacteria, cyanobacteria, and Nitrospira are highlighted in yellow, green, orange, blue, and red, respectively. Asterisks indicate available crystal structures deposited in the Protein Data Bank. (C) Comparison of structures of coproheme and heme b.
Clds are oxidoreductases that are able to convert chlorite into chloride and dioxygen.8 In this reaction, an oxygen–oxygen double bond is formed, a reaction so far described only for the manganese water-splitting complex of Photosystem II.9 DyPs are versatile peroxidases, capable of performing hydrogen peroxide-dependent one-electron oxidations of various aromatic compounds.10,11 In both Clds and DyPs, a heme b is tightly bound to the protein by a proximal histidine, which is part of an extended H-bonding network.5,12−16 In addition, Clds have a catalytic distal arginine,5,17 which is important for stabilization of the transiently produced hypochlorite during the reaction.18,19 In DyPs, the catalytic distal arginine structurally aligns with the distal arginine of Clds, but in addition, a catalytic aspartate is present.20−23
HemQs were demonstrated to bind heme b reversibly with low affinities, and no substantial enzymatic activities toward hydrogen peroxide or chlorite could be detected for heme b-bound HemQs.24−26 Similar to the case in Cld and DyP, in HemQ the proximal ligand is also a histidine (H174, LmHemQ numbering), but the lack of the proximal hydrogen bonding network seems to be responsible for its weak heme b binding properties.27 In contrast to Clds and DyPs, no charged amino acid residue is found distally of the predicted heme binding site. On the basis of structural and sequence alignments, a glutamine is present in HemQs from Firmicutes (Q187) and an alanine in Actinobacteria at the respective position of the catalytic arginine in Clds or DyPs. This might explain the absence of any significant catalase, peroxidase, or chlorite dismutase activity.27 Until now, there have been only apo structures of HemQs available in the Protein Data Bank (PDB) (entries 4WWS and 1T0T).
Dailey and co-workers discovered that HemQ is an essential enzyme in the heme biosynthesis of Firmicutes and Actinobacteria and identified coproheme as its substrate.1,24 Coproheme has four propionate groups located at positions 2, 4, 6, and 7 of the porphyrin ring. Propionates at positions 2 and 4 are decarboxylated by HemQ in a stepwise fashion to form the respective vinyl groups of heme b (Figure 1C).1 The interactions of coproheme with the protein moiety of HemQ as well as the catalytic reaction mechanism of the decarboxylation reactions are unknown. HemQs are attractive targets for the development of new classes of therapeutics, because many organisms with coding sequences for HemQ are pathogens (e.g., Listeria monocytogenes, Staphylococcus aureus, and Mycobacterium tuberculosis); several have developed antibiotic resistances and are a major threat. The equilibrium established by the organisms of iron uptake, heme biosynthesis, heme degradation, and iron release is very sensitive and essential for their viability.28
To understand the enzymology of HemQ, we need to characterize the interactions of coproheme and heme b with HemQ at the biochemical and biophysical level to serve as a starting point for the elucidation of the reaction mechanism and the identification of intermediate redox species. Here we report the biochemical and biophysical properties of coproheme-HemQs from L. monocytogenes and S. aureus, including (i) spectroscopic characterization, (ii) the kinetics of binding of coproheme and hemin to the apo forms, (iii) the kinetics of binding of cyanide to coproheme-HemQ and heme b-HemQ, and (iv) the redox thermodynamics of the Fe(III)/Fe(II) couple. Moreover, we present molecular dynamics simulation data of a coproheme-HemQ model that allow us to discuss the structural differences among apo-HemQ, coproheme-HemQ, and heme b-HemQ, providing an important basis for the elucidation of the mechanism of conversion of coproheme to heme b.
Materials and Methods
Phylogenetic Analysis
A selection of 120 Cld and HemQ sequences were collected from public databases (Uniprot, NCBI). First, multiple-sequence alignments for Clds and HemQs were constructed using MUSCLE29 with the following parameters: gap penalties, −2,9; gap extension, 0; hydrophobicity multiplier, 1.2; maximum number of iterations, 8. From these sequence alignments, a phylogenetic tree of Cld and HemQ proteins was reconstructed with the maximum likelihood algorithm using the Jones–Taylor–Thornton (JTT) model, with the γ parameter set to 3 and 1000 bootstrap replications; complete deletion was used for gaps and/or missing data treatment. From the original sequences, 20 were selected for presentation in this work, covering subfamily I and subfamily II of the Clds and both subfamilies of confirmed HemQs (Firmicutes and Actinobacteria). All tools for sequence alignments and phylogenetic tree reconstruction were embedded in the MEGA5 package.30 The phylogenetic tree was drawn with FigTree version 1.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Expression and Purification of HemQs
Cloning, expression, and purification of the apo form of HemQ from L. monocytogenes (LmHemQ) were described previously.26 We kindly received the plasmid for HemQ from S. aureus (SaHemQ) from L. M. Saraiva [Universidade Nova de Lisboa (ITQB), Lisbon, Portugal]. Cloning, expression, and purification were described recently.3 Briefly, SaHemQ was cloned into a pET-23b vector, with a C-terminal poly-His tag. Heterologous expression was performed at 16 °C overnight with 180 rpm shaking (after induction at an OD600 of approximately 0.6) in Escherichia coli BL21(DE3)pLysS cells (Merck/Novagen). Purification of the apoprotein was performed by affinity chromatography using a HisTrap column (GE Healthcare). Apo-HemQ was stored at −80 °C and, if needed, reconstituted with equimolar concentrations coproheme (Frontiers Scientific) or hemin (Sigma) to yield holo-HemQ. All protein concentrations of pentameric HemQs refer to subunit concentrations.
Kinetics of Binding of Coproheme to HemQ and Cyanide to Coproheme-HemQ
Time-resolved binding of coproheme to apo-HemQ was monitored using a stopped-flow apparatus equipped with a diode array detector (model SX-18MV, Applied Photophysics), in the conventional mode. The optical quartz cell with a path length of 10 mm had a volume of 20 μL. The fastest mixing time was 1 ms. All measurements were performed at 25 °C. Typically, the concentration of coproheme in the cell was 0.25–1.5 μM, and HemQ was present in excess, to ensure pure spectral species of the coproheme-bound proteins. Experiments were conducted in 50 mM phosphate buffer (pH 7.0). Reactions were simulated, and rates were estimated using Pro-Kineticists software (Applied Photophysics). The apparent second-order rate constants, kon, were obtained from the slope of a plot of kobs versus coproheme concentration.
For the studies of binding of cyanide to ferric coproheme-LmHemQ and coproheme-SaHemQ, the increase in absorbance at 410 nm was monitored. In a typical experiment, one syringe contained 2 μM HemQ in 50 mM buffered solution (pH 5.5–10.0) and the second syringe contained an at least 5-fold excess of cyanide in the same buffer. A minimum of three measurements were performed for each ligand concentration. Additionally, binding of cyanide to coproheme-HemQ was also investigated using the diode array detector (Applied Photophysics), which allowed the synthesis of artificial sets of time-dependent spectra as well as spectral analysis of enzyme intermediates.
Heme Transfer
Transfer of the prosthetic group from HemQ to apo-myoglobin (apo-Mb) was followed photometrically over time. Apo-Mb (horse heart, Sigma) was prepared by a modified extraction method of Teale, as described previously.26,31,32 Experiments were performed using a Specord S10 UV–vis diode array spectrophotometer (Zeiss) in the cyclic measurement mode under constant stirring, which allows time-resolved monitoring over time of the entire UV–vis absorption spectrum (200–800 nm). Typically, 1 μM coproheme- or heme b-bound LmHemQ or SaHemQ was incubated with a 10-fold excess of apo-Mb in 50 mM phosphate buffer (pH 7.0). For comparison, binding of 1 μM coproheme or 1 μM hemin to 10 μM apo-Mb was also tested.
Thermal Stability of Apo- and Holo-HemQ
Differential scanning calorimetry (DSC) measurements were performed using a VP-capillary DSC microcalorimeter from Microcal with a cell volume of 137 μL. The measurements were controlled by the VP-viewer program, and the instrument was equipped with an autosampler for 96-well plates. Samples were analyzed using a programmed heating scan rate of 60 °C h–1 over a temperature range from 20 to 100 °C, and the cell pressure was approximately 60 psi (4.136 bar). DSC thermograms were corrected for buffer baseline and protein concentration. Apo-, heme b-, or coproheme-LmHemQ and SaHemQ (5 μM each) in 50 mM phosphate buffer (pH 7.0) were used for each measurement. For data analysis and conversion, the Microcal origin software was used. Heat capacity (Cp) was expressed in kilocalories per mole per kelvin. Data points were fitted to non-two-state equilibrium unfolding models by the Lavenberg–Marquardt (LM) nonlinear least-squares method.
UV–Visible Electronic Absorption Spectroscopy and Electronic Circular Dichroism Spectroscopy
UV–vis spectroscopic studies were performed at 25 °C between 250 and 700 nm on a Hitachi U-3900 UV–vis spectrophotometer. Temperature was controlled with a water bath connected to the cuvette holder. The path length was 10 mm, and the scan rate was 600 nm min–1. Typically, 6–15 μM enzyme (LmHemQ or SaHemQ) was measured. To obtain the ferrous form, samples were chemically reduced by addition of 10 mM freshly prepared sodium dithionite between pH 5.5 and 10.0 (50 mM phosphate-citrate buffer, 50 mM phosphate buffer, or 50 mM glycine/NaOH buffer).
Electronic circular dichroism spectroscopy (ECD) was performed using Chirascan (Applied Photophysics). The instrument was flushed with a nitrogen flow of 5 L min–1 that allowed the simultaneous monitoring of UV–vis electronic absorption and circular dichroism. Coproheme-HemQ at 10 μM was analyzed in the far-UV region (190–260 nm; 5 s nm–1 scan speed, 3 nm bandwidth, 1 mm path length) and in the near-UV and visible region (260–500 nm; 5 s nm–1 scan speed, 1 nm bandwidth, 10 mm path length). All measurements were performed in 50 mM phosphate buffer (pH 7.0).
Electron Paramagnetic Resonance Spectroscopy (EPR)
EPR was performed on a Bruker EMX continuous wave (cw) spectrometer, operating at X-band (9 GHz) frequencies. The instrument was equipped with a high-sensitivity resonator and an Oxford Instruments ESR900 helium cryostat for low-temperature measurements. Spectra were recorded under nonsaturating conditions using a microwave power of 2 mW, a modulation frequency of 100 kHz, a modulation amplitude of 1 mT, a conversion time of 40 ms, a time constant of 40 ms, and 2048 points. Samples (100 μL of a 50 μM solution) of recombinant LmHemQ and SaHemQ were prepared in 100 mM phosphate buffer (pH 7.0), transferred into Wilmad quartz tubes (3 mm inner diameter), and flash-frozen in liquid nitrogen. Cyanide binding was tested in the presence of 1 mM cyanide and HemQ concentrations of 100 μM. To remove O2, the tubes were flushed with argon while the sample was kept frozen on dry ice. Measurements were taken at 10 K. The spectra were simulated with the Easyspin toolbox for Matlab33 and consist of a weighted sum of simulations of the individual high-spin and low-spin species. The rhombicity was obtained from gxeff and gyeff, and the relative intensities were calculated on the basis of the simulations, following the procedure of Aasa and Vanngard to account for the different integral intensity per unit spin of species that display different effective g values (as found in low-spin and high-spin centers).34,35
Spectroelectrochemistry
All experiments were conducted in a homemade optical transparent thin-layer electrochemical (OTTLE) cell with a path length of 0.05 cm. The three-electrode configuration consisted of a platinum gauze working electrode (Goodfellow Cambridge Ltd.), a platinum wire auxiliary electrode (Goodfellow Cambridge Ltd.), and a RE-6 Ag/AgCl reference electrode (BASi). The reference electrode was calibrated against a saturated calomel electrode (HgCl). All potentials are referenced to the standard hydrogen electrode (SHE, +242 mV). Potentials were applied across the OTTLE cell with a Series G 300 Potentiostat/Galvanostat/ZRA (Gamry). UV–vis spectra were recorded with an Agilent 8453 UV–vis Diode Array System (Agilent Technologies). Spectroelectrochemical experiments were performed in an O2-free environment in a thermostated glovebox (Whitley DG250) with 500 μL samples containing 80 μM HemQ in 100 mM phosphate buffer (pH 7.0) in the presence of various mediators: methylviologen (200 μM), indigocarmin (4 μM), methylene blue (4 μM), and phenazine methosulfate (4 μM). The Nernst plot consisted of at least seven points and was linear with a slope consistent with a one-electron reduction process. Variable-temperature experiments were performed using an isothermal cell configuration.36 Parametrization of the entropic component was possible via calculation of ΔS′° from the slope of the plot of E′° versus temperature. ΔS′°rc(reaction) was approximated using the relationship ΔS′°rc = ΔS′° + 65.2 J mol–1 K–1.37 ΔH°′rc was calculated using the following equation38
Experiments with LmHemQ were performed at 20 °C, and experiments with SaHemQ were conducted over a temperature range from 10 to 25 °C.
Molecular Dynamics Simulations
Molecular dynamics simulations were performed for LmHemQ to study how coproheme fits inside the protein and to determine which residues are most likely involved in the interactions. For the simulations, we have used the GROMOS11 software package with the GROMOS 54A7 force field.39,40 Force-field parameters for the coproheme in the ferric form were derived from the heme b parameters described by Zou et al.41
The pentameric LmHemQ crystal structure26 (PDB entry 4WWS) was used, modeling the missing residues (V5, K6, and N112–D122) of chains B–D by superimposing chain A on them. The coproheme molecule was built by adding two propionate groups to the porphyrin system taken from the crystal structure of the closely related chlorite dismutase (Cld) (PDB entry 3NN1).5 Because of the high degree of similarity of HemQ with Cld, the coprohemes were placed into the putative active sites according to the corresponding binding pose of heme b in Cld, manually moving them slightly closer to H174 to facilitate coordination of iron to the Nε atom of H174. The protein was solvated with SPC water molecules42 in a periodic rectangular box of roughly 9.4 nm × 10.8 nm × 11.3 nm, together with 65 sodium ions to ensure a zero net charge for the whole system at pH 7.
The long-range interactions were calculated employing the reaction-field scheme with a cutoff of 1.4 nm and a relative dielectric constant of 61 (as appropriate for the SPC model for water).43,44 Short-range interactions up to 0.8 nm were calculated for every time step from a pairlist that was determined every five steps. Intermediate-range interactions up to 1.4 nm were computed at pairlist updates and kept constant between the updates.
An equilibration phase was conducted for 20 ps, starting from 60 K, keeping the solute positionally restrained. Four subsequent equilibration steps were performed, increasing the temperature by 60 K in each step and relaxing the positional restraints during heating. In the last equilibration step, the system was propagated for 300 ps. A production run was then performed for 30 ns using a Berendsen thermostat and barostat45 with coupling times of 0.1 and 0.5 ps to keep the temperature and pressure constant at 300 K and 1 atm, respectively. The isothermal compressibility was set to 4.575 × 10–4 kJ–1 mol nm3. Simulations were conducted by employing the SHAKE algorithm46 to constrain bond lengths, allowing an integration time step of 2 fs. Coordinate and energy trajectories were printed out every picosecond.
All analyses were performed using the GROMOS++ software package.47
Results
LmHemQ and SaHemQ were heterologously expressed in E. coli as apoproteins (LmHemQ, ∼30 mg L–1E. coli culture; SaHemQ, ∼50 mg L–1E. coli culture) and purified as described recently.26 HemQs are likely to be present as apoproteins in their physiological resting state, because coproheme acts as both a substrate and a redox cofactor and is released after decarboxylation as protoheme IX.1 Phylogenetic analysis revealed the presence of two HemQ subfamilies, one being formed of proteins from Firmicutes and one including HemQs from Actinobacteria (Figure 1B). From the Firmicutes subfamily, the structures of the apoproteins from LmHemQ (PDB entry 4WWS) and HemQ from Geobacillus stearothermophilus (PDB entry 1T0T) have been published. In the study presented here, we focus on HemQs from Firmicutes, namely from L. monocytogenes (LmHemQ) and S. aureus (SaHemQ).
Kinetics of Binding of Coproheme and Hemin to HemQ
As a first step, the kinetics of binding of coproheme (substrate) and hemin (product) to both apoproteins was investigated by stopped-flow spectroscopy to analyze the kinetics of binding and to identify intermediate spectral species. Because of (i) the high absorption of free coproheme in phosphate-buffered solutions (determined molar extinction coefficient, ε390, of 128800 M–1 cm–1) and (ii) the extremely fast binding rate (see below), it was not possible to determine binding rates when the cofactor was present in stoichiometric excess. Therefore, coproheme had to be added at subequimolar concentrations to apo-HemQ to ensure complete binding of coproheme.
Binding of coproheme occurs in a biphasic manner, with the first transition being very fast and accompanied by a decrease in absorbance at 390 nm [free coproheme (blue spectra in Figure 2A,C)] and a shift of the Soret maximum to 399 nm (green spectra). In the second phase, the Soret absorbance peak became asymmetric, having a shoulder at 375 nm and a Soret maximum at 395 nm (red spectra in Figure 2A,C). Both LmHemQ and SaHemQ follow the same transition. Figure S1 shows the calculated spectra of all colored species in this reaction by using the Pro-K software from Applied Photophysics and the following two-step model: (i) a (coproheme) + b (apo-HemQ; colorless in the Soret region) → c and (ii) c → d, with c representing the intermediate species with a Soret maximum at 399 nm and d representing the final spectrum of coproheme-HemQ with a Soret maximum at 395 nm (ε395 = 68000 M–1 cm–1).
Figure 2.

Kinetics of binding of coproheme to apo-HemQs from L. monocytogenes and S. aureus. (A) Spectral transitions upon binding of 1 μM coproheme to 3 μM apo-LmHemQ. The spectrum of apo-LmHemQ is shown as a dashed black line and that of free coproheme as a dashed cyan line. Spectra of a rapidly formed intermediate (75 ms) and of coproheme-LmHemQ (final form, 10 s) are shown as solid green and red lines, respctively. In the inset, the reaction is dissected into two kinetically separated phases, i.e., rapid formation of intermediate species (spectra recorded 1, 5, 15, 35, 45, 55, 65, and 75 ms after mixing) and slow conversion to coproheme-LmHemQ (spectra recorded 85 ms, 128 ms, 254 ms, 483 ms, 899 ms, 3.4 s, 5.6 s, and 10 s after mixing). (C) Spectral transitions upon binding of 1 μM coproheme to 5 μM apo-SaHemQ. The spectrum of apo-SaHemQ is shown as a dashed black line and that of free coproheme as a dashed cyan line. Spectra of a rapidly formed intermediate (21 ms) and of coproheme-LmHemQ (final form, 5 s) are shown as solid green and red lines, respectively. In the inset, the reaction is dissected into two kinetically separated phases, i.e., rapid formation of intermediate species (spectra recorded 1, 3, 4, 6, 10, and 21 ms after mixing) and slow conversion to coproheme-SaHemQ (spectra recorded 29 ms, 49 ms, 101 ms, 189 ms, 338 ms, 592 ms, 1 s, 1.8 s, 3 s, and 5 s after mixing). Conditions: 50 mM phosphate buffer (pH 7.0). (B and D) Time traces followed at 390 nm (fast phase) and 375 nm (slow phase) derived from stopped-flow experiments (left panels): black lines for experimental time traces and red dashed lines for single-exponential fits of time traces. The corresponding plots of calculated kobs values vs ligand concentration are shown at the right.
The time traces at 390 nm depicted in panels B and D of Figure 2 show the rapid kinetics of formation of the intermediate state. Upon single-exponential fitting, kobs values that depended on the coproheme concentration were obtained. This (although it was not possible to provide pseudo-first-order conditions) allowed us to estimate rates of binding of coproheme to apo-HemQ. Obtained kon rates were ∼1.5 × 108 M–1 s–1 (LmHemQ) and ∼2.0 × 108 M–1 s–1 (SaHemQ). Because of the uncertainty of koff values, because of the very fast kon values and the interplay of two consecutive reactions, it was not possible to reliably calculate KD values from these experiments. The second phase followed as the absorbance increase at 375 nm was ∼2 orders of magnitude slower, namely 2.2 × 106 M–1 s–1 (LmHemQ) and 3.7 × 106 M–1 s–1 (SaHemQ) (Figure 2B,D).
Binding of hemin to apo-LmHemQ or apo-SaHemQ was accompanied by spectral transitions from free hemin (Soret maximum at 385 nm, shoulder at 365 nm; ε385 = 58440 M–1 cm–1)48 to the corresponding heme b-HemQs with a Soret maximum of 410 nm (ε410 = 76500 M–1 cm–1) (Figure 3A,C). Binding of hemin to both apo-HemQs was also biphasic but slower compared to coproheme binding. By following the increase in absorbance at 410 nm, we could obtain kobs values of the rapid phase that depended on hemin concentration. Again because of spectral interference, it was not possible to provide a stoichiometric excess of ligand. From the plot of kobs versus hemin concentration, kon and koff were calculated to be 2.2 × 107 M–1 s–1 and 4.2 s–1 (LmHemQ) and 6.0 × 105 M–1 s–1 and 0.4 s–1 (SaHemQ), respectively. This allowed us to calculate KD values of 5.2 and 1.3 μM, respectively (Figure 3B,D).
Figure 3.

Kinetics of binding of hemin to apo-HemQs from L. monocytogenes and S. aureus. (A) Spectral transitions upon binding of hemin to apo-LmHemQ. (C) Spectral transitions upon binding of hemin to apo-SaHemQ. (B and D) Time traces followed at 410 nm upon binding of hemin (0.25–1 μM) to 5 μM apo-LmHemQ or apo-SaHemQ: black lines for experimental time traces and red dashed lines for single-exponential fits of time traces. The right panels show plots of kobs vs ligand concentration.
Because the physiological role of HemQ is to convert coproheme to heme b and deliver the latter for the synthesis of heme proteins, we tested how easily heme b or coproheme bound to HemQ can be extracted by apo-myoglobin (apo-Mb). Recently, apo-Mb was shown to be able to extract bound heme b from LmHemQ26 and also from NdCld variants with a disrupted proximal H-bonding network (thus mimicking the proximal heme cavity of HemQ).27 Spectra of coproheme- and heme b-bound myoglobin are shown in Figure S2. Figure S3A clearly demonstrates that 10 μM apo-myoglobin added to 1 μM coproheme-SaHemQ (black spectrum) does not mediate the transfer of the prosthetic group. The resulting red spectrum simply reflects that of the mixture of both proteins in their initial states (Figure S3A). This indicates that coproheme is not kinetically labile during the time scale of the experiment. By contrast, mixing 1 μM heme b-SaHemQ [black spectrum with a Soret maximum of 411 nm (Figure S3B)] with 10 μM apo-myoglobin leads to the complete transfer of the prosthetic group and the formation of 1 μM myoglobin with a Soret maximum of 409 nm.
Thermal Stability of Apo-, Heme b-, and Coproheme-HemQs
Because binding of a prosthetic group or cofactor and the mode of noncovalent interactions are typically reflected by the thermal stability of the respective proteins, we analyzed the thermostability of LmHemQ and SaHemQ in their apo, coproheme-bound, and heme b-bound states by differential scanning calorimetry. Stabilities of apo and heme b-bound LmHemQ were reported previously26 and determined again in this study for comparison with those of the coproheme-bound species. While temperature-mediated unfolding of apo and heme b-bound LmHemQ clearly follows a non-two-state transition, coproheme-LmHemQ is more stable and shows one major endothermic transition (Tm = 55 °C). At >60 °C, LmHemQ starts to aggregate (Figure S4).
A similar picture is seen with SaHemQ. Both apo-SaHemQ and heme b-SaHemQ show an almost identical unfolding behavior (Tm = 59 °C), suggesting weak interaction of the prosthetic group with the protein. By contrast, when coproheme binds, the Tm value is increased by 14 °C (Figure 4). The difference in thermal stability between SaHemQ and LmHemQ is difficult to rationalize because of the lack of structural data for SaHemQ. Despite the high degree of similarity at the heme cavity, discrepancies in noncovalent interactions at the interfaces between the subunits of the oligomeric proteins could cause these differences as was also observed with related chlorite dismutases.49
Figure 4.

Thermal stability of apo-SaHemQ, heme b-SaHemQ, and coproheme-SaHemQ followed by differential scanning calorimetry. Thermograms (black lines) and fits of the endotherms (gray lines) of apo-SaHemQ, coproheme-bound SaHemQ, and heme b-SaHemQ. Conditions: 50 mM phosphate buffer (pH 7.0).
Spectroscopic Signatures of Ferric and Ferrous Coproheme-HemQs
Next we tested the impact of pH on the spectral features of ferric and ferrous states of both coproheme-HemQs. UV–vis absorption and resonance Raman spectra of coproheme-SaHemQ at pH 7.4 were reported recently.4 However, the impact of pH as well as spectroscopic data for other HemQs is unknown. Here we studied the spectroscopic signatures of LmHemQ and SaHemQ over a broad pH range using UV–vis absorption, circular dichroism (far-UV, near-UV, and visible), and electron paramagnetic resonance spectroscopies. UV–vis spectra of coproheme-SaHemQ were measured between pH 6.5 and 10.0, because the protein started to precipitate below pH 6.5. The observed spectral signatures of the ferric protein are in agreement with previously published data,4 exhibiting a Soret maximum at 395 nm and a distinct shoulder at 375 nm, the latter being verified by evaluation of the second derivative of the spectrum (Figure 5A and Figure S5). Q-Bands are at 495 and 532 nm, and the charge transfer band is at 630 nm. Between pH 6.5 and 9.0, almost identical spectra were obtained. Ferric coproheme-LmHemQ shows spectral signatures identical to those of coproheme-SaHemQ in the pH range of pH 5.0–9.0 (Figure 5A and Figure S5). However, at pH 10.0, both proteins undergo a transition to a distinct species that has its Soret maximum still at 395 nm but exhibits a blue-shift of the Soret shoulder to 350 nm and of the CT band to 601 nm (LmHemQ) or 605 nm (SaHemQ). The spectra of ferrous coproheme-LmHemQ and coproheme-SaHemQ are very similar, having Soret maxima at 425 nm and a prominent band at 552 nm (Figure 5A, red spectra).
Figure 5.
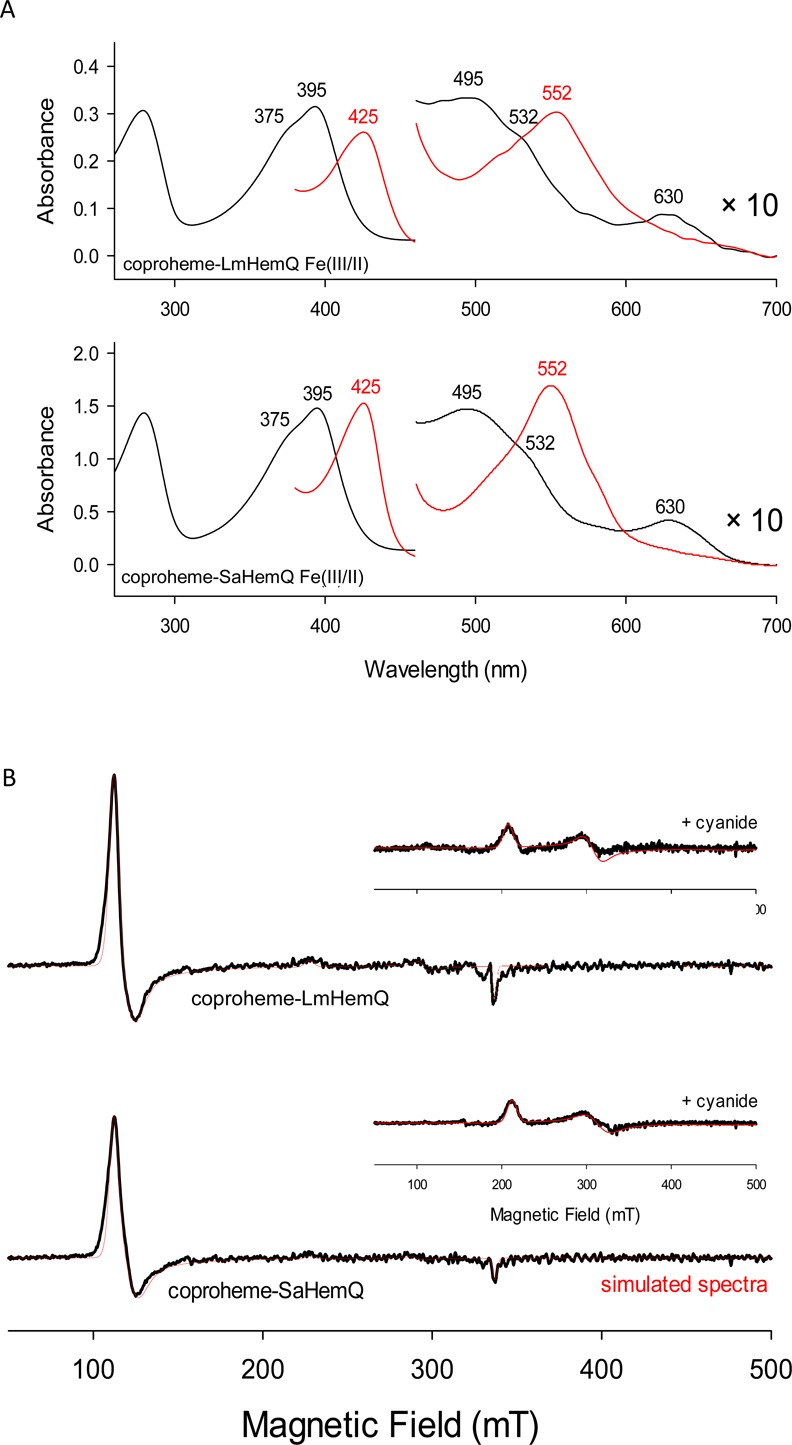
Spectral signatures of ferric and ferrous coproheme-HemQs. (A) UV–vis absorption spectra of LmHemQ and SaHemQ in their ferric (black line) and ferrous (red line) states. Conditions: 50 mM phosphate buffer (pH 7.0). (B) EPR spectra of 50 μM coproheme-LmHemQ and coproheme-SaHemQ. Experimental traces are shown as solid black lines, and simulated spectra (using EasySpin software) are shown as red lines. The inset depicts EPR spectra recorded in the presence of 1 mM cyanide. Conditions: 10 K and 200 mM phosphate buffer (pH 7.0).
Spectra recorded by electron paramagnetic spectroscopy (EPR) demonstrate the existence of a predominant high-spin state of both coproheme-LmHemQ and coproheme-SaHemQ at pH 7.0 (Figure 5B). With both proteins, simulation of the spectra (Table 1) suggests the presence of a dominating slightly rhombic axial high-spin species together with some low-spin form. Very similar high-spin spectra were seen between pH 6.0 and 9.0, whereas at pH 10.0, a pure low-spin form was present (data not shown). Upon addition of 1 mM cyanide, both proteins were completely converted to the corresponding low-spin complexes (insets of Figure 5B).
Table 1. EPR Parameters of HemQs at pH 7.0.
|
g strain |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| protein | species | g1 | g2 | g3 | Ra (%) | I (%) | g1 | g2 | g3 |
| SaHemQ | HS | 5.95 | 5.55 | 1.99 | 2.5 | 87 | 0.30 | 0.60 | 0.02 |
| LS | 2.95 | 2.30 | ≤1 | 13 | 0.07 | 0.05 | 0.08 | ||
| LmHemQ | HS | 5.95 | 5.63 | 1.99 | 2.0 | 87 | 0.30 | 0.60 | 0.01 |
| LS | 2.95 | 2.27 | ≤1 | 13 | 0.08 | 0.05 | 0.08 | ||
| SaHemQ-CN | LS | 3.16 | 2.13 | ≤1 | 100 | 0.20 | 0.20 | 0.20 | |
| LmHemQ-CN | LS | 3.22 | 2.17 | ≤1 | 100 | 0.20 | 0.15 | 0.20 | |
R is the rhombicity of HS signals calculated according to the method of Peisach et al.34 (Δg/16).
Electronic circular dichroism (ECD) spectroscopy of apo-HemQ, coproheme-HemQ, and heme b-HemQ suggests similar secondary structure compositions. The minima at 209 and 221 nm (Figure S6) reflect the presence of α-helices and β-sheets as predicted by examination of the X-ray structures of apo-LmHemQ (PDB entry 4WWS) and HemQ from G. stearothermophilus (PDB entry 1T0T). The observed differences are small, e.g., reflected by ratio of ellipticities at 220 and 209 nm, but followed the same trend in LmHemQ and SaHemQ. Importantly, the spectrum of heme b-SaHemQ (which is highly similar to that of heme b-LmHemQ) is significantly different from that previously reported, where the apoprotein exhibits a minimum at 227 nm and the heme b-bound form at 234 nm, which is unusual and might indicate misfolding.25,50
ECD spectroscopy in the near-UV and visible region revealed interesting distinct spectral features of coproheme-HemQ and heme b-HemQ. Free coproheme is an achiral compound but upon binding to apo-HemQ becomes chiral. Both coproheme-HemQs exhibited a minimum in ellipticity at 388 nm (Figure S6). By contrast, heme b-HemQs showed a maximum in ellipticity at 414–416 nm. The cyanide complexes of both coproheme-HemQs have distinct maxima at 328 and 385 nm and minima at 360 and 421 nm, respectively. The insets of Figure S6 show the corresponding UV–vis spectra for comparison.
Ligand Binding
The binding of cyanide to heme proteins is often used to probe the accessibility of the ligand to the active site as well as the architecture of the distal heme cavity. As demonstrated by spectroscopic means, cyanide is able to enter the active site of copropheme-HemQs and bind to the iron. To probe the kinetics and thermodynamics of this process, kon, koff, and KD (=koff/kon) were determined by stopped-flow spectroscopy. Upon addition of cyanide, both HemQs were directly converted to their low-spin complex (S = 1/2), exhibiting a red-shifted Soret maximum at 410 nm (LmHemQ) and 411 nm (SaHemQ) (isosbestic point of transition at 402 nm) (Figure 6A,D). With both proteins, ligand binding followed at 410 nm was monophasic and kobs values could be obtained from single-exponential fits (Figure 6B,E). The apparent second-order rate constants for cyanide binding (kon) at pH 7.0 were calculated from the slope of the linear plots of kobs versus cyanide concentration (Figure 6C,F), whereas the dissociation rate constant (koff) was determined from the intercepts of those linear plots. Both HemQs exhibited similar kinetics and thermodynamics of cyanide binding with a kon of 1.0 × 104 M–1 s–1, a koff of 0.17 s–1, and a KD (dissociation constant) of 16.4 μM (LmHemQ) and a kon of 9.2 × 103 M–1 s–1, a koff of 0.09 s–1, and a KD of 9.9 μM (SaHemQ). Binding of cyanide to coproheme-LmHemQ and coproheme-SaHemQ did not depend on pH (data not shown), indicating that there is no proton acceptor involved in cyanide (i.e., HCN) binding close to the active site. Around pH 9, there was a slight increase in kon. Note that the pKa of HCN is 9.2.
Figure 6.
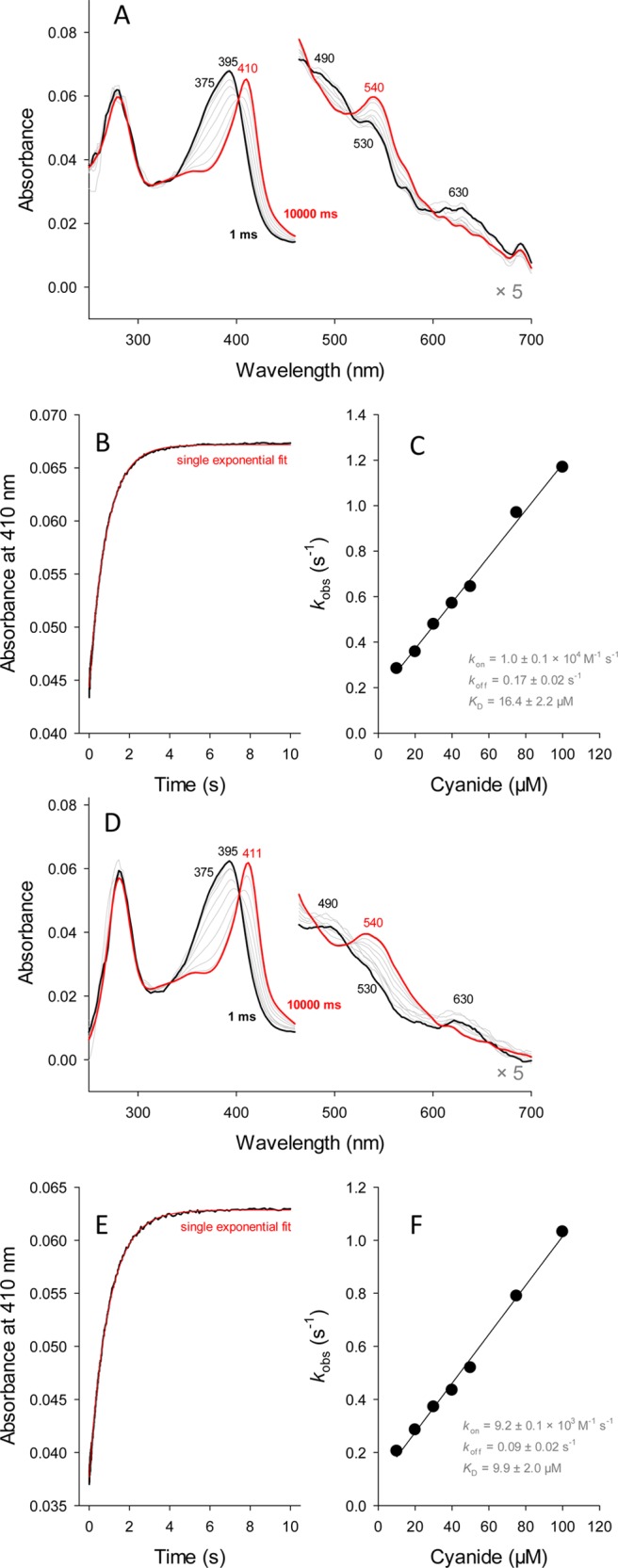
Kinetics and thermodynamics of binding of cyanide to coproheme-HemQs. Spectral changes upon reaction of 1 μM ferric coproheme-LmHemQ (A) and coproheme-SaHemQ (D) with 100 μM cyanide measured in the conventional stopped-flow mode. Typical time traces at 410 nm with a single-exponential fit for LmHemQ (B) and SaHemQ (E). Linear dependence of kobs values from the cyanide concentration for LmHemQ (C) and SaHemQ (F). Conditions: 50 mM phosphate buffer (pH 7.0).
Spectroelectrochemistry
Next we performed temperature-controlled spectroelectrochemical redox titrations between 10 and 25 °C to analyze the thermodynamics of the reduction of ferric coproheme-HemQ to ferrous coproheme-HemQ. At 20 °C, the standard reduction potentials, E°′, of the Fe(III)/Fe(II) couple of coproheme-LmHemQ and coproheme-SaHemQ were determined to be 205 ± 3 and 207 ± 3 mV, respectively, versus the SHE. The redox reactions showed a clear isosbestic point at 405 nm (Figure 7A).
Figure 7.
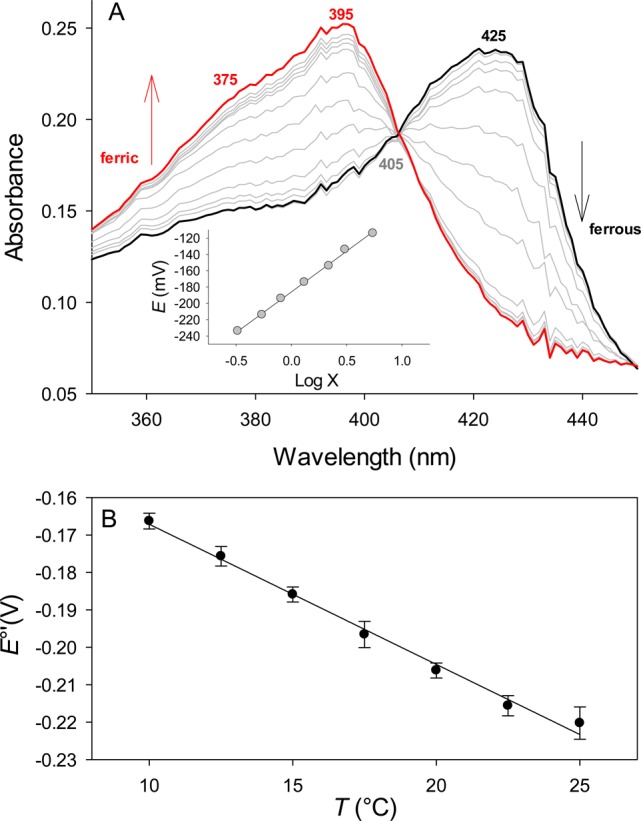
Spectroelectrochemistry and redox thermodynamics of coproheme-SaHemQ. (A) Data for ferrous SaHemQ (at −414 mV vs the SHE) are colored black and data for ferric SaHemQ (at −34 mV vs the SHE) red. Spectra recorded at various potentials between −414 and −34 mV vs the SHE are colored gray. The Nernst plot is shown as an inset, where log X is log[(A°425 – A425)/(A°395 – A395)]. (B) Redox thermodynamics of SaHemQ; temperature dependence of the reduction potential for SaHemQ. The slope of the plot yields ΔS°′/F. Solid lines are least-squares fits to the data points.
To study the mechanism of E°′ modulation, we investigated its temperature dependence. This allows parametrization of the corresponding enthalpic (ΔH°′rc) and entropic (ΔS°′rc) contributions of the Fe(III) to Fe(II) reduction reaction.37 Reduction of coproheme-SaHemQ from Fe(III) to Fe(II) is enthalpically favorable (ΔH°′rc = −67 ± 4 kJ mol–1) but entropically disfavored (ΔS°′rc = −296 ± 14 J mol–1 K–1) (Figure 7B and Table 2). As a consequence, the resulting enthalpic contribution to E°′ at 25 °C (−ΔH°′rc/F = 694 ± 43 mV) partially compensates for the entropic stabilization of the ferric state (TΔS°′rc/F = −915 ± 41 mV). Table 2 compares these thermodynamic data with those of homologous pentameric (NdCld) and dimeric (NwCld) chlorite dismutases.51
Table 2. Standard Reduction Potentials (vs SHE) of Coproheme-HemQs and Chlorite Dismutases.
| E°′ (mV) | ΔS°′ (J mol–1 K–1) | ΔH°′rc (kJ mol–1) | ΔS°′rc (J mol–1 K–1) | –ΔH°′rc/F (mV) | TΔS°′rc/F (mV) | –FE°′ [=ΔH°′rc(int)] (kJ mol–1) | ref | |
|---|---|---|---|---|---|---|---|---|
| SaHemQ | –207 ± 3 | –361 ± 14 | –67 ± 4 | –296 ± 14 | 694 ± 43 | –915 ± 41 | 21.3 ± 0.4 | this study |
| LmHemQ | –205 ± 3 | NDa | NDa | NDa | NDa | NDa | NDa | this study |
| NdCld | –113 ± 5 | 29 ± 6 | 63 ± 20 | –305 ± 60 | 194 ± 60 | 10.9 ± 0.1 | (50) | |
| NwCld | –119 ± 5 | 40 ± 4 | 95 ± 13 | –413 ± 40 | 292 ± 40 | 11.5 ± 0.1 | (50) |
Not determined.
Molecular Dynamics Simulation of Coproheme-LmHemQ
To rationalize the biochemical and biophysical observations described above, we performed MD simulations of apo-HemQ, heme b-HemQ, and coproheme-HemQ. Coproheme was placed in silico at the predicted heme binding site of all five subunits of apo-LmHemQ (PDB entry 4WWS).26 In the starting position, H174 acted as the proximal ligand and the propionates at positions 6 and 7 were positioned as in the heme b enzymes NdCld and NwCld. During the 30 ns simulation, the overall structure remained intact (Figure 8A). In agreement with the DSC experiments, the apo structure seems less stable than the coproheme-HemQ. This is most pronounced for simulations of the isolated monomers, which start to unfold at residues 150–175 in the absence of a substrate, while the coproheme-HemQ monomer remains as stable as the corresponding chain in the pentameric structures (Figure S8). The greatest structural variability was seen in the flexible loop (V107–K141) (Figure 8F) at the entrance of the substrate channel and in the α-helix (M164–Y181) on the proximal heme side that harbors H174 (Figure 8G and Figure S7). H174 coordinates the coproheme for the vast majority of the time, while a water molecule serves as a sixth ligand, as expected for a high-spin complex. Interestingly, R179 of the α-helix (M164–Y181) shows the same structural variability in the simulation of apo-LmHemQ and coproheme-LmHemQ, in contrast to other residues in this helix (Figure 8G). In apo-LmHemQ, R179 is blocking the potential coproheme/substrate binding site, whereas in coproheme-LmHemQ (after a 30 ns simulation), R179 adopts a completely different conformation; therefore, the coproheme/substrate binding site is highly accessible (Figure S9).
Figure 8.

Proposed interactions of coproheme with HemQ based on MD simulations. (A) Overlay of subunit structures of apo-LmHemQ (yellow, PDB entry 4WWS) and coproheme-LmHemQ (gray, chains A–E) from MD simulations (last frames). (B) Hydrogen bonding network of coproheme in LmHemQ (last frame, chain A), in which positions of porphyrin substituents are numbered in white. (C) H-Bonding interaction of atoms of respective amino acids and coproheme propionates (for details, see Table S1) and for heme b propionates. H-Bonds formed (in percent) throughout the simulation time as averages over all five subunits are represented as dark (coproheme) and light (heme b) gray bars (residues H-bonded to propionate at position 7), dark (coproheme) and light (heme b) blue bars (p6), red bars (p4), and green bars (p2). H-Bonding of (D) p2 to coproheme (last frame, chain A) and (E) p4 to coproheme (last frame, chain C). (F) Root-mean-square fluctuations (in angstroms) for all atoms (averages over all five chains) of the flexible loop on the entrance to the coproheme/heme b binding site between residues V107 and K141 in apo-LmHemQ (black) and coproheme-LmHemQ (red) over 30 ns simulations. (G) Root-mean-square fluctuation analysis as in panel F for the α-helix (M164–Y181) that harbors the proximal histidine (H174).
The MD data show that all four propionates interact with the protein (Figure 8B,C). The most prominent H-bonding partners for the propionates at positions 6 (p6) and 7 (p7), which are not modified during the enzymatic reaction, are the side chains of Y113 and K151 for p6 and the side chain of S111 and the backbone of Y113 for p7. Those propionates that are decarboxylated, namely, p2 and p4, interact with G183 and Q187 as well as Y147 and S225, respectively. Analysis of H-bond formation (average over all five subunit chains) over the entire simulation time is depicted in Figure 8C, whereas Table S1 shows all hydrogen bonds throughout the 30 ns simulation over all five subunits.
The propionates p2 and p4 that are cleaved off during catalysis are thus of significant interest. Molecular dynamics simulation suggests that the propionate at position 2 can form H-bonds for more than 5% of the simulation time, with only G183 and Q187 (Figure 8D), whereas p4 builds significant H-bonds with Y147 and S225, respectively (Figure 8E and Table S1). The most prominent noncovalent interaction is formed between the O1 or O2 atom of p4 and the oxygen of Y147 (∼100%), whereas the S225 side chain shows H-bonding to p4 for 41%; it must be mentioned that in SaHemQ S225 (in LmHemQ) aligns with T223 (Figure 1A). Therefore, we can conclude that Y147 is the primary and most important H-bonding partner of p4.
Propionate at position 2 (p2) is the closest to the protein surface and partly solvent-exposed. It can form H-bonds with the G183 main chain and Q187 side chain. The strongest H-bond connections are formed by Q187 (81%) and by the backbone N atom of G183 (61%) (Table S1).
Other data derived from MD simulations of heme b-HemQ show that p6 and p7 interact with the same amino acid residues as in coproheme-HemQ (Figure 8 C). The significance of amino acid residues in coordinating p6 and p7 of heme b and coproheme is similar; only in heme b-HemQ is the interaction of A115 with p7 less important, whereas the H-bond between K151 and p6 is even more dominant.
Discussion
Following the availability of large numbers of microbial genomes and comprehensive evaluation of heme biosynthesis pathways together with biochemical analysis, it became clear that chlorite dismutase-like proteins in Firmicutes and Actinobacteria are essential for heme biosynthesis.1,2 These proteins, finally named HemQs,24 were proposed to oxidatively decarboxylate coproheme (formed by HemH, which inserts ferrous iron into coproporphyrin) into protoheme (heme b).1 This conversion was shown in vitro to follow two consecutive decarboxylation steps that were shown to depend on an excess of hydrogen peroxide or peracetic acid.4In vitro, the reaction occurs with the conversion of p2 to the vinyl group at pyrrole ring A forming the monovinyl, monopropionate deuteroheme isomer as an intermediate (incorrectly named three-propionate harderoheme isomer III in the literature), which can be released from HemQ. In a slower reaction, the latter is decarboxylated, thereby converting p4 to the vinyl substituent at pyrrole ring B.4 It is not known whether these reactions occur in vivo.
So far, there are no Actinobacterial HemQ structures in the PDB, but four homopentameric structures of (initially annotated as chlorite dismutases) HemQs from Firmicutes and Archaea do exist, namely from G. stearothermophilus (1TOT), Thermoplasma acidophilum (3DTZ), Thermus thermophilus (1VDH), and L. monocytogenes (4WWS). None of those structures possess bound heme or coproheme. As a consequence, the position of the substrate coproheme and the product protoheme can be presumed only by analogy to known Clds. It is not known if coproheme binding and/or heme b release involves conformational changes or structural rearrangements like in ferrochelatase.52 This lack of knowledge motivated us to study the mode of binding and interactions of coproheme and heme b with the HemQ from two troublesome pathogens, namely, S. aureus (SaHemQ) and L. monocytogenes (LmHemQ).
We showed that both apo-SaHemQ and apo-LmHemQ behave identically and bind coproheme very rapidly in a biphasic reaction with (i) a spectrally clearly visible intermediate with a Soret maximum at 399 nm and (ii) kinetically well separated phases (∼1.5–2 × 108 M–1 s–1 vs 2.2–3.3 × 106 M–1 s–1). Because of the high binding rate and the spectral interference with the ligand coproheme, it was not possible to determine an exact bimolecular rate constant for the fast phase. However, the findings clearly demonstrate that (i) apo-HemQ efficiently binds coproheme even in the presence of a subequimolar or equimolar ligand concentration and (ii) this suggests that coproheme released by HemH will not accumulate in vivo.
Binding of coproheme to apo-HemQ includes coordination by the proximal histidine (H174) and establishment of many noncovalent interactions, especially between the four propionate side chains and the protein. The far-UV ECD spectra suggest only very small but evident differences in the overall secondary structure composition of apo-HemQ and coproheme-HemQ, which is indeed observed in the MD simulations. Apo-HemQ shows 34.0% α-helical and 26.2% β-sheet conformations, averaged over the simulation and the five chains, while for coproheme-HemQ, these values are 33.6 and 26.3%, respectively, according to the DSSP classification.53 However, the MD simulations clearly demonstrate structural variability in regions that are involved in ligand accessibility and binding. HemQs of Firmicutes (in contrast to Actinobacterial counterparts) have an additional loop at the mouth of the (putative) active site (V107–K141 in LmHemQ) (Figure 8F). The α-helix (M164–Y181) that harbors H174 exhibits significantly increased flexibility in apo-HemQ compared to that in coproheme-HemQ and seems to have a mobile arginine (R179), which possibly has a gating function (Figure 8G and Figure S9). In contrast to the case for Clds, this proximal histidine is not part of an extended H-bonding network that decreases the flexibility of the corresponding helix in Clds. The biphasic behavior of binding of coproheme (but also hemin) to apo-HemQ could be explained by initial rapid binding of the prosthetic group by H174 followed by a slower rearrangement of this α-helix and establishment of noncovalent interactions between the prosthetic group and the protein.
The UV–vis spectrum of ferric coproheme-HemQ exhibits an asymmetric Soret band with a maximum at 395 nm and a prominent shoulder at 375 nm. The CT band at 630 nm together with the EPR spectra clearly suggests the presence of predominating high-spin heme between pH 5 and 9. At higher pH, low-spin coproheme-HemQ dominates, most probably reflecting the alkaline transition and the presence of an OH– ligand at the sixth coordination site. The origin of the asymmetric Soret peak was hypothesized to be partial conversion of coproheme to monovinyl, monopropionate deuteroheme,4 but this reaction would need hydrogen peroxide (which was not present in our experiments) and as a consequence should lead to heterogeneity of the active site structure, which should be reflected by the kinetics of ligand binding. However, the latter was monophasic and very similar to both proteins (kon = 0.9–1.0 × 104 M–1 s–1; KD = 9.9–16.4 μM). On the basis of (i) the symmetric spectrum of the intermediate formed during coproheme binding and (ii) the heme b-HemQ spectra that also lost the shoulder at 375 nm, it is reasonable to assume that the asymmetry of the Soret band in coproheme-HemQ is related to specific interactions of p2 and p4 with the protein that are not yet established in the intermediate species and already absent in heme b-HemQ.
Compared to those of homologous Clds (with heme b at the active site), the cyanide binding rate in coproheme-HemQ is ∼2 orders of magnitude slower, suggesting a more restricted accessibility of the ligand to the heme cavity. This would also hold for the putative substrate hydrogen peroxide. Additionally, the calculated KD values of cyanide complexes in Clds are 3–5 times smaller than those of coproheme-HemQ.51 In structures of cyanide–Cld complexes, the conserved distal arginine binds to ligands like cyanide, azide, and thiocyanate.13,14,54 In HemQs from Firmicutes, this arginine is missing and exchanged with a glutamine, which, because of the chemical nature of its side chain, has a lower capacity to electrostatically attract and bind the negatively charged ions.
Recently, we have demonstrated that both lineages of homologous and heme b-carrying Clds that differ in oligomeric state and subunit architecture exhibit an almost identical standard reduction potential (E°′) of the Fe(III)/Fe(II) couple of −113 to −119 mV at pH 7.0.51 This compares with values of −205 and −207 mV versus the SHE for coproheme-LmHemQ and coproheme-SaHemQ, respectively. This clearly suggests that the native stable oxidation state of coproheme-HemQ is Fe(III), which is important for reaction with hydrogen peroxide. The question of how Fe(II)-coproheme delivered by HemH1,2 is oxidized to Fe(III)-coproheme before incorporation into HemQ remains.
Besides the more negative E°′ value, the mechanism of modulation of E°′ by temperature was completely different in coproheme-HemQ and Clds. In the latter, reduction of the ferric protein to the ferrous state was shown to be entropically favored but enthalpically disfavored (Table 2), whereas the transition of ferric coproheme-SaHemQ to ferrous coproheme-SaHemQ is enthalpically favored but entropically disfavored. Reduction-induced changes in entropy (ΔS°′rc) reflect changes in the conformational degree of freedom of the polypeptide chain(s) and solvent reorganization effects. Negative ΔS°′rc values are often associated with proteins undergoing redox-induced conformational changes, such as adrenodoxin.55 When reduction-induced three-dimensional structural changes are small (ΔS°′rc,int ≈ 0), as observed for many heme proteins, the change in entropy would mainly reflect solvent reorganization (ΔS°′rc,solv).56−59 In these proteins, negative ΔS°′rc,solv values indicate a reduction-induced increase in the level of solvent ordering, in particular within the heme cavity, and are generally observed in systems containing buried metal sites, characterized by a limited solvent accessibility.56−59 Therefore, the obtained negative values of ΔS°′rc (≈ΔS°′rc,solv) in coproheme-SaHemQ might suggest limited solvent accessibility and mobility at the heme cavity. Limited accessibility to the distal heme cavity is also reflected by the significantly slower kinetics of cyanide binding in coproheme-HemQ compared to Clds.
An important contribution to the difference existing between the ΔS°′rc (≈ΔS°′rc,solv) values of coproheme-HemQs and Clds could derive from the two additional carboxylate groups of coproheme, which change the electrostatic charge of the corresponding fully deprotonated ferrous and ferric forms to −4 and −3, respectively. The negative ΔS°′rc,solv values of coproheme-HemQ are consistent with the enhanced electrostatic interaction between the ferrous coproheme (overall charge of −4) and the water molecules in the distal cavity as compared with that of the ferric form (overall charge of −3), leading to an increased level of solvent ordering upon reduction.56−59 Moreover, the two additional carboxylates in coproheme-HemQ could strengthen the H-bond network compared to that of Cld, significantly altering the reduction-induced solvent reorganization within the heme cavity.
Reduction-induced solvent reorganization effects usually induce compensatory enthalpy and entropy changes. The corresponding enthalpic contribution can be factored out from the measured enthalpy change56−59 and allows estimation of the protein-based contribution (ΔH°′rc,int) to ΔG°′rc = −nFE°′ = ΔH°′rc – TΔS°′rc = ΔH°′rc,int + ΔH°′rc,solv – TΔS°′rc,solv. Solvent reorganization effects typically cancel in enthalpy and entropy; therefore, it follows that ΔG°′rc = −nFE°′ = ΔH°′rc,int – TΔS°′rc,int ≈ ΔH°′rc,int. As a consequence, ΔH°′rc,int corresponds to 23.3 kJ/mol, which is more positive compared to that of Clds (Table 2). Primarily, ΔH°′rc,int is determined by metal–ligand binding interactions and the electrostatics among the metal, the protein environment, and the solvent. Whereas Clds have a proximal histidine with a pronounced imidazolate character and a mobile positively charged distal arginine, in coproheme-HemQ the basic character of the proximal histidine is less pronounced and no charged amino acid is found at the distal heme cavity in neighborhood of the heme iron. On the other hand, the higher negative charge of coproheme compared to that of heme b distinguishes coproheme-HemQ from Cld (or heme b-HemQ) and could contribute to the selective enthalpic stabilization of the less charged ferric form (Table 2). Unfortunately, it was not possible to perform spectroelectrochemical studies of heme b-HemQ, because the prosthetic group was partly lost during the redox reactions and the establishment of equilibria.
In any case, during transformation of coproheme to heme b, p2 and p4 are converted to vinyl groups and the H-bonds to G183 and Q187 (p2) as well as Y147 and S225 (p4) are lost. This loss of noncovalent interactions was nicely reflected by the significantly decreased thermal stability of heme b-HemQ compared to that of coproheme-HemQ. As a consequence, heme b binding is significantly weakened and the reaction product can easily be extracted by another protein with a high affinity for heme b. Here we have probed this transfer with apo-myoglobin that is known to bind the prosthetic group strongly with a KD value in the low nanomolar range,60 which is ∼3 orders of magnitude lower than the KD value determined for heme b-HemQ.26
In conclusion, we show that apo-HemQs from Firmicutes rapidly and efficiently bind the substrate and redox cofactor coproheme in a biphasic reaction that results in the formation of a ferric high-spin resting state with a standard reduction potential around −206 mV versus the SHE. The α-helix that carries the proximal ligand histidine seems to play a crucial role in capturing the substrate, which was reflected by the observed differences in its flexibility in the apoprotein and the loaded forms. Binding of coproheme significantly stabilizes the protein and restricts access to the distal heme cavity as suggested by the slower (but monophasic) binding of cyanide and the change in entropy during reduction of Fe(III) to Fe(II). Hydrogen peroxide, which is suggested to initiate the conversion of coproheme to heme b, should also have possibly restricted access to the heme cavity and react with the ferric protein, thereby converting the coproheme protein into a higher oxidation state, e.g., a high-valent iron intermediate. Because of the absence of catalytic amino acids at the distal side, it is not clear how the cleavage of H2O2 is catalyzed (homolytically vs heterolytically?) and how this oxidized enzyme intermediate subsequently mediates the decarboxylation reactions at p2 and p4. The possibility of releasing the intermediate monovinyl, monoproprionate deuteroheme followed by subsequent rebinding to HemQ (to allow decarboxylation at a single active site) is unlikely as MD simulations and heme transfer experiments did not suggest spontaneous release of either the substrate or the product. Celis et al.4 have reported that the reaction needs an amount of peroxide that is significantly greater than the expected amount of 2 equiv and that the conversions were often accompanied by heme bleaching and destruction. In any case, conversion of p2 and p4 to vinyl groups decreases the number of H-bond and polar interactions with the protein and consequently the overall stability of heme b-HemQ. In addition, it increases the flexibility of the proximal heme cavity and the access channel. As a consequence, the reaction product can be released for incorporation into a target protein. Because of the importance of this enzyme for the survival of Firmicutes and Actinobacteria,2 it is reasonable to assume that the study of this protein will rapidly expand.
Glossary
Abbreviations
- Cld
chlorite dismutase
- coproheme
coproporphyrin III
- DSC
differential scanning calorimetry
- DyP
dye-decolorizing peroxidase
- ECD
electronic circular dichroism
- EPR
electron paramagnetic resonance
- LmHemQ
HemQ from L. monocytogenes
- NdCld
chlorite dismutase from “Candidatus Nitrospira defluvii”
- NwCld
chlorite dismutase from Nitrobacter winogradskyi
- OTTLE
optical transparent thin-layer electrochemical
- p2
propionate in coproheme at position 2
- p4
propionate in coproheme at position 4
- p6
propionate in coproheme and heme b at position 6
- p7
propionate in coproheme and heme b at position 7
- SaHemQ
HemQ from S. aureus
- rc
reaction
- ΔH°′rc
change in reaction enthalpy under standard conditions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.6b00701.
Data from simulation of the kinetics of binding of coproheme to apo-HemQ (Figure S1), binding of coproheme and heme b to apo-myoglobin (Figure S2), transfer of heme to apo-myoglobin (Figure S3), thermal stability of LmHemQ (Figure S4), pH dependence of UV–vis absorption spectra of LmHemQ and SaHemQ (Figure S5), circular dichroism spectra of LmHemQ and SaHemQ (Figure S6), root-mean-square fluctuations of the protein backbones of HemQ (Figure S7), root-mean-square deviation and fluctuation of the protein backbone (Figure S8), accessibility to the coproheme/substrate binding site in apo- and coproheme-LmHemQ (Figure S9), and detailed listing of H-bonds of coproheme propionates to the protein moiety derived from MD simulations (Table S1) (PDF)
This project was supported by the Austrian Science Fund, FWF [Doctoral Program BioToP-Molecular Technology of Proteins (W1224) and Projects P25270, P29099, and I2429] and the Federal Ministry of Economy, Family and Youth through the initiative “Laura Bassi Centres of Expertise”, funding the Center of Optimized Structural Studies (N° 253275).
The authors declare no competing financial interest.
Supplementary Material
References
- Dailey H. A.; Gerdes S.; Dailey T. A.; Burch J. S.; Phillips J. D. (2015) Noncanonical coproporphyrin-dependent bacterial heme biosynthesis pathway that does not use protoporphyrin. Proc. Natl. Acad. Sci. U. S. A. 112, 2210–2215. 10.1073/pnas.1416285112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailey H. A.; Gerdes S. (2015) HemQ: An iron-coproporphyrin oxidative decarboxylase for protoheme synthesis in Firmicutes and Actinobacteria. Arch. Biochem. Biophys. 574, 27–35. 10.1016/j.abb.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo S. A.; Scott A.; Videira M. A.; Winpenny D.; Gardner M.; Palmer M. J.; Schroeder S.; Lawrence A. D.; Parkinson T.; Warren M. J.; Saraiva L. M. (2015) Staphylococcus aureus haem biosynthesis: characterisation of the enzymes involved in final steps of the pathway. Mol. Microbiol. 97, 472–487. 10.1111/mmi.13041. [DOI] [PubMed] [Google Scholar]
- Celis A. I.; Streit B. R.; Moraski G. C.; Kant R.; Lash T. D.; Lukat-Rodgers G. S.; Rodgers K. R.; DuBois J. L. (2015) Unusual Peroxide-Dependent, Heme-Transforming Reaction Catalyzed by HemQ. Biochemistry 54, 4022–4032. 10.1021/acs.biochem.5b00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostan J.; Sjöblom B.; Maixner F.; Mlynek G.; Furtmüller P. G.; Obinger C.; Wagner M.; Daims H.; Djinović-Carugo K. (2010) Structural and functional characterisation of the chlorite dismutase from the nitrite-oxidizing bacterium ″Candidatus Nitrospira defluvii″: identification of a catalytically important amino acid residue. J. Struct. Biol. 172, 331–342. 10.1016/j.jsb.2010.06.014. [DOI] [PubMed] [Google Scholar]
- Goblirsch B.; Kurker R. C.; Streit B. R.; Wilmot C. M.; DuBois J. L. (2011) Chlorite dismutases, DyPs, and EfeB: 3 microbial heme enzyme families comprise the CDE structural superfamily. J. Mol. Biol. 408, 379–398. 10.1016/j.jmb.2011.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zámocký M.; Hofbauer S.; Schaffner I.; Gasselhuber B.; Nicolussi A.; Soudi M.; Pirker K. F.; Furtmüller P. G.; Obinger C. (2015) Independent evolution of four heme peroxidase superfamilies. Arch. Biochem. Biophys. 574, 108–119. 10.1016/j.abb.2014.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ginkel C. G.; Rikken G. B.; Kroon A. G.; Kengen S. W. (1996) Purification and characterization of chlorite dismutase: a novel oxygen-generating enzyme. Arch. Microbiol. 166, 321–326. 10.1007/s002030050390. [DOI] [PubMed] [Google Scholar]
- Lee A. Q.; Streit B. R.; Zdilla M. J.; Abu-Omar M. M.; DuBois J. L. (2008) Mechanism of and exquisite selectivity for O-O bond formation by the heme-dependent chlorite dismutase. Proc. Natl. Acad. Sci. U. S. A. 105, 15654–15659. 10.1073/pnas.0804279105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugano Y. (2009) DyP-type peroxidases comprise a novel heme peroxidase family. Cell. Mol. Life Sci. 66, 1387–1403. 10.1007/s00018-008-8651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T.; Sugano Y. (2015) A structural and functional perspective of DyP-type peroxidase family. Arch. Biochem. Biophys. 574, 49–55. 10.1016/j.abb.2015.01.022. [DOI] [PubMed] [Google Scholar]
- Mlynek G.; Sjöblom B.; Kostan J.; Füreder S.; Maixner F.; Gysel K.; Furtmüller P. G.; Obinger C.; Wagner M.; Daims H.; Djinović-Carugo K. (2011) Unexpected diversity of chlorite dismutases: a catalytically efficient dimeric enzyme from Nitrobacter winogradskyi. J. Bacteriol. 193, 2408–2417. 10.1128/JB.01262-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goblirsch B. R.; Streit B. R.; Dubois J. L.; Wilmot C. M. (2010) Structural features promoting dioxygen production by Dechloromonas aromatica chlorite dismutase. J. Biol. Inorg. Chem. 15, 879–888. 10.1007/s00775-010-0651-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Geus D. C.; Thomassen E. A.; Hagedoorn P. L.; Pannu N. S.; van Duijn E.; Abrahams J. P. (2009) Crystal structure of chlorite dismutase, a detoxifying enzyme producing molecular oxygen. J. Mol. Biol. 387, 192–206. 10.1016/j.jmb.2009.01.036. [DOI] [PubMed] [Google Scholar]
- Yoshida T.; Tsuge H.; Konno H.; Hisabori T.; Sugano Y. (2011) The catalytic mechanism of dye-decolorizing peroxidase DyP may require the swinging movement of an aspartic acid residue. FEBS J. 278, 2387–2394. 10.1111/j.1742-4658.2011.08161.x. [DOI] [PubMed] [Google Scholar]
- Linde D.; Pogni R.; Canellas M.; Lucas F.; Guallar V.; Baratto M. C.; Sinicropi A.; Sáez-Jiménez V.; Coscolin C.; Romero A.; Medrano F. J.; Ruiz-Dueñas F. J.; Martinez A. T. (2015) Catalytic surface radical in dye-decolorizing peroxidase: A computational, spectroscopic and site-directed mutagenesis study. Biochem. J. 466, 253–262. 10.1042/BJ20141211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc B.; Mayfield J. A.; McDonald C. A.; Lukat-Rodgers G. S.; Rodgers K. R.; DuBois J. L. (2012) Understanding how the distal environment directs reactivity in chlorite dismutase: spectroscopy and reactivity of Arg183 mutants. Biochemistry 51, 1895–1910. 10.1021/bi2017377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer S.; Gruber C.; Pirker K. F.; Sündermann A.; Schaffner I.; Jakopitsch C.; Oostenbrink C.; Furtmüller P. G.; Obinger C. (2014) Transiently produced hypochlorite is responsible for the irreversible inhibition of chlorite dismutase. Biochemistry 53, 3145–3157. 10.1021/bi500401k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffner I.; Hofbauer S.; Krutzler M.; Pirker K. F.; Furtmüller P. G.; Obinger C. (2015) Mechanism of chlorite degradation to chloride and dioxygen by the enzyme chlorite dismutase. Arch. Biochem. Biophys. 574, 18–26. 10.1016/j.abb.2015.02.031. [DOI] [PubMed] [Google Scholar]
- Singh R.; Grigg J. C.; Armstrong Z.; Murphy M. E.; Eltis L. D. (2012) Distal heme pocket residues of B-type dye-decolorizing peroxidase: arginine but not aspartate is essential for peroxidase activity. J. Biol. Chem. 287, 10623–10630. 10.1074/jbc.M111.332171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugano Y.; Muramatsu R.; Ichiyanagi A.; Sato T.; Shoda M. (2007) DyP, a unique dye-decolorizing peroxidase, represents a novel heme peroxidase family: ASP171 replaces the distal histidine of classical peroxidases. J. Biol. Chem. 282, 36652–36658. 10.1074/jbc.M706996200. [DOI] [PubMed] [Google Scholar]
- Strittmatter E.; Liers C.; Ullrich R.; Wachter S.; Hofrichter M.; Plattner D. A.; Piontek K. (2013) First crystal structure of a fungal high-redox potential dye-decolorizing peroxidase: substrate interaction sites and long-range electron transfer. J. Biol. Chem. 288, 4095–4102. 10.1074/jbc.M112.400176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.; Eltis L. D. (2015) The multihued palette of dye-decolorizing peroxidases. Arch. Biochem. Biophys. 574, 56–65. 10.1016/j.abb.2015.01.014. [DOI] [PubMed] [Google Scholar]
- Dailey T. A.; Boynton T. O.; Albetel A. N.; Gerdes S.; Johnson M. K.; Dailey H. A. (2010) Discovery and Characterization of HemQ: an essential heme biosynthetic pathway component. J. Biol. Chem. 285, 25978–25986. 10.1074/jbc.M110.142604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield J. A.; Hammer N. D.; Kurker R. C.; Chen T. K.; Ojha S.; Skaar E. P.; DuBois J. L. (2013) The chlorite dismutase (HemQ) from Staphylococcus aureus has a redox-sensitive heme and is associated with the small colony variant phenotype. J. Biol. Chem. 288, 23488–23504. 10.1074/jbc.M112.442335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer S.; Hagmüller A.; Schaffner I.; Mlynek G.; Krutzler M.; Stadlmayr G.; Pirker K. F.; Obinger C.; Daims H.; Djinović-Carugo K.; Furtmüller P. G. (2015) Structure and heme-binding properties of HemQ (chlorite dismutase-like protein) from Listeria monocytogenes. Arch. Biochem. Biophys. 574, 36–48. 10.1016/j.abb.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer S.; Howes B. D.; Flego N.; Pirker K. F.; Schaffner I.; Mlynek G.; Djinovic-Carugo K.; Furtmüller P. G.; Smulevich G.; Obinger C. (2016) From chlorite dismutase towards HemQ-the role of the proximal H-bonding network in haeme binding. Biosci. Rep. 36, e00312. 10.1042/BSR20150330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon J. R.; Heinrichs D. E. (2015) Recent developments in understanding the iron acquisition strategies of gram positive pathogens. FEMS Microbiol Rev. 39, 592–630. 10.1093/femsre/fuv009. [DOI] [PubMed] [Google Scholar]
- Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K.; Peterson D.; Peterson N.; Stecher G.; Nei M.; Kumar S. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teale F. W. (1959) Cleavage of the haem-protein link by acid methylethylketone. Biochim. Biophys. Acta 35, 543. 10.1016/0006-3002(59)90407-X. [DOI] [PubMed] [Google Scholar]
- Le P.; Zhao J.; Franzen S. (2014) Correlation of heme binding affinity and enzyme kinetics of dehaloperoxidase. Biochemistry 53, 6863–6877. 10.1021/bi5005975. [DOI] [PubMed] [Google Scholar]
- Stoll S.; Schweiger A. (2006) EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178, 42–55. 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Peisach J.; Blumberg W. E.; Ogawa S.; Rachmilewitz E. A.; Oltzik R. (1971) The effects of protein conformation on the heme symmetry in high spin ferric heme proteins as studied by electron paramagnetic resonance. J. Biol. Chem. 246, 3342–3355. [PubMed] [Google Scholar]
- Aasa R.; Vänngard T. (1975) EPR signal intensity and powder shapes. Reexamination. J. Magn. Reson. 19, 308–315. 10.1016/0022-2364(75)90045-1. [DOI] [Google Scholar]
- Battistuzzi G.; Borsari M.; Ranieri A.; Sola M. (2002) Redox thermodynamics of the Fe(3+)/Fe(2+) couple in horseradish peroxidase and its cyanide complex. J. Am. Chem. Soc. 124, 26–27. 10.1021/ja017188m. [DOI] [PubMed] [Google Scholar]
- Battistuzzi G.; D’Onofrio M.; Borsari M.; Sola M.; Macedo A. L.; Moura J. J.; Rodrigues P. (2000) Redox thermodynamics of low-potential iron-sulfur proteins. J. Biol. Inorg. Chem. 5, 748–760. 10.1007/s007750000164. [DOI] [PubMed] [Google Scholar]
- Varadarajan R.; Zewert T. E.; Gray H. B.; Boxer S. G. (1989) Effects of buried ionizable amino acids on the reduction potential of recombinant myoglobin. Science 243, 69–72. 10.1126/science.2563171. [DOI] [PubMed] [Google Scholar]
- Schmid N.; Eichenberger A. P.; Choutko A.; Riniker S.; Winger M.; Mark A. E.; van Gunsteren W. F. (2011) Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 40, 843–856. 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- Schmid N.; Christ C.; Christen M.; Eichenberger A.; van Gunsteren W. (2012) Architecture, implementation and parallelisation of the GROMOS software for biomolecular simulation. Comput. Phys. Commun. 183, 890–903. 10.1016/j.cpc.2011.12.014. [DOI] [Google Scholar]
- Zou C.; Larisika M.; Nagy G.; Srajer J.; Oostenbrink C.; Chen X.; Knoll W.; Liedberg B.; Nowak C. (2013) Two-Dimensional Heterospectral Correlation Analysis of the Redox-Induced Conformational Transition in Cytochrome c Using Surface-Enhanced Raman and Infrared Absorption Spectroscopies on a Two-Layer Gold Surface. J. Phys. Chem. B 117, 9606–9614. 10.1021/jp404573q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., and Hermans J. (1981) Interaction models for water in relation to protein hydration, Reidel, Dordrecht, The Netherlands. [Google Scholar]
- Tironi I. G.; Sperb R.; Smith P. E.; van Gunsteren W. F. (1995) A generalized reaction field method for molecular-dynamics simulations. J. Chem. Phys. 102, 5451–5459. 10.1063/1.469273. [DOI] [Google Scholar]
- Heinz T. N.; van Gunsteren W. F.; Hünenberger P. H. (2001) Comparison of four methods to compute the dielectric permittivity of liquids from molecular dynamics simulations. J. Chem. Phys. 115, 1125–1136. 10.1063/1.1379764. [DOI] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. (1984) Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. C. (1977) Numerical integration of cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Eichenberger A. P.; Allison J. R.; Dolenc J.; Geerke D. P.; Horta B. A.; Meier K.; Oostenbrink C.; Schmid N.; Steiner D.; Wang D.; van Gunsteren W. F. (2011) GROMOS++ Software for the Analysis of Biomolecular Simulation Trajectories. J. Chem. Theory Comput. 7, 3379–3390. 10.1021/ct2003622. [DOI] [PubMed] [Google Scholar]
- Deniau C.; Gilli R.; Izadi-Pruneyre N.; Létoffé S.; Delepierre M.; Wandersman C.; Briand C.; Lecroisey A. (2003) Thermodynamics of heme binding to the HasA(SM) hemophore: effect of mutations at three key residues for heme uptake. Biochemistry 42, 10627–10633. 10.1021/bi030015k. [DOI] [PubMed] [Google Scholar]
- Hofbauer S.; Gysel K.; Mlynek G.; Kostan J.; Hagmüller A.; Daims H.; Furtmüller P. G.; Djinović-Carugo K.; Obinger C. (2012) Impact of subunit and oligomeric structure on the thermal and conformational stability of chlorite dismutases. Biochim. Biophys. Acta, Proteins Proteomics 1824, 1031–1038. 10.1016/j.bbapap.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly S. M.; Jess T. J.; Price N. C. (2005) How to study proteins by circular dichroism. Biochim. Biophys. Acta, Proteins Proteomics 1751, 119–139. 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Hofbauer S.; Bellei M.; Sündermann A.; Pirker K. F.; Hagmüller A.; Mlynek G.; Kostan J.; Daims H.; Furtmüller P. G.; Djinovic-Carugo K.; Oostenbrink C.; Battistuzzi G.; Obinger C. (2012) Redox Thermodynamics of High-Spin and Low-Spin Forms of Chlorite Dismutases with Diverse Subunit and Oligomeric Structures. Biochemistry 51, 9501–9512. 10.1021/bi3013033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailey H. A.; Wu C. K.; Horanyi P.; Medlock A. E.; Najahi-Missaoui W.; Burden A. E.; Dailey T. A.; Rose J. (2007) Altered orientation of active site residues in variants of human ferrochelatase. Evidence for a hydrogen bond network involved in catalysis. Biochemistry 46, 7973–7979. 10.1021/bi700151f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W.; Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637. 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- De Schutter A.; Correia H. D.; Freire D. M.; Rivas M. G.; Rizzi A.; Santos-Silva T.; González P. J.; Van Doorslaer S. (2015) Ligand Binding to Chlorite Dismutase from Magnetospirillum sp. J. Phys. Chem. B 119, 13859–13869. 10.1021/acs.jpcb.5b04141. [DOI] [PubMed] [Google Scholar]
- Lambeth J. D.; Kamin H. (1979) Adrenodoxin reductase.adrenodoxin complex. Flavin to iron-sulfur electron transfer as the rate-limiting step in the NADPH-cytochrome c reductase reaction. J. Biol. Chem. 254, 2766–2774. [PubMed] [Google Scholar]
- Battistuzzi G.; Bellei M.; Bortolotti C. A.; Sola M. (2010) Redox properties of heme peroxidases. Arch. Biochem. Biophys. 500, 21–36. 10.1016/j.abb.2010.03.002. [DOI] [PubMed] [Google Scholar]
- Bellei M.; Jakopitsch C.; Battistuzzi G.; Sola M.; Obinger C. (2006) Redox thermodynamics of the ferric-ferrous couple of wild-type synechocystis KatG and KatG(Y249F). Biochemistry 45, 4768–4774. 10.1021/bi0517943. [DOI] [PubMed] [Google Scholar]
- Battistuzzi G.; Bellei M.; Zederbauer M.; Furtmüller P. G.; Sola M.; Obinger C. (2006) Redox thermodynamics of the Fe(III)/Fe(II) couple of human myeloperoxidase in its high-spin and low-spin forms. Biochemistry 45, 12750–12755. 10.1021/bi061647k. [DOI] [PubMed] [Google Scholar]
- Battistuzzi G.; Bellei M.; Borsari M.; Di Rocco G.; Ranieri A.; Sola M. (2005) Axial ligation and polypeptide matrix effects on the reduction potential of heme proteins probed on their cyanide adducts. J. Biol. Inorg. Chem. 10, 643–651. 10.1007/s00775-005-0014-4. [DOI] [PubMed] [Google Scholar]
- Gryczynski Z.; Lubkowski J.; Bucci E. (1995) Heme-protein interactions in horse heart myoglobin at neutral pH and exposed to acid investigated by time-resolved fluorescence in the pico- to nanosecond time range. J. Biol. Chem. 270, 19232–19237. 10.1074/jbc.270.33.19232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.