

Abstract
Lung cancer is a heterogeneous disease consisting of multiple histological subtypes each driven by unique genetic alterations. Despite the development of targeted therapies that inhibit the oncogenic mutations driving a subset of lung cancer cases, there is a paucity of effective treatments for the majority of lung cancer patients and new strategies are urgently needed. In recent years, the concept of synthetic lethality has been established as an effective approach for discovering novel cancer-specific targets as well as a method to improve the efficacy of existing drugs which provide partial but insufficient benefits for patients. In this review, we discuss the concept of synthetic lethality, the various types of synthetic lethal interactions in the context of oncology and the approaches used to identify these interactions, including recent advances that have transformed the ability to discover novel synthetic lethal combinations on a global scale. Lastly, we describe the specific synthetic lethal interactions identified in lung cancer to date and explore the pharmacological challenges and considerations in translating these discoveries to the clinic.
Keywords: Lung cancer, Synthetic lethality, Combination treatments, Synergy, Drug-drug interactions
Background: lung cancer – a need for new treatment strategies
Lung cancer is the leading cause of cancer mortality worldwide suffering from a late stage of disease at the time of diagnosis and a lack of effective therapeutic strategies available to treat lung tumours [1]. Lung cancer is comprised of two main subtypes: Small Cell Lung Cancer (SCLC) and Non-Small Cell Lung Cancer (NSCLC), which correspond to ~20 % and ~80 % of cases, respectively [2]. Lung adenocarcinoma (LAC) is the most common type of NSCLC, responsible for ~40 % of all lung cancer cases and, unlike other subtypes, is associated with both smokers and never smokers [2, 3]. Squamous Cell Carcinoma (SqCC) is the other major NSCLC subtype and, along with SCLC, is characterized by its development in the central airways and close association with smoking [2]. The different lung cancer subtypes develop from unique cells of origin, involve the deregulation of specific oncogenic pathways and have diverse responses to conventional chemotherapies, demonstrating the importance of considering histology in the clinical management of this disease [4].
Recently, large-scale genomics studies have revealed the genetic changes driving the development of lung cancer subtypes. Activating mutations in EGFR and KRAS as well as translocations involving ALK and RET are common in LAC while SqCC contain frequent mutations in PIK3CA and amplification of FGFR1 [5]. Meanwhile, SCLCs are characterized by the dual inactivation of the tumour suppressor genes RB and TP53 and, less frequently PTEN [6]. With this increasing understanding of lung cancer biology has come the advent of targeted therapies to combat this devastating disease. These therapies target mutated components of key cellular pathways on which tumours cells have become dependent on for survival, a phenomena known as oncogene addiction [7]. For example, tyrosine kinase inhibitors (TKIs) targeting LACs driven by mutant EGFR or ALK rearrangements have been clinically successful, highlighting the potential of designing drugs to specifically target the molecular mechanisms driving cancer development, a concept often described as “personalized medicine” [7–9]. However, despite these encouraging developments, significant problems remain. First, the majority of LAC patients are not candidates for these therapies as they have tumours without mutations in targetable genes, owing either to the lack of an identified driver or mutation in drivers such as mutant KRAS for which the development of inhibitors have proven elusive. Second, all patients eventually develop resistance to treatment with these targeted agents, either through secondary mutation of the target gene or downstream activation of their signalling pathways that sustain tumour growth. Furthermore, although targeted therapies have been successfully employed in the treatment of LAC, advances have lagged in SCLC and SqCC. In SCLC, the causative genetic changes involve inactivation of tumour suppressor genes - which are notoriously difficult to exploit therapeutically - while in SqCC, FGFR1 inhibitors have demonstrated mixed success, likely due to additional genetic determinants regulating response or the presence of alterative oncogene targets attributed to the amplified chromosome region [10]. In addition to the traditional targeted therapies described above, the recent efficacy and approval of inhibitors targeting the PD-1 (Programmed T cell death 1)/PD-L1 immune checkpoint in subsets of NSCLC patients has highlighted the promise of using immunotherapeutics for lung cancer treatment. However, as with kinase inhibitors, many patients do not respond to these treatments and those that do often develop resistance and efforts are already being made to understand the mechanisms regulating sustained response [11]. Thus, while undoubtedly a major advancement in improving lung cancer patient outcomes, current therapeutic approaches have failed to achieve the major goal of increasing long-term survival rates and new strategies to treat lung cancers –perhaps combining kinase inhibitors and immunotherapies - are urgently needed.
Main text
The concept of synthetic lethality and approaches for uncovering interactions in cancer cells
Synthetic lethality is traditionally defined as a condition where simultaneous mutation in two genes – but not either alone - leads to cell death [12, 13]. Where mutation in both genes impairs cellular fitness but does not cause lethality, this is described as a synthetic sick interaction [12–14]. Calvin Bridges first described synthetic lethality in 1922 when he observed that combinations of mutations in fruit flies lead to lethality (Fig. 1) [14–16]. The term itself was later coined by Theodore Dobzhansky who made the same observations in 1946 [17]. For decades, synthetic lethal interactions were studied mainly in fruit flies; however, starting in the 1980s, the search for synthetic lethal interactions expanded to other model systems including algae, yeast, and the nematode C. elegans [18–21]. These studies contributed significantly to our understanding of gene function, biological pathways, and genetic robustness, and identified many interactions potentially important in human cancer.
Fig. 1.
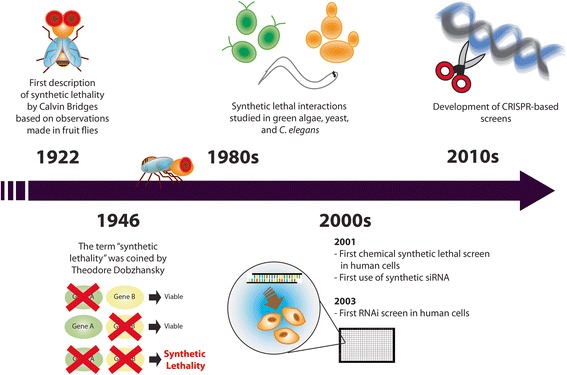
Synthetic Lethality: History and Evolution. The timeline indicates the major events that took place over the last century, from the first description of synthetic lethality to the recent development of technologies for high-throughput discoveries of synthetic lethal interactions
The initial concept of screening for drugs that can specifically kill cancer cells carrying defined genetic changes was originally conceived using yeast as a model system. This basic approach combined with improvements in screening platforms subsequently allowed chemical compound libraries to be assessed in human cancer cell lines, the first foray into exploring synthetic lethal targets for cancer causing genetic alterations [22]. However, chemical libraries suffer from difficulties in target identification, especially for larger libraries of diverse compounds, limiting their effectiveness in defining new synthetic lethal interactions on a global scale. Hypothesis-based assessment can alleviate these problems, as demonstrated by the successful validation of BRCA-deficiency and PARP inhibitor sensitivity in breast cancer [23]. However, the advent and rapid development of RNAi technology in the early 2000s [18, 19] allowed the first high-throughput genetic screens to be performed in human cancer cells driven by specific oncogenic mutations [24–26]. Consisting of well-based screens using transfection of individual siRNAs or pooled drop-out/enrichment screens employing transduction of lentiviral shRNA libraries, this approach has proven invaluable for identifying synthetic lethal interactions for various oncogenes/tumour suppressor genes as well as chemosensitizing genes in lung and other cancers. Recently, major advancements in the generation of RNAi libraries, sequencing, high throughput screening platforms, and the recent development of CRISPR technology have further expanded the capacity to uncover synthetic lethal interactions in cancer [27–30]. In addition, contemporary screens are now relying more on computational and bioinformatics approaches such as statistically inferring synthetic lethal interaction pairs from cancer genomic data [119]. These synthetic lethal screening approaches have been exhaustively reviewed recently [31–33].
While synthetic lethality can occur at the cellular or organismal level depending on the model being used, the goal in cancer treatment is to specifically eliminate a population of malignant cells, which is heterogeneous in nature [34]. With that perspective, drug combinations are designed with the ultimate goal of curing the disease, but more frequently, significant improvements in treatment outcomes are achieved through synergy, which we argue is equivalent to a synthetic sick effect at the cell population level in the context of cancer biology. Hence, the concept of synthetic lethality encompasses a wide range of interactions involving genetic variations as a result of single-gene mutations, chromosomal translocation and deletions, as well as cellular responses to different cytotoxic and targeting pharmaceuticals. In this review, our discussion begins with the various types of synthetic lethal interactions uncovered in the context of lung cancer. We then discuss consideration for the application and translation of these findings to treating lung cancer patients, an area that has proven challenging for the development of therapeutics based on synthetic lethal interactions to date and yet to be directly addressed in the literature.
Types of synthetic lethality and identified interactions in lung cancer
The transformation of normal cells to cancer cells involves a step-wise evolution: a progressive series of genetic mutations that allow cells to acquire the hallmarks of cancer over time and become malignant [35]. These genetic changes cause deficiencies in, or addiction to, certain cellular processes and biological pathways that initiate transformation and are thus, prime targets for therapeutic intervention. However, cancer cells often develop resistance to broad spectrum and targeted treatments. This may first arise through cytoprotective responses and the presence of redundant (“back-up”) proteins and pathways. However, resistance will eventually arise through selection of clones that exhibit at least one mechanism required for inhibition of the drug’s action. This genetic robustness is another factor that can be targeted when using synthetic lethal strategies [13]. Here, we describe the different types of synthetic lethal interactions in two broad categories: 1) interactions that are purely based on genetic mutations and, 2) interactions that involve existing cytotoxic agents that are known to be effective in cancer patients (Fig. 2). Furthermore, we highlight specific examples of synthetic lethality that have been identified to date in lung cancer. We believe these will provide new strategies for therapeutic intervention (Table 1).
Fig. 2.
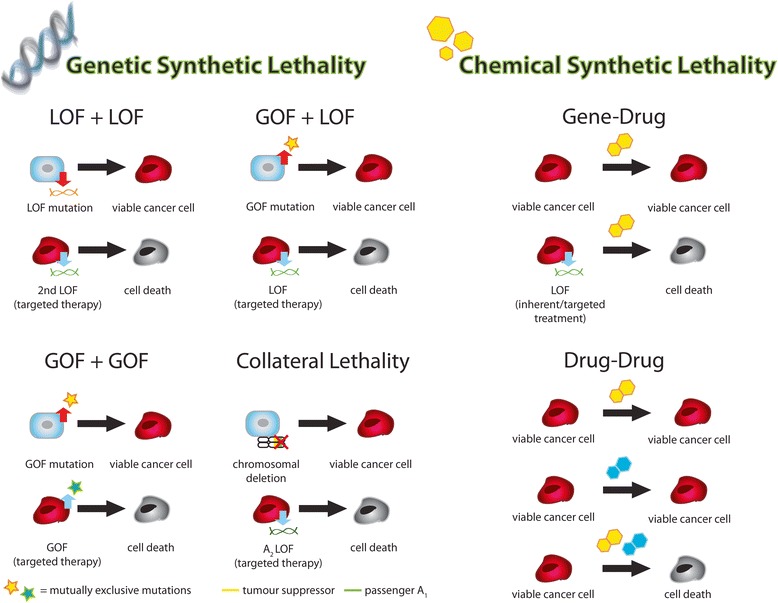
Types of Synthetic Lethal Interactions in the Context of Cancer. The various types of synthetic lethal interactions can be grouped into two categories: genetic-based and chemical-based. Genetic synthetic lethality is primarily based on cancer-specific genetic alterations (blue normal cells undergo genetic changes that result in transformation to red cancer cells) that become susceptible to further induced changes in gene expression resulting in synthetic lethality. Chemical synthetic lethality describes synthetic lethal interactions between inherent or induced genetic alterations and broad-spectrum therapeutics (chemosensitization) as well as synergistic outcomes from the use of two or more chemotherapeutics. Please see text for full description of each type of interaction. (LOF = loss-of-function, GOF = gain-of-function, passenger A1 = passenger gene deletion, A2 = isoform of deleted passenger A1, blue cell = normal cell, red cell = cancer cell, grey cell = dead cancer cell)
Table 1.
Synthetic Lethal Interactions Identified in Lung Cancer
| Interactor 1 | Interactor 2 | Type of Interaction | Method of Discovery | Lung Cancer Subtype | Year discovered | First Author | PMID |
|---|---|---|---|---|---|---|---|
| PAPSS1 | Cisplatin (or other DNA damaging agents) | Chemo-sensitization | RNAi Screen (siRNA) + Low-Dose Cisplatin | LAC | 2015 | Leung | 26220590 |
| CABYR | Cisplatin | Chemo-sensitization | RNAi Screen (siRNA) + Cisplatin | LAC | 2014 | Qian Leung |
24362251 26938915 |
| dUTPase | FUdR/pemetrexed | Chemo-sensitization | Hypothesis Based | LAC; bronchioalveolar carcinoma; LCC | 2012 | Wilson | 22172489 |
| mKRAS | PKCi Aggegation (via Oncrasin-1 treatment) | GOF + GOF | Chemical library Compound Screen | LAC | 2008 | Guo | 18794128 |
| mKRAS | mEGFR | GOF + GOF | Computational - Mutual Exclusivity Analysis in Lung Cancer Genomic Data | LAC | 2015 | Unni/Lockwood | 26047463 |
| mKRAS | mBRAF | GOF + GOF | Hypothesis Based | LAC | 2016 | Cisowski | 26028035 |
| mEGFR | ARHG5 | GOF + LOF | Computational - EGFR Interactome Mapping | LAC | 2013 | Li | 24189400 |
| mKRAS | GATA2 | GOF + LOF | RNAi Screen (siRNA) | LAC | 2012 | Kumar | 22541434 |
| mKRAS | STK33 | GOF + LOF | RNAi Screen (shRNA) | LAC | 2009 | Scholl | 19490892 |
| mKRAS | TBK1 | GOF + LOF | RNAi Screen (shRNA) | LAC | 2009 | Barbie | 19847166 |
| mKRAS | PLK1 | GOF + LOF | RNAi Screen (shRNA) | LAC | 2009 | Luo | 19490893 |
| mKRAS | WT1 | GOF + LOF | RNAi Screen (shRNA) | LAC | 2010 | Vicent | 20972333 |
| mKRAS | CDK4 | GOF + LOF | RNAi Screen (shRNA) | LAC | 2010 | Puyol | 20609353 |
| MYC | PRKDC | GOF + LOF | RNAi Screen (shRNA) | SCLC | 2014 | Zhou | 25495526 |
| mEGFR | PRKCSH + EGFR-i | GOF + LOF + LOF | RNAi Screen (shRNA) + Gefitinib | LAC | 2014 | Sudo | 25528770 |
| mEGFR | NF-kB + EGFR-i | GOF + LOF + LOF | RNAi Screen (shRNA/siRNA) + EGFR TKI | LAC | 2011;2015 | Bivona Blakely |
21430781 25843712 |
| mKRAS | BCL-XL + MEK-i | GOF + LOF + LOF | RNAi Screen (shRNA) + Selumetinib | LAC | 2013 | Corcoran | 23245996 |
| ATM | DNAPK | LOF + LOF | Hypothesis Based | LAC | 2013 | Riabinska | 23761041 |
| ATM/p53 | ATR (under DNA damaging conditions) | LOF + LOF | Hypothesis Based | LAC | 2011 | Reaper | 21490603 |
| EGFR | Tankyrase 1 | LOF + LOF | RNAi Screen (shRNA) + Gefitinib | LAC | 2012 | Casas-Selves | 22738915 |
| MAX | BRG1 | LOF + LOF | Computational - Global gene expression analysis; cancer databases | SCLC | 2014 | Romero | 24362264 |
| RB1 | CDKN2A | LOF + LOF | Computational - Proteome/transcriptome profiling | LAC; SCLC | 2016 | Kim | 26647789 |
| LKB1 | phenformin | LOF + LOF | Compound library screen | LAC | 2013 | Shackelford | 23352126 |
| BRM/SMARCA2 | BRG1 | LOF + LOF/collateral | RNAi Screen (shRNA) | LAC | 2014 | Hoffman Orvis Wilson Oike |
24520176 25115300 24421395 23872584 |
| PSMA1 (proteosome subunit) | Radiation | Radio-sensitization | RNAi Screen (shRNA) | LAC; LCC | 2013 | Cron | 24040035 |
| Cisplatin | Irinotecan | Synergistic Interaction | Hypothesis Based | SCLC | 2002 | Noda | 11784874 |
GOF gain-of-function, LOF loss-of-function, LAC lung adenocarincoma, SCLC small-cell lung cancer, LCC large cell carcinoma, m-(gene) mutant variant of the gene, (gene)-i inhibitor of gene product
Genetic-based synthetic lethality
As mentioned above, tumour cells acquire mutations that allow them to grow rapidly over time. While these characteristics give cancerous cells advantages in proliferation and survival, they also become therapeutic targets. The differential regulation of genes between cancer cells and their corresponding normal cells allow researchers to identify those that can be targeted to induce synthetic lethality in a cancer-specific manner. The various types of interactions are described below.
Loss-of-function/Loss-of function
Loss-of-function (LOF) mutations in tumour suppressors are extremely common in human cancers. One such example is the genome guardian p53, the most frequently mutated gene in human cancers, ranging from 25 to 50 % in various tumour types including ovarian, breast, colorectal, head and neck, and lung cancers [36]. Although p53 mutations have been studied extensively, it has been a difficult target as it has no enzymatic activity and primarily functions through protein-protein interactions [37]. One approach to target this “undruggable” target has focused on strategies to restore the wild-type function of mutated p53 [38]. Another therapeutic approach exploits the vulnerabilities or genetic dependencies arising from loss of wild-type p53 functions [39]. For example, based on differential gene expression analyses, Wang and Simon proposed a list of 98 candidate genes that, when suppressed, may induce synthetic lethality in p53-deficient cancers [40]. We define such a strategy as LOF/LOF as it involves inhibiting a gene/pathway/function in the background of genetic or pharmacological inactivation of another gene to induce synthetic lethality.
To date, the biggest clinical success in synthetic lethal targeting is the use of PARP (poly (ADP-ribose) polymerase) inhibitors against tumours with BRCA mutations. BRCA1 and BRCA2 deficiencies cause defects in homologous recombination (HR) and have a significant role in hereditary breast and ovarian cancers [41]. It is known that mice lacking PARP expression are viable but are defective in repairing single-stranded DNA breaks. Loss of PARP function results in the dependence on the use of homologous recombination (HR) to repair DNA damage [42]. Based on these observations, Bryant et al. hypothesized and then demonstrated that BRCA-deficient cancers are hypersensitive to PARP inhibitors [42]. The discovery of this truly synthetic lethal interaction (by definition) has revolutionized our approach to the treatment of BRCA-associated cancers and served as the paradigm for uncovering other synthetic lethal interactions.
PTEN, the second most frequently mutated tumour suppressor in cancer, is a similar therapeutic target [43]. Inspired by the synthetic lethality induced by loss of BRCA2 and PARP functions, Mendes-Pereira et al. demonstrated that cancers with PTEN deficiency, which also cause HR defects, are sensitive to PARP inhibitors [44]. This synthetic lethal interaction was validated in vitro and in vivo. As a result, PARP inhibitors are currently in Phase II clinical trials for treatment of PTEN-deficient cancers. Exploiting deficiencies in DNA repair proteins has since revealed additional synthetic lethal interactions with particular relevance to lung cancer. Similar to the case of PARP and BRCA, Riabinska et. al. observed that cancers with ATM-deficiency are defective in HR and dependent on nonhomologous end joining (NHEJ) for DNA repair and cell survival [45]. Through genetic and pharmacological methods, they subsequently demonstrated that inhibition of the catalytic subunit of an essential NHEJ protein, DNA-dependent protein kinase (PRKDC), induces synthetic lethality specifically in ATM-deficient cancers, but not normal cells or cancer cells with active ATM. Likewise, after developing a potent and selective inhibitor of the DNA damage response (DDR) kinase ATR, Reaper and colleagues found that it induced synthetic lethality specifically in ATM- or p53-deficient cancers in the context of genotoxic stress. Since ATM is mutated and inactivated in ~10 % of LACs, these findings provide a potential therapeutic strategy for a large subset of patients, one which is especially resistant to standard chemotherapeutics.
While the above examples were mainly identified in other cancer types, many of the described genes are also disrupted in lung cancer suggesting that similar synthetic lethal interactions may also exist in this context. However, screens for lung cancer specific LOF/LOF synthetic lethal interactions have also been performed. For example, through a chemical library screen, Shackelford and colleagues found that LKB1 (STK11) deficient lung cancer cell lines were acutely sensitive to phenformin, an inhibitor of mitochondrial function [46]. LKB1 is mutated in ~20 % of NSCLC and this work, subsequently validated in Lkb1 mutant mouse models, suggests that metabolism based therapeutics may be effective in this subset of cancer patients. Using a loss-of-function whole-genome shRNA screen, inhibition of the canonical Wnt pathway, specifically the positive regulators tankyrase 1 and 2, was found to induce cell death in LAC cells only in the context of EGFR inhibition [47]. Furthermore, computational approaches have revealed LOF/LOF synthetic lethality of MAX and BRG1 in SCLC and RB1 and CDKN2A in both SCLC and NSCLC [48, 49].
Lastly, it is important to note a unique subcategory of LOF/LOF interactions that only occur in the presence of a gain-of-function (GOF) oncogene mutation. These interactions typically involve inhibiting the mutant oncogene with a small molecule inhibitor and another gene with pharmacological or genetic methods. Since the inhibitors only work – and hence synthetic lethality is only induced - in the context of the GOF mutation, we term this association GOF/LOF/LOF, even though only two genes are involved. Examples of this association include the use of EGFR kinase inhibitors in the background of mutant EGFR (mEGFR). For instance, using RNAi screening in the presence of the EGFR kinase inhibitor Erlotinib, Bivona and colleagues found that inhibiting FAS and other components of the NF-kB pathway specifically enhanced cell death in mEGFR LAC cell lines [50, 51]. Likewise, Sudo et. al. found a similar association between NF-kB inhibition and Gefitinib (another EGFR kinase inhibitor) and also identified a novel gene candidate, PRKCSH, that induced cell death in Gefitnib-treated mEGFR LAC cells [52]. Finally, a pooled shRNA screen in KRAS mutant cancer cells, which are dependent on MEK activation, found that inhibition of BCL-XL cooperated with MEK inhibitors to induce cell death [53]. These findings were validated using the MEK inhibitor selumetinib and the BCL-XL inhibitor ABT-263 in a genetically engineered KRAS-driven mouse model of lung cancer; highlighting the potential clinical relevance of this interaction.
Gain-of-function/Loss-of function (synthetic dosage lethality)
While loss of tumour suppressor functions is a common characteristic in cancer cells, activation of various oncogenes through gain-of-function (GOF) mutations is another major contributor to tumour development. Like LOF mutations, GOF mutations rewire cancer cells, making them susceptible to additional changes in cellular functions that would cause little or no harm to normal cells. When lethality occurs as a result of one gene being genetically activated (GOF) and another being inactivated through genetics or drug targeting (LOF), the interaction is known as synthetic dosage lethality.
As an example, constitutive activation of RAS signaling, particularly KRAS, is known to be the oncogenic driver for approximately 20 % of all human cancers [54]. Cancers of the pancreas, colon, and lung are known to have high frequencies of KRAS mutations [55]. KRAS has long been considered an “undruggable” target due to the lack of suitable binding pockets for small molecule inhibitors [56]. To target KRAS mutants, several groups have utilized high throughput RNAi screening to identify synthetic lethal partners of activated mutant KRAS. Luo et al. performed a genome-wide shRNA screen in colorectal cancer cells and found that KRAS mutants are hypersensitive to PLK1, APC/C, and proteasome inhibition relative to isogenic cells with wild-type KRAS [57]. Other synthetic lethal partners that have been identified through RNAi screening within an activated KRAS background include STK33, TBK1, WT1, CDK1, CDK4, GATA2, and Snail2, studies conducted primarily using colon and lung cancer cells [58–64]. These interactions have been reviewed elsewhere.
Hyperactivation of members of the MYC protein family is very common in a wide range of cancers. c-MYC is amplified in about 10 % of LAC and c-MYC, MYCL or MYCN amplification/overexpression occurs in >20 % of SCLC. MYCs are transcription factors - and thus, difficult to inhibit directly – thus, synthetic lethal strategies are being used to define novel treatments for cancers driven by the activation of these proteins. Inhibition of mTOR, Aurora A/B, SAE2, or CDK1 has been shown to induce synthetic lethality in MYC-driven cancers [65–68]. Synthetic lethal relationships between hyperactivated MYCN and suppression of BRD4 and CSNK1e have also been demonstrated in acute myeloid leukaemia and neuroblastoma, respectively [69, 70]. Importantly these interactions are being tested clinically where Phase II clinical trials are assessing therapeutic benefit of inhibitors targeting CDK1 or BRD4 in patients with MYC-amplified tumours. Recently, through a pooled shRNA screen, inhibition PRKDC was found to specifically induce synthetic lethality in SCLC cell lines with amplification/overexpression of MYC family members but not in those without [71]. This was attributed to the role of PRKDC in controlling MYC protein levels as well as facilitating repair of MYC-induced DNA damage. These results could be recapitulated with the PRKDC inhibitor, NU-7441, suggesting a possible therapeutic avenue for this aggressive lung cancer subtype.
Lastly, although EGFR kinase inhibitors have proven effective at improving outcomes of LAC patients with activating EGFR mutations, all patients treated with these inhibitors eventually develop resistance and relapse. Identifying the key factors necessary for mEGFR-mediated tumorigenesis may offer avenues for combination based therapies that can overcome EGFR kinase inhibitor resistance. Through charactering the LAC‐specific mEGFR interactome through global analysis of protein–protein interactions and phosphorylation, Li and collegues identified 8 key proteins necessary for survival mEGFR lung cancer cell lines in addition to EGFR itself: GRB2, MK12, SHC1, ARAF, CD11B, ARHG5, GLU2B, and CD11A [72]. When inhibited through siRNAs, these genes induced cell death specifically in mEGFR, but not EGFR wild-type, LAC cells. This clearly suggested a synthetic lethal relationship. Indeed, ablation of ARHG5 in mEGFR LAC cells induced apoptosis and was shown to interact with downstream EGFR signalling proteins including SHC1 and GRB2, highlighting its potential as a target for combination based therapy.
Gain-of-function/Gain-of-function
With the tools currently available, it is relatively easy to identify synthetic lethal interactions using gene knockdown or knockout approaches. In contrast, there has yet to be a large-scale GOF screen in human cells to search for synthetic lethality in cancers driven by known oncogenes. It is, however, not impossible to define synthetic lethal interactions that are based on two gain-of-function mutations using information derived from studies assessing human tumour evolution. For instance, it has been known for years that oncogenic mutations in KRAS and EGFR are not only common, but also mutually exclusive in LAC [73–75]. It was typically assumed that since the two oncogenes work through the activation of similar pathways, mutation in both genes was functionally redundant and thus, not positively selected during tumour development. However, in a recent study, Unni et al. discovered that these two mutations are mutually exclusive because activation of both genes in lung cells induces synthetic lethality. This explained why there is selectedion against cells expressing both mutations together during tumour evolution [76]. Expressing both mutant oncogenes in a mouse model of lung cancer led to the selection of tumours expressing a single oncogene while forced expression of mEGFR or mKRAS in LAC cells with endogenous mutations in the reciprocal oncogene induced cell death through uncontrolled macropinocytosis and catastrophic cell vacuolization. The later events were likely controlled through increased MAPK signalling. From a clinical perspective, while it is challenging to increase expression of a gene product, this study suggests that there may be opportunities to activate combinations of pathways to induce lethality. The method through which this interaction was revealed and validated suggests that additional synthetic lethal interactions may be identified through exploring co-activation of mutually exclusive driver mutations and their respective pathways. Indeed, a subsequent study revealed a similar association between mutant BRAF and mKRAS in LAC, demonstrating the validity of this approach.
Although difficult to categorize, chemical compound screens have also revealed interactions suggestive of synthetic lethality induced by GOF/GOF associations. As an example, Guo et al. identified a small molecular weight compound (oncrasin-1) that selectively and effectively killed human lung cancer cells with mKRAS through induction of apoptosis [77]. A search for a target of this novel compound revealed no changes to the activity of known RAS signalling pathway components. However, treatment with oncrasin-1 led to abnormal aggregation of PKCι in the nucleus of sensitive, but not in resistant cells. While oncrasin-1 did not change the activity of PKCι, it did induce a GOF change in terms of modifying PKCι subcellular localization. This was, in turn, associated with cell death in KRAS mutant cells. It is conceivable that compounds that stimulate or modify pathway signalling/function may also provide an opportunity to induce synthetic lethality in cancer cells in the context of GOF/GOF interactions.
Collateral lethality
A more recent concept of synthetic lethality has been termed “collateral lethality” [78]. In the process of malignant transformation, some tumour suppressor genes may be inactivated through chromosomal deletions. Loss of heterozygosity or homozygous deletions results in complete inactivation of a tumour suppressor; which in itself enables cancer-specific targeting of synthetic lethal partners as described above. However, collateral lethality takes advantage of “passenger” or “neighboring” genes that are co-deleted “unintentionally” in this process [78, 79]. These passenger genes may encode for housekeeping functions that are essential to cell viability but are masked by the presence of redundant genes encoded elsewhere to complement for the loss. Targeting homologues of these passengers forms the basis of collateral lethality. As an example, ENO1 is a gene that is homozygously deleted in glioblastoma (GBM) as a result of deletion in the 1p36 tumour-suppressor locus [79]. This gene encodes for enolase, an enzyme that is essential for glycolysis. Although ENO1 accounts for up to 90 % of the enolase activity in GBM, its loss is tolerated through the expression of ENO2 which is exclusively expressed in neural cells [55]. Loss of ENO2 does not cause reduced viability in the presence of ENO1 expression, but GBM cells with deletion in 1p36 are highly sensitive to ENO2 inhibition due to collateral lethality [79]. Muller et al. have identified other homozygously deleted essential genes that have potential collateral lethal partners in GBM [79].
To date, the only described instance of collateral lethality in lung cancer involves SMARCA4 (a chromatin remodelling helicase) and its paralogue, SMARCA2. Unlike the ENO1/ENO2 example, SMARCA4 is a true tumour suppressor gene and is directly inactivated through mutations or deletions in approximately 10–15 % of LACs. However, unlike other tumour suppressors, SMARCA4 and SMARCA2 perform cellular housekeeping functions rather than preventing abnormal cell growth. Thus, inhibition of SMARCA2 in the background of SMARCA4 genetic inactivation leads to synthetic lethality as the cell loses the ability to complete an essential function necessary for survival, as is the case with ENO1/ENO2 in GBM. Inhibitors targeting SMARCA2 may therefore prove effective in the large subset of LAC patients with SMARCA4 mutations [80–83].
Synthetic lethal interactions involving broad-spectrum pharmaceuticals
As introduced earlier, the traditional definition of synthetic lethality/sickness involves defects (hyperactivation or inactivation) in a pair of genes. In the context of cancer, however, synthetic lethal/sick interactions also apply to gene-drug and drug-drug interactions. To date, cancer treatments rely heavily on the use of broad-spectrum therapeutics that target essential cellular processes such as DNA replication and mitosis; processes that are particularly important to rapidly dividing cells. Although these cytotoxic agents are highly effective, their uses have been limited by narrow therapeutic windows. Effective doses can result in severe and potentially life-threatening toxicities. Further, cancer cells have a remarkable capability to develop resistance against these agents over time. Nonetheless, these agents have provided significant therapeutic benefits to patients and their use makes sense even today given the nature of intra- and inter-tumoural heterogeneity. It will not be easy to supplant these agents used as standard of care for many cancers. With a better understanding of cancer genetics and the mechanisms of drug action, a tremendous amount of effort is being placed on improving the efficacy of existing cytotoxic agents through synthetic lethal approaches. From our perspective this may involve selecting the proper drug based on tumour-specific defects (chemosensitization) or developing drug combinations to achieve synergy (synthetic sickness).
Gene-drug interactions
Cisplatin is undoubtedly one of the most successful chemotherapeutics ever discovered for cancer treatments. While its use and clinical success is widespread, it causes severe side-effects including nephrotoxicity, neurotoxicity, and ototoxicity [84]. Furthermore, resistance to platinum based drugs is common. To overcome these challenges, many groups have attempted to develop combination products comprising cisplatin and a targeted treatment. The later can be uncovered by performing high-throughput RNAi synthetic lethal screens to better understand the mechanisms of resistance and the genetics underlying the sensitivity of cancer cells to cisplatin and other DNA damaging agents. For instance, cancers with defects in DNA damage repair pathways, such as BRCA mutations, are hypersensitive to DNA cross-linkers including cisplatin, carboplatin, and mitomycin C [85]. Other genes that have been identified as potential therapeutic targets for sensitization to cisplatin treatment include AMBRA, and PRKAB1 [86, 87]. In fact, numerous genes have been identified as chemosensitizing targets where gene knockdown or target inhibition via small molecules sensitizes tumours to multiple chemotherapeutics. One such target is CABYR, a cancer testis antigen in lung cancer that sensitizes NSCLC to paclitaxel and cisplatin [88, 89]. Inhibition of components of the ATR-CHK1 checkpoint signaling pathway appears to sensitize ovarian cancer cells to multiple DNA damaging agents [86, 90]. PAPSS1, one target that our group identified recently, is a nuclear enzyme that produces the obligate substrate for sulfonation reactions which when inhibited, sensitizes NSCLC cells to a wide range of DNA damaging agents [91]. Swanton et al. have also identified CERT, a ceramide-binding protein, as a target that enhances the activity of cisplatin and paclitaxel in lung cancer cells, paclitaxel and 5-Fu in colorectal carcinoma cells, and paclitaxel and doxorubicin in breast cancer cells [92]. As already established through the existing principles of combination chemotherapy for treatment of cancer, maximal therapeutic effects will almost certainly involve multiple targets. This is exemplified by the studies of De et al., who demonstrated that optimal treatment outcomes were achieved in vivo when triple negative breast cancer were treated with carboplatin in combination with both an mTOR and a PARP inhibitor [93]. Similar to these studies on chemotherapeutics, groups have also aimed to identify genes that modify the sensitivity of lung cancer cells to radiation, which is commonly used in the treatment of lung cancer patients. For example, a whole genome RNAi screen found that inhibition of proteasome subunits (e.g. PSMA1) worked synergistically with ionizing radiation to induce enhanced killing of lung cancer cells [94].
Drug-drug interactions
Traditionally, drug combinations have been defined by simply combining two or more drugs with known single agent activity, different mechanisms of action and non-overlapping toxicities. This approach maximized potential therapeutic benefits while reducing the potential for development of resistance. These broad spectrum therapeutic approaches have led to numerous successes, but also significant failures. It is now apparent that some drug combinations work synergistically through synthetic lethality while others can result in antagonism, where the combinatorial effects turn out to be worse than what can be achieved with the single agents [96]. So the goal should be to focus on synergistic combinations; but this can be challenging as synergy can be dependent on a number of factors including drug-drug ratio, exposure time, and sequencing [95–98]. Regardless many examples of synergistic drug-drug interactions have been published and some of these have even been put into clinical practice. Peters et al. found that the combination of cisplatin and gemcitabine is synergistic in vitro and in vivo using ovarian, head and neck, and colorectal cancer models [99]. This drug combination is currently used for the treatment for at least seven different cancers types including ovarian and NSCLC. Likewise, irinotecan plus cisplatin was demonstrated to provide a synergistic benefit in the treatment of metastatic small-cell lung cancer as compared to etoposide plus cisplatin, which was the standard of care [100]. For in vitro and in vivo studies, a number of approaches have been used to assess drug-drug interactions [101, 102]. A common method uses a fixed ratio experimental designed and subsequent calculation of combination indices using the Chou-Talalay method [103, 104]. As an interesting alternative method Kang et al. analyzed over 1000 Phase II clinical trials to determined clinical synergy and antagonism of different combinations of two drugs; where overall response rates were used as the therapeutic outcome [105].
Conclusions
Perspectives: translating synthetic lethality for clinical applications
Although numerous synthetic lethal interactions have been discovered through genetic and chemical screening approaches, many have yet to translate into clinical successes. Translating synthetic lethal/sick discoveries to the clinic can be challenging depending on a host of factors ranging from tumor biology to the ability to access proprietary drugs own by different pharmaceutical companies. This is further complicated by the current clinical trial paradigms that typically require new treatments to be tested in patient populations that have failed standard of care therapies. An example of the latter concern is highlighted by our screen looking for cisplatin sensitizers; a screen that relies on use of lung cancer cell lines that were derived from patients that were chemo-naive. The rationale was based on the believe that optimal treatment outcomes with a combination of cisplatin and a targeted agent known to be a sensitizer would only be achieved in the first line setting. This, however, would be a clinically challenging study to complete.
Below, we have identified several factors that should be considered when translating synthetic lethal interactions to therapeutic strategies and have included pharmacological considerations that may be important when trying to achieve synthetic lethality interactions targeting tumours in cancer patients. These factors are summarized in Figs. 3 and 4.
Fig. 3.
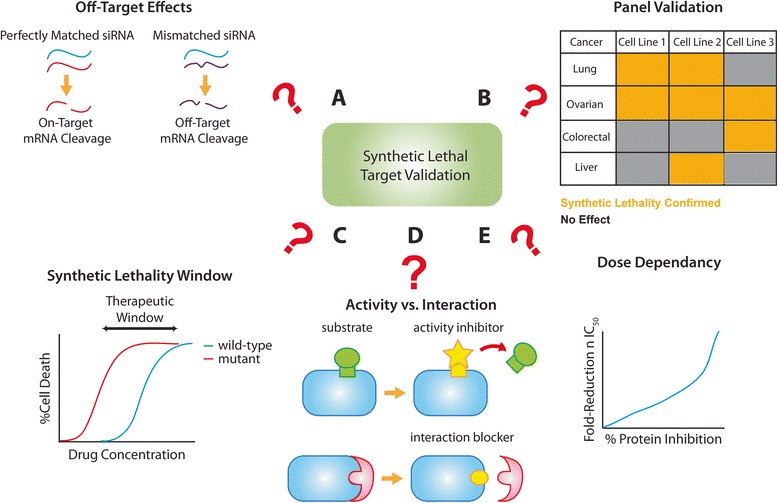
Considerations when validating synthetic lethal targets. Several factors should be considered when deciding whether or not to translate a synthetic lethal discovery to therapeutics. If the target was discovered from an RNAi screen, off-target effects should be eliminated by testing individual siRNA duplexes, using pools of siRNAs, or even testing the interaction using small molecules if available (a). Secondly, the synthetic lethality should be verified in a panel of cell lines for the indication(s) of interest to assess potential applications of the therapeutic strategy of interest (b). The therapeutic window should also be assessed to ensure that synthetic lethality occurs in a cancer-specific manner (c). When developing pharmaceuticals for the target of interest, it is crucial to understand whether it is the enzymatic activity or a specific interaction that is responsible for the synthetic lethality observed (d). Finally, synthetic lethality might be dependent on the extent of genetic alteration. This dose dependency should be explored and addressed when designing and developing therapeutics for synthetic lethal targets (e)
Fig. 4.
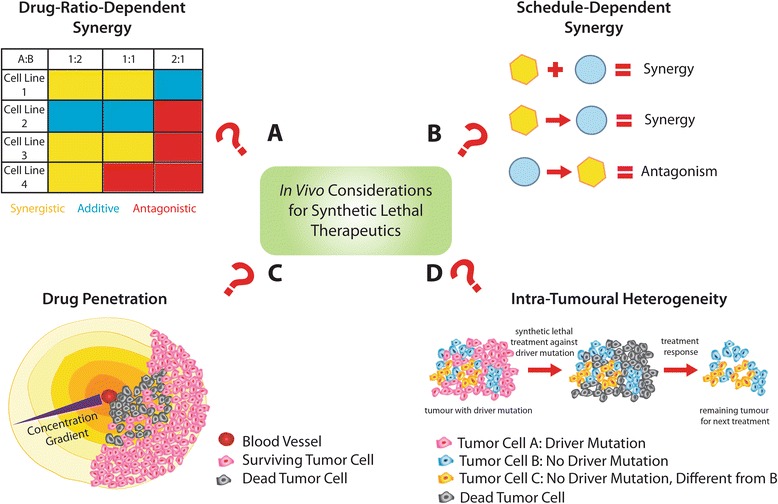
In Vivo Considerations for Synthetic Lethal Therapeutics. When using two or more therapeutics, it is important to determine the drug combination ratios at which synergy occur (a). This should be done in a panel of cell lines for the indication(s) of interest. Synergism may also be dependent on the timing of the administration of the different therapeutics (b). Another challenge that needs to be addressed is the issue associated with drug penetration into the entire tumour (c). As a result of poorly organized vasculature, concentration gradients will be generated upon treatment and outcomes of synthetic lethal approaches may be limited by the inability to induce sufficient genetic alterations in all cells of the targeted population. Finally, while synthetic lethal approaches are promising, certain populations of the tumour may survive treatment due to intra-tumoural heterogeneity which makes them insensitive to the specific treatment regimen (d)
Considerations for translating synthetic lethal interactions to therapeutic strategies
When validating hits from RNAi screens, it is important to ensure that the phenotypic observations from gene-knockdown are not due to off-target effects (Fig. 3a). While mRNA sequences that perfectly match the siRNA guide strand are cleaved by the RNAi machinery, off-target silencing could also occur where the mRNA has slight mismatches with the siRNA template, particularly when the siRNA targets the 3'-UTR regions, giving rise to false positives due to unintended microRNA-like activities. These off-target effects could be minimized through strategies such as chemical modification of the siRNA duplexes and utilization of pooled sequences [106]. Validation of the on-target effects is also necessary through the use of multiple RNAi sequences to eliminate sequence-specific effects and “rescue” experiments using cDNAs. The cDNA used should lack siRNA-binding sites so that the putative target can be exogenously expressed in the presence of endogenous gene knockdown. Once the on-target effects are confirmed, it is crucial to test for synthetic lethality in a range of cell lines and relevant disease models (Fig. 3b). The goal of these validation studies is to assess the potential of synthetic lethal interaction in different contexts represented by tumour subtypes, tumours of different origin and different genetic backgrounds. It is just as important to assess the therapeutic window associated with the synthetic lethal interaction. For example, in cases where the synthetic lethal interaction is specific to an oncogenic or loss-of-function genetic background, it would be ideal that treatment causes little or no effect on cells expressing the wild-type version of the gene. The BRCA and PARP interaction highlights this point; PARP inhibitors are particularly effective against BRCA2-deficient tumours as demonstrated by Bryant et al. [42]. However, tumours harbouring wild-type BRCA2 or BRCA2-deficient tumours with BRCA2 overexpression did not respond to the treatment.
Widening of the synthetic lethality window (Fig. 3c) should also be carefully examined when validating chemo-sensitizing targets where a genetic deficiency is introduced globally to enhance the activity of another drug. Ideally, gene knockdown alone should not be deleterious to the viability of normal cells. Further, chemosensitization should be selective for only cancer cells. As an example, the target that we recently identified (PAPSS1) was found to enhance the activity of various DNA damaging agents in NSCLC cells [55] when PAPSS1 was depleted. We demonstrated that knockdown of the gene did not sensitize normal bronchial epithelial cells to cisplatin treatment while there was a > five-fold reduction in the cisplatin IC50 achieved when PAPSS1 was depleted in NSCLC cells.
When developing small molecules based on the findings from an RNAi screen, it is important to understand the specific function of the gene product (Fig. 3d) involved in the synthetic lethal interaction. Since transcription factors and other non-enzyme targets are generally considered “undruggable”, candidates with enzymatic activities such as kinases are prioritized for target development with the assumption that the enzymatic activity is responsible for the synthetic lethality observed in an RNAi screen. However, it is a possibility that the effect is the result of interactions between proteins rather than enzymatic function. In 2009, Scholl et al. discovered from an RNAi screen that KRAS-driven cancers are dependent on a gene that encodes for a serine/threonine protein kinase STK33 [58]. In 2012, Luo et al. developed a potent and selective kinase inhibitor for STK33 which failed to reproduce the synthetic lethality observed in the original screen [107]. It can be argued that this is due to the fact that STK33 has other non-kinase functions that are critical to the viability of KRAS-driven cancer cells, for example protein scaffolding. Such protein-protein interactions could be explored through co-immunoprecipitation studies or through more sophisticated approaches such as tandem affinity purification during target validation [108]. While small molecule inhibitors may be developed to inhibit the enzymatic activity of a candidate target, peptides or peptide mimetics could be developed to inhibit specific protein-protein interactions important for synthetic lethality [109]. This rather novel therapeutic area also opens up opportunities for targeting the traditionally “undruggable” hits from synthetic lethal screens.
It should be noted that synthetic lethal targets discovered in yeast studies are based on complete gene knockouts, therefore these interaction are “definitive.” In contrast, RNAi screens utilizing siRNA or shRNA rarely eliminate the gene product completely and therefore the amount of depletion achieved may be critical to defining the interaction. For this reason, it is important to determine the minimal level of protein depletion necessary to achieve the desired synthetic lethal/sick effect. For instance, our studies on PAPSS1 showed that sensitization to cisplatin treatment occurred in a siRNA dose-dependent manner [91]. At the protein level, at least 80 % inhibition relative to scramble controls was necessary to achieve a meaningful improvement in cisplatin activity. While the extent to which the protein activity/interaction is inhibited can be determined in vitro and perhaps adjusted through a medicinal chemistry campaign. This dose dependency (Fig. 3e) will, however, be more challenging to address in vivo, as discussed in the following section.
Optimizing multidrug combinations to induce synthetic lethality
As indicated previously, the success of multidrug combinations is largely dependent on ensuring, in vivo, that the combinatorial effect is maintained at the site where the cancer resides. The factors influencing synergistic or synthetic sick interactions can be better understood through carefully designed in vitro studies that consider drug concentrations, exposure time, sequence and dosing parameters (e.g. drug/drug ratio). However, in the context of achieving a synthetic lethal interaction in vivo, one must have an excellent understanding of the drug pharmacokinetics and biodistribution attributes. Traditionally, drug combinations were given at maximally tolerated doses to achieve the greatest therapeutic effects. However, studies in the last decade have found that drug combinations display drug-ratio-dependent synergy (Fig. 4a) [110]. For instance, the combination of cisplatin and irinotecan, which is an approved combination for the treatment of lung cancer, was screened by Tardi et al. in a panel of 20 cell lines over a range of drug ratios [111]. Their study indicated that an antagonistic region (irinotecan/cisplatin molar ratios 1:2 to 4:1) was consistently detected in these cell lines. Importantly, the regions where synergy was observed (<1:2 and >4:1) were conserved in vivo. These results raise an important consideration that some drug combinations may need to be administered in a manner that can maintain an optimal synergistic ratio. This, however, can be challenging as different drugs exhibit different adsorption, distribution, and metabolism profiles. One approach to address this has been through the use of drug delivery technology. By co-encapsulating the two drugs at the optimally synergistic ratio into nanocarriers such as liposomes, the pharmacokinetic profile of both drugs can be controlled to maintain the drug ratio in vivo [110]. As an example, VYXEOS (CPX-351) is a liposomal formulation comprising cytarabine and daunorubicin (5:1 molar ratio) that was developed using the Combiplex® technology for the treatment of acute myeloid leukemia (AML) [97, 112, 96]. The Phase III clinical trial with VYXEOS has recently been completed and the results indicated that this formulation comprising two drugs was more effective than the standard of care 7 + 3 cytarabine/daunorubicin treatment [113].
While drug-drug ratios can be optimized when drug combinations are given concurrently, if the drug interactions are schedule-dependent (Fig. 4b), then one may have to optimize how treatments are sequenced. For instance, in a study conducted by Li et al., combinations of pemetrexed and erlotinib where synergistic when the two drugs concurrently and also when permetrexed was administered first followed by erlotinib [114]. However, the same two drug combination was antagonistic when erlotinib was given before pemetrexed. In another previous study, the use of the liposomal irinotecan formulation Irinophore C™ in combination with 5-FU concurrently resulted in high levels of toxicity. However this toxicity was reduced substantially when 5-FU was administered sequentially following Irinophore C™ in colorectal cancer models [115, 97]. Similarly, a phase III node-positive breast cancer trial demonstrated significantly better overall survival when patients were given doxorubicin and docetaxel sequentially relative to concurrent chemotherapy [116]. Although these examples represent classic drug-drug combinations, it is very likely that combinations arising from synthetic lethal/sick screens will be influenced by the similar factors.
Finally, when translating synthetic lethality to therapeutic strategies, it is imperative to consider intra- and inter-tumoural heterogeneity as well as the tumour microenvironment. The tumour microenvironment has long been known to significantly limit drug penetration (Fig. 4c). Tumor cells are exposed to sub-lethal doses of drug and this contributes to treatment failures [117]. Due to the poorly organized vasculature in tumours, drug treatment with small molecules create concentration gradients that lead to reduced drug exposure at certain regions of the tumour. While screening strategies have been used to identify synthetic lethal gene partners that can be inhibited to enhance the cytotoxic effects of low-dose chemotherapeutics, these therapeutic strategies may again be limited by the same drug penetration issues, the level of hypoxia, as well as the nutritional status of the target cells. If, as indicated earlier, a minimal level of target inhibition/depletion is needed to achieve a desired synthetic lethal or synthetic sick effect, then this must be achieved throughout the tumour. Further it will be important to validate that the interaction still occurs under conditions where cells may be stressed due to lack of oxygen or nutrients. In the context of the target identified in our studies, PAPSS1, a significant challenge associated with the future development of gene, peptide, or small molecule therapy against this target would be the need to achieve 80 % PAPSS1 knockdown in all regions of tumours that are exposed to low-dose cisplatin. Although there will always be a drug concentration gradient, the effects could potentially be mitigated through the use of drug delivery systems. As an example, liposomal formulations of doxorubicin, which are clinically approved, have shown to be efficacious through increased circulation lifetime leading to increased drug accumulation at the tumour site [118, 120]. Although concentration gradients would still be generated in that case, a greater amount of drug would be available even at hypoxic or nutrient-deprived regions due to greater overall exposure to the therapeutic agent.
In terms of future perspectives, synthetic lethality is a promising approach at the cellular level and even at the population level if the tumour population is clonal. In reality, however, intra-tumoural heterogeneity (Fig. 4d) contributes tremendously to the challenge of curing cancer. By use of chemotherapy and/or targeted agents, one treatment regimen could potentially eradicate a large population of tumour cells. At the same time, this treatment may remove selective pressures against existing dormant tumour cells. Even in the context of tumours with driver mutations, which could potentially be targeted using synthetic lethal strategies, not all tumour cells would harbour the specific driver mutation simply due to the countless mutations acquired through numerous generations. Nonetheless, we believe that treatments arising out of the principles of synthetic lethality, if applied early, should extend patient survival. With the use of other advanced technologies, such as post-treatment sequencing of tumour biopsies, continuous applications of synthetic lethal strategies will allow the disease to be managed for a longer period of time, ultimately making aggressive and difficult to treat cancers, such as lung cancer, a chronic disease instead of a terminal, illness.
Acknowledgments
Funding
The authors thank the Canadian Institutes for Health Research (CIHR, MOPs 89938 and 142313) and British Columbia Cancer Foundation for their financial support. AL received financial support from the CIHR (Vanier Canada Graduate Scholarship) and the University of British Columbia (Four-Year Doctoral Fellowship). WWL is supported by Michael Smith Foundation for Health Research Scholar and CIHR New Investigator Awards.
Availability of data and materials
Yes.
Authors’ contributions
AWYL and WWL were major contributors in writing the manuscript. TDS was involved in the design and the research in lung cancer-related synthetic lethal interactions. MBB made substantial contributions to the design of the manuscript and revised it critically for important intellectual content. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
N/A.
Ethics approval and consent to participate
N/A.
Abbreviations
- AML
Acute myeloid leukemia
- DDR
DNA damage response
- GBM
Glioblastoma multiforme
- GOF
Gain-of-function
- HR
Homologous recombination
- LAC
Lung adenocarcinoma
- LOF
Loss-of-function
- NHEJ
Non-homologous end joining
- NSCLC
Non-small cell lung cancer
- SCLC
Small cell lung cancer
- SqCC
Squamous cell carcinoma
- TKI
Tyrosine kinase inhibitors
Contributor Information
Ada W. Y. Leung, Email: aleung@bccrc.ca
Tanya de Silva, Email: tdesilva@bccrc.ca.
Marcel B. Bally, Email: mbally@bccrc.ca
William W. Lockwood, Email: wlockwood@bccrc.ca
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Sullivan JP, Minna JD, Shay JW. Evidence for self-renewing lung cancer stem cells and their implications in tumor initiation, progression, and targeted therapy. Cancer Metastasis Rev. 2010;29:61–72. doi: 10.1007/s10555-010-9216-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12:175–80. doi: 10.1016/S1470-2045(10)70087-5. [DOI] [PubMed] [Google Scholar]
- 4.Lockwood WW, Wilson IM, Coe BP, Chari R, Pikor LA, Thu KL, Solis LM, Nunez MI, Behrens C, Yee J, et al. Divergent genomic and epigenomic landscapes of lung cancer subtypes underscore the selection of different oncogenic pathways during tumor development. PLoS One. 2012;7:e37775. doi: 10.1371/journal.pone.0037775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammerman PS, Lawrence MS, Voet D, Jing R, Cibulskis K, Sivachenko A, Stojanov P, McKenna A, Lander ES, Gabriel S, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, Leenders F, Lu X, Fernandez-Cuesta L, Bosco G, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53. doi: 10.1038/nature14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 8.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mok T, Wu YL, Zhang L. A small step towards personalized medicine for non-small cell lung cancer. Discov Med. 2009;8:227–31. [PubMed] [Google Scholar]
- 10.Lockwood W, Politi K. MYCxing it up with FGFR1 in squamous cell lung cancer. Cancer Discov. 2014;4:152–4. doi: 10.1158/2159-8290.CD-13-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–98. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 13.Nijman SM. Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011;585:1–6. doi: 10.1016/j.febslet.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brough R, Frankum JR, Costa-Cabral S, Lord CJ, Ashworth A. Searching for synthetic lethality in cancer. Curr Opin Genet Dev. 2011;21:34–41. doi: 10.1016/j.gde.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 15.Lucchesi JC. Synthetic lethality and semi-lethality among functionally related mutants of Drosophila melanfgaster. Genetics. 1968;59:37–44. doi: 10.1093/genetics/59.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benedict RC. The origin of new varieties of nephrolepis by orthogenetic saltation II. Regressive variation or reversion from the primary and secondary sports of bostoniensis. Am J Bot. 1922;9:140–57. doi: 10.2307/2435486. [DOI] [Google Scholar]
- 17.Dobzhansky T. Genetics of natural populations. Xiii. Recombination and variability in populations of drosophila pseudoobscura. Genetics. 1946;31:269–90. doi: 10.1093/genetics/31.3.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.James SW, Silflow CD, Thompson MD, Ranum LP, Lefebvre PA. Extragenic suppression and synthetic lethality among Chlamydomonas reinhardtii mutants resistant to anti-microtubule drugs. Genetics. 1989;122:567–77. doi: 10.1093/genetics/122.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodson HV, Anderson BL, Warrick HM, Pon LA, Spudich JA. Synthetic lethality screen identifies a novel yeast myosin I gene (MYO5): myosin I proteins are required for polarization of the actin cytoskeleton. J Cell Biol. 1996;133:1277–91. doi: 10.1083/jcb.133.6.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997;278:1064–8. doi: 10.1126/science.278.5340.1064. [DOI] [PubMed] [Google Scholar]
- 21.Johnson CD, Rand JB, Herman RK, Stern BD, Russell RL. The acetylcholinesterase genes of C. elegans: identification of a third gene (ace-3) and mosaic mapping of a synthetic lethal phenotype. Neuron. 1988;1:165–73. doi: 10.1016/0896-6273(88)90201-2. [DOI] [PubMed] [Google Scholar]
- 22.Simons A, Dafni N, Dotan I, Oron Y, Canaani D. Establishment of a chemical synthetic lethality screen in cultured human cells. Genome Res. 2001;11:266–73. doi: 10.1101/gr.154201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 24.Paddison PJ, Silva JM, Conklin DS, Schlabach M, Li M, Aruleba S, Balija V, O’Shaughnessy A, Gnoj L, Scobie K, et al. A resource for large-scale RNA-interference-based screens in mammals. Nature. 2004;428:427–31. doi: 10.1038/nature02370. [DOI] [PubMed] [Google Scholar]
- 25.Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B, et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428:431–7. doi: 10.1038/nature02371. [DOI] [PubMed] [Google Scholar]
- 26.Aza-Blanc P, Cooper CL, Wagner K, Batalov S, Deveraux QL, Cooke MP. Identification of modulators of TRAIL-induced apoptosis via RNAi-based phenotypic screening. Mol Cell. 2003;12:627–37. doi: 10.1016/S1097-2765(03)00348-4. [DOI] [PubMed] [Google Scholar]
- 27.Moutsatsos IK, Parker CN. Recent advances in quantitative high throughput and high content data analysis. Expert Opin Drug Discov. 2016;11:415–423. doi: 10.1517/17460441.2016.1154036. [DOI] [PubMed] [Google Scholar]
- 28.Park SJ, Saito-Adachi M, Komiyama Y, Nakai K. Advances, practice, and clinical perspectives in high-throughput sequencing. Oral Diseases. 2016;22:353–364. doi: 10.1111/odi.12403. [DOI] [PubMed] [Google Scholar]
- 29.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–4. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xue HY, Ji LJ, Gao AM, Liu P, He JD, Lu XJ. CRISPR-Cas9 for medical genetic screens: applications and future perspectives. J Med Genet. 2016;53:91–7. doi: 10.1136/jmedgenet-2015-103409. [DOI] [PubMed] [Google Scholar]
- 31.Basu B, Yap TA, Molife LR, de Bono JS. Targeting the DNA damage response in oncology: past, present and future perspectives. Curr Opin Oncol. 2012;24:316–24. doi: 10.1097/CCO.0b013e32835280c6. [DOI] [PubMed] [Google Scholar]
- 32.Jackson RA, Chen ES. Synthetic lethal approaches for assessing combinatorial efficacy of chemotherapeutic drugs. Pharmacol Ther. 2016;162:69–85. doi: 10.1016/j.pharmthera.2016.01.014. [DOI] [PubMed] [Google Scholar]
- 33.Horn CR, Cochrane GM. An audit of morbidity associated with chronic asthma in general practice. Respir Med. 1989;83:71–5. doi: 10.1016/S0954-6111(89)80063-0. [DOI] [PubMed] [Google Scholar]
- 34.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12:323–34. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 35.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 36.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mandinova A, Lee SW. The p53 pathway as a target in cancer therapeutics: obstacles and promise. Sci Transl Med. 2011;3:64rv1. doi: 10.1126/scitranslmed.3001366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lazo JS, Sharlow ER. Drugging undruggable molecular cancer targets. Annu Rev Pharmacol Toxicol. 2016;56:23–40. doi: 10.1146/annurev-pharmtox-010715-103440. [DOI] [PubMed] [Google Scholar]
- 39.Gurpinar E, Vousden KH. Hitting cancers’ weak spots: vulnerabilities imposed by p53 mutation. Trends Cell Biol. 2015;25:486–95. doi: 10.1016/j.tcb.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 40.Wang XS, Simon R. Identification of potential synthetic lethal genes to p53 using a computational biology approach. BMC Med Genomics. 2013;6:30. doi: 10.1186/1755-8794-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72:1117–30. doi: 10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 43.Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27:5443–53. doi: 10.1038/onc.2008.241. [DOI] [PubMed] [Google Scholar]
- 44.Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, Waldman T, Lord CJ, Ashworth A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1:315–22. doi: 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Riabinska A, Daheim M, Herter-Sprie GS, Winkler J, Fritz C, Hallek M, Thomas RK, Kreuzer KA, Frenzel LP, Monfared P, et al. Therapeutic targeting of a robust non-oncogene addiction to PRKDC in ATM-defective tumors. Sci Transl Med. 2013;5:189ra178. doi: 10.1126/scitranslmed.3005814. [DOI] [PubMed] [Google Scholar]
- 46.Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, Wei L, Fishbein MC, Czernin J, Mischel PS, Shaw RJ. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013;23:143–58. doi: 10.1016/j.ccr.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Casas-Selves M, Kim J, Zhang Z, Helfrich BA, Gao D, Porter CC, Scarborough HA, Bunn PA, Jr, Chan DC, Tan AC, DeGregori J. Tankyrase and the canonical Wnt pathway protect lung cancer cells from EGFR inhibition. Cancer Res. 2012;72:4154–64. doi: 10.1158/0008-5472.CAN-11-2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Romero OA, Torres-Diz M, Pros E, Savola S, Gomez A, Moran S, Saez C, Iwakawa R, Villanueva A, Montuenga LM, et al. MAX inactivation in small cell lung cancer disrupts MYC-SWI/SNF programs and is synthetic lethal with BRG1. Cancer Discov. 2014;4:292–303. doi: 10.1158/2159-8290.CD-13-0799. [DOI] [PubMed] [Google Scholar]
- 49.Kim N, Song M, Kim S, Seo Y, Kim Y, Yoon S. Differential regulation and synthetic lethality of exclusive RB1 and CDKN2A mutations in lung cancer. Int J Oncol. 2016;48:367–75. doi: 10.3892/ijo.2015.3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bivona TG, Hieronymus H, Parker J, Chang K, Taron M, Rosell R, Moonsamy P, Dahlman K, Miller VA, Costa C, et al. FAS and NF-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature. 2011;471:523–6. doi: 10.1038/nature09870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blakely CM, Pazarentzos E, Olivas V, Asthana S, Yan JJ, Tan I, Hrustanovic G, Chan E, Lin L, Neel DS, et al. NF-kappaB-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer. Cell Rep. 2015;11:98–110. doi: 10.1016/j.celrep.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sudo M, Mori S, Madan V, Yang H, Leong G, Koeffler HP. Short-hairpin RNA library: identification of therapeutic partners for gefitinib-resistant non-small cell lung cancer. Oncotarget. 2015;6:814–24. doi: 10.18632/oncotarget.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, Greninger P, Brown RD, Godfrey JT, Cohoon TJ, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23:121–8. doi: 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rajalingam K, Schreck R, Rapp UR, Albert S. Ras oncogenes and their downstream targets. Biochim Biophys Acta-Mol Cell Res. 2007;1773:1177–95. doi: 10.1016/j.bbamcr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 55.Barault L, Lamba S, Di Nicolantonio F. Ras Mutations in Cancer. In eLS. John Wiley & Sons, Ltd; 2001.
- 56.McCormick F. KRAS as a therapeutic target. Clin Cancer Res. 2015;21:1797–801. doi: 10.1158/1078-0432.CCR-14-2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, Silver SJ, Tamayo P, Wadlow RC, Ramaswamy S, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821–34. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 59.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108-U122. [DOI] [PMC free article] [PubMed]
- 60.Wang Y, Ngo VN, Marani M, Yang Y, Wright G, Staudt LM, Downward J. Critical role for transcriptional repressor Snail2 in transformation by oncogenic RAS in colorectal carcinoma cells. Oncogene. 2010;29:4658–70. doi: 10.1038/onc.2010.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Costa-Cabral S, Brough R, Konde A, Aarts M, Campbell J, Marinari E, Riffell J, Bardelli A, Torrance C, Lord CJ, Ashworth A. CDK1 is a synthetic lethal target for KRAS mutant tumours. PLoS One. 2016;11:e0149099. doi: 10.1371/journal.pone.0149099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vicent S, Chen R, Sayles LC, Lin C, Walker RG, Gillespie AK, Subramanian A, Hinkle G, Yang X, Saif S, et al. Wilms tumor 1 (WT1) regulates KRAS-driven oncogenesis and senescence in mouse and human models. J Clin Invest. 2010;120:3940–52. doi: 10.1172/JCI44165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Puyol M, Martin A, Dubus P, Mulero F, Pizcueta P, Khan G, Guerra C, Santamaria D, Barbacid M. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18:63–73. doi: 10.1016/j.ccr.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 64.Kumar MS, Hancock DC, Molina-Arcas M, Steckel M, East P, Diefenbacher M, Armenteros-Monterroso E, Lassailly F, Matthews N, Nye E, et al. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell. 2012;149:642–55. doi: 10.1016/j.cell.2012.02.059. [DOI] [PubMed] [Google Scholar]
- 65.Pourdehnad M, Truitt ML, Siddiqi IN, Ducker GS, Shokat KM, Ruggero D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc Natl Acad Sci U S A. 2013;110:11988–93. doi: 10.1073/pnas.1310230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang D, Liu H, Goga A, Kim S, Yuneva M, Bishop JM. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proc Natl Acad Sci U S A. 2010;107:13836–41. doi: 10.1073/pnas.1008366107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, Schmitt EM, Skinner SO, Xu Q, Li MZ, Hartman ZC, et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 2012;335:348–53. doi: 10.1126/science.1212728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goga A, Yang D, Tward AD, Morgan DO, Bishop JM. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat Med. 2007;13:820–7. doi: 10.1038/nm1606. [DOI] [PubMed] [Google Scholar]
- 69.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–8. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Toyoshima M, Howie HL, Imakura M, Walsh RM, Annis JE, Chang AN, Frazier J, Chau BN, Loboda A, Linsley PS, et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc Natl Acad Sci U S A. 2012;109:9545–50. doi: 10.1073/pnas.1121119109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou Z, Patel M, Ng N, Hsieh MH, Orth AP, Walker JR, Batalov S, Harris JL, Liu J. Identification of synthetic lethality of PRKDC in MYC-dependent human cancers by pooled shRNA screening. BMC Cancer. 2014;14:944. doi: 10.1186/1471-2407-14-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li J, Bennett K, Stukalov A, Fang B, Zhang G, Yoshida T, Okamoto I, Kim JY, Song L, Bai Y, et al. Perturbation of the mutated EGFR interactome identifies vulnerabilities and resistance mechanisms. Mol Syst Biol. 2013;9:705. doi: 10.1038/msb.2013.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Suda K, Tomizawa K, Mitsudomi T. Biological and clinical significance of KRAS mutations in lung cancer: an oncogenic driver that contrasts with EGFR mutation. Cancer Metastasis Rev. 2010;29:49–60. doi: 10.1007/s10555-010-9209-4. [DOI] [PubMed] [Google Scholar]
- 74.Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–46. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 75.Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res. 2004;64:8919–23. doi: 10.1158/0008-5472.CAN-04-2818. [DOI] [PubMed] [Google Scholar]
- 76.Unni AM, Lockwood WW, Zejnullahu K, Lee-Lin SQ, Varmus H. Evidence that synthetic lethality underlies the mutual exclusivity of oncogenic KRAS and EGFR mutations in lung adenocarcinoma. Elife. 2015;4:e06907. doi: 10.7554/eLife.06907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo W, Wu S, Liu J, Fang B. Identification of a small molecule with synthetic lethality for K-ras and protein kinase C iota. Cancer Res. 2008;68:7403–8. doi: 10.1158/0008-5472.CAN-08-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Muller FL, Aquilanti EA, DePinho RA. Collateral lethality: a new therapeutic strategy in oncology. Trends Cancer. 2015;1:161–73. doi: 10.1016/j.trecan.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Muller FL, Colla S, Aquilanti E, Manzo VE, Genovese G, Lee J, Eisenson D, Narurkar R, Deng P, Nezi L, et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature. 2012;488:337–42. doi: 10.1038/nature11331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hoffman GR, Rahal R, Buxton F, Xiang K, McAllister G, Frias E, Bagdasarian L, Huber J, Lindeman A, Chen D, et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc Natl Acad Sci U S A. 2014;111:3128–33. doi: 10.1073/pnas.1316793111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Orvis T, Hepperla A, Walter V, Song S, Simon J, Parker J, Wilkerson MD, Desai N, Major MB, Hayes DN, et al. BRG1/SMARCA4 inactivation promotes non-small cell lung cancer aggressiveness by altering chromatin organization. Cancer Res. 2014;74:6486–98. doi: 10.1158/0008-5472.CAN-14-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wilson BG, Helming KC, Wang X, Kim Y, Vazquez F, Jagani Z, Hahn WC, Roberts CW. Residual complexes containing SMARCA2 (BRM) underlie the oncogenic drive of SMARCA4 (BRG1) mutation. Mol Cell Biol. 2014;34:1136–44. doi: 10.1128/MCB.01372-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oike T, Ogiwara H, Tominaga Y, Ito K, Ando O, Tsuta K, Mizukami T, Shimada Y, Isomura H, Komachi M, et al. A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1. Cancer Res. 2013;73:5508–18. doi: 10.1158/0008-5472.CAN-12-4593. [DOI] [PubMed] [Google Scholar]
- 84.Barabas K, Milner R, Lurie D, Adin C. Cisplatin: a review of toxicities and therapeutic applications. Vet Comp Oncol. 2008;6:1–18. doi: 10.1111/j.1476-5829.2007.00142.x. [DOI] [PubMed] [Google Scholar]
- 85.Martin RW, Connell PP, Bishop DK. The yin and yang of treating BRCA-deficient tumors. Cell. 2008;132:919–20. doi: 10.1016/j.cell.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 86.Arora S, Bisanz KM, Peralta LA, Basu GD, Choudhary A, Tibes R, Azorsa DO. RNAi screening of the kinome identifies modulators of cisplatin response in ovarian cancer cells. Gynecol Oncol. 2010;118:220–7. doi: 10.1016/j.ygyno.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 87.Li X, Zhang L, Yu L, Wei W, Lin X, Hou X, Tian Y. shRNA-mediated AMBRA1 knockdown reduces the cisplatin-induced autophagy and sensitizes ovarian cancer cells to cisplatin. J Toxicol Sci. 2016;41:45–53. doi: 10.2131/jts.41.45. [DOI] [PubMed] [Google Scholar]
- 88.Qian Z, Li M, Wang R, Xiao Q, Wang J, He D, Xiao X. Knockdown of CABYR-a/b increases chemosensitivity of human non-small cell lung cancer cells through inactivation of Akt. Mol Cancer Res. 2014;12:335–47. doi: 10.1158/1541-7786.MCR-13-0391. [DOI] [PubMed] [Google Scholar]
- 89.Leung AW, Hung SS, Backstrom I, Ricaurte D, Kwok B, Poon S, McKinney S, Segovia R, Rawji J, Qadir MA, et al. Combined use of gene expression modeling and siRNA screening identifies genes and pathways which enhance the activity of cisplatin when added at no effect levels to non-small cell lung cancer cells in vitro. PLoS One. 2016;11:e0150675. doi: 10.1371/journal.pone.0150675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huntoon CJ, Flatten KS, Wahner Hendrickson AE, Huehls AM, Sutor SL, Kaufmann SH, Karnitz LM. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013;73:3683–91. doi: 10.1158/0008-5472.CAN-13-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Leung AW, Dragowska WH, Ricaurte D, Kwok B, Mathew V, Roosendaal J, Ahluwalia A, Warburton C, Laskin JJ, Stirling PC, et al. 3′-Phosphoadenosine 5′-phosphosulfate synthase 1 (PAPSS1) knockdown sensitizes non-small cell lung cancer cells to DNA damaging agents. Oncotarget. 2015;6:17161–77. doi: 10.18632/oncotarget.3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Swanton C, Marani M, Pardo O, Warne PH, Kelly G, Sahai E, Elustondo F, Chang J, Temple J, Ahmed AA, et al. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell. 2007;11:498–512. doi: 10.1016/j.ccr.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 93.De P, Sun Y, Carlson JH, Friedman LS, Leyland-Jones BR, Dey N. Doubling down on the PI3K-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia. 2014;16:43–72. doi: 10.1593/neo.131694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cron KR, Zhu K, Kushwaha DS, Hsieh G, Merzon D, Rameseder J, Chen CC, D’Andrea AD, Kozono D. Proteasome inhibitors block DNA repair and radiosensitize non-small cell lung cancer. PLoS One. 2013;8:e73710. doi: 10.1371/journal.pone.0073710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Neijzen R, Wong MQ, Gill N, Wang H, Karim T, Anantha M, Strutt D, Waterhouse D, Bally MB, Tai IT, et al. Irinophore C™, a lipid nanoparticulate formulation of irinotecan, improves vascular function, increases the delivery of sequentially administered 5-FU in HT-29 tumors, and controls tumor growth in patient derived xenografts of colon cancer. J Control Release. 2015;199:72–83. doi: 10.1016/j.jconrel.2014.11.031. [DOI] [PubMed] [Google Scholar]
- 96.Mayer LD, Harasym TO, Tardi PG, Harasym NL, Shew CR, Johnstone SA, Ramsay EC, Bally MB, Janoff AS. Ratiometric dosing of anticancer drug combinations: Controlling drug ratios after systemic administration regulates therapeutic activity in tumor-bearing mice. Mol Cancer Ther. 2006;5:1854–63. doi: 10.1158/1535-7163.MCT-06-0118. [DOI] [PubMed] [Google Scholar]
- 97.Ramsay EC, Dos Santos N, Dragowska WH, Laskin JJ, Bally MB. The formulation of lipid-based nanotechnologies for the delivery of fixed dose anticancer drug combinations. Curr Drug Deliv. 2005;2:341–51. doi: 10.2174/156720105774370294. [DOI] [PubMed] [Google Scholar]
- 98.Patankar NA, Pritchard J, van Grinsven M, Osooly M, Bally MB. Topotecan and doxorubicin combination to treat recurrent ovarian cancer: the influence of drug exposure time and delivery systems to achieve optimum therapeutic activity. Clin Cancer Res. 2013;19:865–77. doi: 10.1158/1078-0432.CCR-12-2459. [DOI] [PubMed] [Google Scholar]
- 99.Peters GJ, Bergman AM, van Haperen VW R, Veerman G, Kuiper CM, Braakhuis BJ. Interaction between cisplatin and gemcitabine in vitro and in vivo. Semin Oncol. 1995;22:72–9. [PubMed] [Google Scholar]
- 100.Noda K, Nishiwaki Y, Kawahara M, Negoro S, Sugiura T, Yokoyama A, Fukuoka M, Mori K, Watanabe K, Tamura T, et al. Irinotecan plus cisplatin compared with etoposide plus cisplatin for extensive small-cell lung cancer. N Engl J Med. 2002;346:85–91. doi: 10.1056/NEJMoa003034. [DOI] [PubMed] [Google Scholar]
- 101.Harasym TO, Tardi PG, Johnstone SA, Bally MB, Janoff A, Mayer L. Fixed drug ratio liposome formulations of combination cancer therapeutics. Liposome Technology. 2006;3:25–48. [Google Scholar]
- 102.Harasym TO, Liboiron BD, Mayer LD. Drug ratio-dependent antagonism: a new category of multidrug resistance and strategies for its circumvention. Methods Mol Biol. 2010;596:291–323. doi: 10.1007/978-1-60761-416-6_13. [DOI] [PubMed] [Google Scholar]
- 103.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–6. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 104.Chang TT, Chou TC. Rational approach to the clinical protocol design for drug combinations: a review. Acta Paediatr Taiwan. 2000;41:294–302. [PubMed] [Google Scholar]
- 105.Kang W, DiPaola RS, Vazquez A. Inference of synergy/antagonism between anticancer drugs from the pooled analysis of clinical trials. BMC Med Res Methodol. 2013;13:1–8. doi: 10.1186/1471-2288-13-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- 107.Luo T, Masson K, Jaffe JD, Silkworth W, Ross NT, Scherer CA, Scholl C, Frohling S, Carr SA, Stern AM, et al. STK33 kinase inhibitor BRD-8899 has no effect on KRAS-dependent cancer cell viability. Proc Natl Acad Sci U S A. 2012;109:2860–5. doi: 10.1073/pnas.1120589109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Burckstummer T, Bennett KL, Preradovic A, Schutze G, Hantschel O, Superti-Furga G, Bauch A. An efficient tandem affinity purification procedure for interaction proteomics in mammalian cells. Nat Methods. 2006;3:1013–9. doi: 10.1038/nmeth968. [DOI] [PubMed] [Google Scholar]
- 109.Tsomaia N. Peptide therapeutics: targeting the undruggable space. Eur J Med Chem. 2015;94:459–70. doi: 10.1016/j.ejmech.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 110.Mayer LD, Janoff AS. Optimizing combination chemotherapy by controlling drug ratios. Mol Interv. 2007;7:216–23. doi: 10.1124/mi.7.4.8. [DOI] [PubMed] [Google Scholar]
- 111.Tardi PG, Dos Santos N, Harasym TO, Johnstone SA, Zisman N, Tsang AW, Bermudes DG, Mayer LD. Drug ratio-dependent antitumor activity of irinotecan and cisplatin combinations in vitro and in vivo. Mol Cancer Ther. 2009;8:2266–75. doi: 10.1158/1535-7163.MCT-09-0243. [DOI] [PubMed] [Google Scholar]
- 112.Liboiron BD, Mayer LD. Nanoscale particulate systems for multidrug delivery: towards improved combination chemotherapy. Ther Deliv. 2014;5:149–71. doi: 10.4155/tde.13.149. [DOI] [PubMed] [Google Scholar]
- 113.Stein EM, Tallman MS. Emerging therapeutic drugs for AML. Blood. 2016;127:71–8. doi: 10.1182/blood-2015-07-604538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li T, Ling YH, Goldman ID, Perez-Soler R. Schedule-dependent cytotoxic synergism of pemetrexed and erlotinib in human non-small cell lung cancer cells. Clin Cancer Res. 2007;13:3413–22. doi: 10.1158/1078-0432.CCR-06-2923. [DOI] [PubMed] [Google Scholar]
- 115.Hare JI, Neijzen RW, Anantha M, Dos Santos N, Harasym N, Webb MS, Allen TM, Bally MB, Waterhouse DN. Treatment of colorectal cancer using a combination of liposomal irinotecan (Irinophore C) and 5-fluorouracil. PLoS One. 2013;8:e62349. doi: 10.1371/journal.pone.0062349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Oakman C, Francis PA, Crown J, Quinaux E, Buyse M, De Azambuja E, Vila MM, Andersson M, Nordenskjold B, Jakesz R, et al. Overall survival benefit for sequential doxorubicin-docetaxel compared with concurrent doxorubicin and docetaxel in node-positive breast cancer-8-year results of the Breast International Group 02–98 phase III trial. Ann Oncol. 2013;24:1203–11. doi: 10.1093/annonc/mds627. [DOI] [PubMed] [Google Scholar]
- 117.Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–92. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 118.Mayer LD, Dougherty G, Harasym TO, Bally MB. The role of tumor-associated macrophages in the delivery of liposomal doxorubicin to solid murine fibrosarcoma tumors. J Pharmacol Exp Ther. 1997;280:1406–14. [PubMed] [Google Scholar]
- 119.Chari R, Thu KL, Wilson IM, Lockwood WW, Lonergan KM, Coe BP, Malloff CA, Gazdar AF, Lam S, Garnis C, MacAulay CE, Alvarez CE, Lam LL. Integrating the multiple dimensions of genomic and epigenomic landscapes of cancer. Cancer and Metastasis Reviews. 2010;29(1):73–93. doi: 10.1007/s10555-010-9199-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Abraham SA, Waterhouse DN, Mayer LD, Cullis PR, Madden TD, Bally MB. The liposomal formulation of doxorubicin. Methods Enzymol. 2005;391:71–97. doi: 10.1016/S0076-6879(05)91004-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Yes.