


Abstract
Bookmarking factors are transcriptional regulators involved in the mitotic transmission of epigenetic information via their ability to remain associated with mitotic chromatin. The mechanisms through which bookmarking factors bind to mitotic chromatin remain poorly understood. HNF1β is a bookmarking transcription factor that is frequently mutated in patients suffering from renal multicystic dysplasia and diabetes. Here, we show that HNF1β bookmarking activity is impaired by naturally occurring mutations found in patients. Interestingly, this defect in HNF1β mitotic chromatin association is rescued by an abrupt decrease in temperature. The rapid relocalization to mitotic chromatin is reversible and driven by a specific switch in DNA-binding ability of HNF1β mutants. Furthermore, we demonstrate that importin-β is involved in the maintenance of the mitotic retention of HNF1β, suggesting a functional link between the nuclear import system and the mitotic localization/translocation of bookmarking factors. Altogether, our studies have disclosed novel aspects on the mechanisms and the genetic programs that account for the mitotic association of HNF1β, a bookmarking factor that plays crucial roles in the epigenetic transmission of information through the cell cycle.
INTRODUCTION
Mitosis is a moment of drastic changes in the organization of the cell. At nuclear envelope breakdown, the loss of nuclear compartmentalization leads to a massive release and diffusion of most of the soluble nuclear constituents in the cytoplasm (1). At the same time, the typical progressive chromatin compaction, starting at the beginning of prophase (2), leads to the characteristic transcriptionally inactive status of condensed mitotic chromosomes. The faithful transmission and reactivation of genetic programs after mitosis is considered a key issue in regard to the epigenetic inheritance of the cellular identity.
While most nuclear factors dissociate from mitotic chromosomes, the observation that certain transcription factors remain associated with mitotic chromatin suggested the idea that mitotically retained factors could represent or deliver an epigenetic memory signal (bookmarking) throughout mitosis (3). In recent years, it has been demonstrated that the impairment of the function of specific bookmarking factors can result in delayed (4–7) or impaired (8) re-expression of target genes after a cell cycle. Relatively little is known about the mechanisms through which specific transcription factors are able to persist on mitotic chromatin, and thus act as bookmarking factors.
We have previously demonstrated that Hepatocyte Nuclear Factor 1 β (HNF1β) is a bookmarking factor (8). HNF1β (also known as vHNF1 or LF-B3) is a POU-homeobox transcription factor that is expressed since the first steps of kidney, pancreas and liver development (9–11). Mutations in HNF1B, the gene encoding for HNF1β, have been shown to be responsible for ‘Maturity Onset Diabetes of the Young 5’ (MODY5) (12), a monogenic form of type 2 diabetes. More recently, a number of studies have shown that mutations in HNF1B are the first genetic cause of Congenital Abnormalities of Kidney and Urogenital Tract (CAKUT) (13,14). With the use of mouse models, we have previously shown that the inactivation of HNF1β (Hnf1b) during the intense proliferation phase that accompanies the tubular elongation in kidneys of perinatal mice, results in severe polycystic disease. In this model, the formation of cysts is due to the loss of expression of several target genes that are often mutated in patients suffering from renal cysts (8,15,16). Interestingly, when Hnf1b is inactivated in quiescent renal cells (starting from postnatal-day 10) the expression of its typical cystic target genes is maintained at normal levels and animals do not develop cysts (8). However, HNF1β cystic target genes become transcriptionally silent when HNF1β-deficient cells are forced to pass through mitosis (8). The analysis of histone methylation and acetylation status in post-mitotic cells indicated that the chromatin of HNF1β target genes acquires heterochromatin marks (8). The observation that HNF1β proteins remain associated with mitotic chromatin indicated that HNF1β may act as a bookmarking factor. The mechanisms that drive the specific mitotic chromatin association of HNF1β are still poorly understood.
In the present study, we attempted to characterize the nature of the interactions that ensure the retention of HNF1β on mitotic chromatin. For this purpose, we characterized several mutations occurring in patients. Our results showed that the DNA binding ability of HNF1β is absolutely required for proper mitotic retention. Interestingly, we have shown that the loss of the mitotic binding of specific mutants is temperature-sensitive since a sudden drop of temperature can restore mitotic localization. In addition, we demonstrated that the mitotic localization is mediated by the importin-β system. Our results disclose an unexpected highly dynamic nature of the binding of HNF1β to mitotic chromatin.
MATERIALS AND METHODS
Cell culture
mIMCD3 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) medium (Life Technologies) supplemented with 10% fetal bovine serum (FBS) serum (Life Technologies) and 1% penicillin/streptomycin (Life Technologies) and transfected with JetPei (Polyplus Transfection). MDCK cells stably expressing inducible Nter-HNF1β-GFP wild-type and mutants were generated using a Tet-ON system. MDCK cells expressing the repressor pcDNA6/TR, a kind gift of Yasuyuki Fujita (University College, London, UK) (17), were maintained in DMEM medium containing 10% FBS serum, 1% pen/strep and blasticidin (5 μg/ml) (Sigma). pcDNA4/TO/HNF1b-GFP constructs were transfected in MDCK-pcDNA6/TR cells using JetPei. After selection with blasticidin (5 μg/ml) (Sigma) and zeocin (400 μg/ml) (Life Technologies), single clones were isolated using cloning cylinders (Fisher Scientific). GFP expression was induced by adding 1 μg/ml Doxycycline (Sigma) to the culture medium.
Plasmids
The sequence of oligonucleotide primers is indicated in Supplementary Table S1. For the construction of the Nter-HNF1β-GFP fusion plasmid (house name 10p5), the N-terminal part of the human HNF1β isoform A (amplification codon 1–313) was amplified by polymerase chain reaction (PCR) on the 6p15 plasmid, containing the human cDNA of HNF1B isoform A (18), with 10i1 and 10i2 primers. The amplicon was ligated with the pEGFP-N1 plasmid (Roche) after digestion by Asp718 and NheI. For constructing ΔHom (house name 10p3), a PCR was carried out on the 6p15 plasmid, containing the human cDNA of HNF1B isoform A (18), with the primers 10i1 and 10i3 (amplification from codon 1 to 236). The obtained amplicon was ligated with the pEGFP-N1 plasmid (Roche) after digestion by Asp718 and NheI. For the construction of ΔH3 (house name 10p7), a PCR was carried out on the 6p15 plasmid, containing the human cDNA of HNF1B isoform A (18) with the primers 10i1 and 10i4 (amplification from codon 1 to 291). The obtained amplicon was ligated with the pEGFP-N1 plasmid (Roche) after digestion by Asp718 and NheI. For the construction of the pcDNA4/TO/Nter-HNF1β-GFP (house name 11p40), the ORF of HNF1β-GFP from 10p5 into pcDNA4/TO was digested by NheI and NotI. Blunt ends were created with the Klenow polymerase. pcDNA4/TO was digested by HindIII and ApaI, and blunt ends were created by using the T4 DNA polymerase (Promega). After ligation and transformation in DH5alpha cells (Life technologies), clones were tested by PCR. Positives clones were sequenced on the full ORF of HNF1β-GFP. For the construction of Nter-HNF1α-GFP (house name 09p23), a PCR was carried out on the 09p19 plasmid, containing the human cDNA of HNF1A (isoform A) with the primers 10i5 and 10i6. The amplicon was ligated with the pEGFP-N1 plasmid (Roche) after digestion by Asp718 and NheI.
Mutagenesis
To introduce point mutations in Nter-HNF1β-GFP, we used the QuickChange II protocol (Agilent). In summary, 20 cycles of PCR were carried on pcDNA4TO/Nter-HNF1β with Pfu polymerase (Promega) and a specific set of primers (P256S: 11i122 and 11i123; V265L: 13i94 and 13i95; G287S: 12i64 and 12i65, Supplementary Table S1). Amplicons were ethanol-precipitated and digested over-night by 1 Unit of DpnI at 37°C, and then transformed into DH5alpha cells (Life technologies). Positive clones were sequenced on the full Nter-HNF1β-GFP ORF, and subcloned by a SpeI/BamHI digestion in the original pcDNA4TO/Nter-HNF1β.
Immunofluorescence
mIMCD3 cells were plated on coverslips in 6-well plates and fixed with 4% formaldehyde (Electron Microscopy Sciences) for 15 min at room temperature or with cold methanol for 15 min at −20°C. After fixation, cells were rinsed three times in phosphate buffered saline (PBS), blocked in 3% bovine serum albumin for 1 h at room temperature and incubated with HNF1b antibodies (sc22840, Santa Cruz, 1/1000) in blocking solution overnight at 4°C. For cells fixed in formaldehyde, a step of permeabilization (5 min with 0.2% Triton X-100) was performed before blocking. After washing in PBS, cells were incubated with secondary antibodies (Alexa Fluor donkey anti-rabbit 488, Life Technologies, 1/1000) for 1 h at room temperature, counterstained with DAPI (Life Technologies) and mounted with Fluoromount G (Interchim). MDCK cells over-expressing Nter-HNF1β-GFP wild-type and mutants were processed as above, except that no antibody incubation was performed. Confocal images were acquired with a Leica DMI6000 spinning disk using a CoolSNAP HQ2 camera (Photometrics).
Videomicroscopy
For live-cell imaging and time-lapse experiments, cells were grown on IBIDI dishes (35 mm, IBIDI), counterstained with Hoechst 33342 (Life Technologies) and imaged with an Axiovert 200M microscope (Zeiss) equipped with a CoolSNAP HQ camera (Photometrics) in a chamber maintained at 37°C and with 5% CO2. For temperature shift experiments, IBIDI dishes (35 mm) with 1 ml of medium at either 20°C or 37°C were left to reach thermic equilibrium and then imaged. Temperature shifts were obtained by adding 1 ml of medium at either 4°C or 50°C and rapidly mixed. Real-time temperature measurements were simultaneously obtained using three probes (Lm35dz) immersed in the cell culture medium and connected to a ATmega328P microcontroller (Arduino card). Importazole (50 μM) or vehicle (DMSO) (Sigma) were diluted in regular culture medium and added to the cells 20 min before cold shock.
Western blot
A total of 30 μg of nuclear extracts were run on 4–15% precast gels (Biorad), transferred on nitrocellulose membranes (GE Healthcare) and immunoblotted using antibodies against GFP at 1/5000 (ab290, Abcam), or Pol2 at 1/2000 (8WG16, BioLegend). Detection was performed with ECL Prime reagent (GE Healthcare) and the Fusion FX imaging system (Vilber Lourmat).
EMSA
Two or three micrograms of nuclear extracts prepared as described in (19) were used for each binding reaction, in 10 mM HEPES, 30 mM KCl, 5 mM DTT, Glycerol 10%, 9 mM Spermidine, 9 mM MgCl2 and variable amounts of 32P-labelled double stranded HNF1probe (CTTTAGTTAATATTTGACAGTTT/ AAACTGTCAAATATTAACTAAAG (20)), in a total volume of 20 μl. Homologous/Heterologous competition controls were realized by adding a 30-fold excess of double stranded cold HNF1 probe, or a double stranded oligonucleotide with an arbitrarily chosen non-related sequence (GGCTGAAGTCCAAAGTTCAGTCCCTTCGCTA/TAGCGAAGGGACTGAACTTTGGACTTCAGCC corresponding to the HNF4alpha binding site in the proximal promoter of Hnf1a), respectively. Binding reactions were incubated on ice or at 38°C for 15–20 min. Gels, 6% acrylamide-0.25× TBE, were pre-run at 6V/cm for 15–20 min, at 4°C or at 38°C. Binding reactions were loaded into the gels and run in an electric field of 6V/cm for 1 h. Gels were fixed in a 10% Ethanol 10% Acetic-Acid solution for 10 min, dried, exposed with a phosphor-imager screen and revealed with a Typhoon phosphor-imager scanner. For 4°C experiments, the binding reactions were established on ice, and the electrophoretic mobility shift assays (EMSAs) were carried out at 4°C, with pre-cooled acrylamide gels and running buffer. For 38°C experiments, the binding reactions were incubated at 38°C and the electrophoresis was carried out in an incubator at 38°C, with pre-heated acrylamide gels and running buffer. The intensity of the bands was measured with ImageJ. Saturation curves were analysed with GraphPad Prism version 6 (Graphpad software, La Jolla, CA, USA, www.graphpad.com).
Chromatin immunoprecipitation
MDCK cells stably expressing inducible Nter-HNF1β-GFP wild-type and mutants were induced with 50–100 ng/ml Doxycycline for 3 h and crosslinked with 0.5% Formaldehyde (Sigma) at 37°C for 10 min. Crosslinking was stopped by the addition of 125 mM Glycine (final concentration). After washing with ice-cold PBS, cells were collected in Lysis buffer (25 mM Tris–HCl pH 8.0, 2 mM ethylenediaminetetraacetic acid (EDTA), 150 mM NaCl, 1% Triton X100, 0.3% sodium dodecyl sulphate (SDS), Protease inhibitors (Roche)). Sonication was performed on a Bioruptor Pico (Diagenode). A total of 7–10 μg of chromatin were used for immunoprecipitation in ChIP buffer (25 mM Tris–HCl pH 8.0, 2 mM EDTA, 150 mM NaCl, 1% Triton X100, 0.1% SDS, protease inhibitors) with 2 μg of GFP antibody (ab290, Abcam) or rabbit IgG (sc-2027X, Santa Cruz), bound to 50 μl of Dynabeads protein A (Life Technologies), overnight at 4°C. Beads were washed with five successive different washing buffers (I:1% TRITON, 0.1% Sodium Deoxycholate, 150 mM NaCl, 10 mM TRIS–HCl pH8; II: 1% NP-40, 1% Sodium Deoxycholate, 150 mM KCl, 10 mM TRIS–HCl pH8; III: 0.5% TRITON, 0.1% Sodium Deoxycholate, 500 mM NaCl, 10 mM TRIS–HCl pH8; IV: 0.5% NP-40, 0.5% Sodium Deoxycholate, 250 mM LiCl, 20 mM TRIS–HCl pH8, 1 mM EDTA; V: 0.1% NP-40, 150 mM NaCl, 20 mM TRIS–HCl pH8, 1 mM EDTA) and twice with TE buffer (10 mM Tris–HCl pH8, 1 mM EDTA pH8). Beads were then incubated with Elution Buffer (1% SDS, 0.1M NaHCO3, 100 mM NaCl) for 1 h at room temperature and overnight at 65°C, after addition of 20 μg/ml RNase A, along with the Inputs. De-crosslinked extracts were treated with 100 μg/ml proteinase K at 55°C for 1–2 h. DNA was extracted with QIAquick PCR Purification Kit (Qiagen) and analysed by qRT-PCR with an MX3005P machine (Stratagene) using FastStart SYBR Green (Roche). Primer sequences are provided in Supplementary Table S2.
Primary urinary renal epithelial cell (UREC) culture preparation
Subjects included a patient carrying a heterozygous point mutation (c.[766C > T];[ = ] p.[Pro256Ser];[ = ]) in exon 3 of HNF1B gene and a healthy subject. The patient presented with normal-sized kidneys with bilateral cortical cysts and a normal glomerular filtration rate (14). Patient urine sample was collected after written informed consent (from patient and parents) through the Reference Center for Rare Diseases MARHEA (Maladies Rénales Héréditaires de l'Enfant et de l'Adulte), in the frame of our collection ‘Néphropathies héréditaires—Antignac/Saunier’ declared to the French Ministry of Research in March 2015 under the number DC 2014–2272 and which obtained an approval of the ethical Committee ‘CPP Ile-de-France II’ on 10 June 2015. The cells were prepared according to the protocol described in (21) and (22). After the second passage, cells were plated for the immunofluorescence experiments on 12 mm coverslips in two 24-well plates. After 2 days of culture, for the temperature shift experiments some plates were incubated at room temperature for 30 min and processed for immunostaining with methanol fixation as described above, along with the control plates (kept at 37°C).
Statistical analysis
All statistical tests (Mann–Whitney, paired t-test and Kruskal–Wallis) were performed on GraphPad Prism version 6 (Graphpad software, La Jolla, CA, USA, www.graphpad.com).
RESULTS
The DNA-binding domain of HNF1β is essential for mitotic chromatin association
HNF1β together with HNF1α constitute a digenic family of dimeric POU-Homeobox transcription factors, structured in three functional parts: an N-terminal dimerization domain, a DNA-binding domain and a C-terminal transactivation domain (Figure 1A). The DNA-binding domain consists of a POU-specific domain (POUs), composed of five α-helices and of a homeodomain (POUh) (23,24). We have previously shown that the N-terminal part of HNF1β (including the dimerization and the DNA-binding domain), when fused to GFP (Nter-HNF1β-GFP), is capable of driving a robust mitotic chromosomal localization ((8), Figure 1B and Supplementary Movie S1). The nature of the interaction of HNF1β with mitotic chromatin could have been potentially ascribed to a spurious association of the protein with the highly condensed mitotic chromatin structure, not necessarily relying on a functional DNA-binding capacity. In order to verify this possibility, we monitored the effect of deletions known to disrupt the ability of HNF1β to bind to DNA. First, we constructed a GFP-fusion version of HNF1β carrying a deletion of the entire homeodomain (ΔHom) (Figure 1C and Supplementary Figure S1). It is known that the homeodomain of HNF1β plays an essential role in DNA binding (23). This deletion did not perturb the sub-cellular localization of the GFP-fusion protein in interphase, suggesting that it retained an efficient nuclear localization signal. In fact, the fluorescence signal was tightly localized in the nucleus, to an extent that was perfectly comparable to the degree of nuclear localization of the wild-type GFP-fusion protein (Figure 1C). Using time-lapse microscopy, we showed that the ΔHom mutant suddenly becomes cytoplasmic in <30 s following the nuclear envelope breakdown at prometaphase (Figure 1C and Supplementary Movie S2). Next, we tested the effect of a more limited deletion specifically affecting the third helix of the homeodomain (ΔH3) (Supplementary Figure S1). This helix is known to specifically interact with nucleotides in the major groove of the DNA (23). Again, our results showed that the deletion of these 22 amino acids dramatically impaired mitotic chromatin-binding of the protein (Figure 1D and Supplementary Movie S3).
Figure 1.

Effect of deletion mutations of the DNA-binding domain on mitotic chromatin binding of HNF1β. (A) Scheme of the structural organization of HNF1β. DIM: dimerization domain, POUs: POU-specific domain, POUh: homeodomain. The coordinates of the amino-acid sequence are indicated in the scheme. (B–D) Time-lapse microscopy of cells expressing Nter-HNF1β-GFP wild-type (in MDCK) (B) and ΔHOM-GFP (C), or ΔH3-GFP (D) in IMCD3 transfected cells. For each fusion protein, green fluorescence (GFP) and phase contrast (PC) are shown for crucial time points of mitosis. NEB is the moment of the nuclear envelope breakdown. Time (in minutes) is indicated for each frame at the bottom right corner. White arrows indicate the cell undergoing mitosis, and the nuclei of the daughter cells after cytokinesis. Scale bars, 10 μm.
In order to verify that the observed mitotic localization was also a characteristic of the endogenously expressed protein, we monitored the sub-cellular localization of the endogenously expressed HNF1β in renal cell lines (Figure 2). Surprisingly, our results showed that in formaldehyde-fixed cells, the immunofluorescence signal corresponding to the endogenous protein was completely excluded from the mitotic chromatin (Figure 2B, upper panel). This apparent discrepancy led us to assess the possible effect of fixation on the mitotic localization of the Nter-HNF1β-GFP. Indeed, we found that when cells expressing the GFP-fusion protein were fixed under the same conditions (4% formaldehyde fixation for 15 min at room temperature), we observed the systematic exclusion of the GFP signal from mitotic chromosomes and dispersal of the protein in the cytoplasm (Figure 2B, lower panel). This suggested that a formaldehyde-based fixation disrupted the association of the protein with the chromosomes during mitosis. We therefore tried to find an alternative fixation protocol that could preserve the localization of Nter-HNF1β-GFP as observed in live cells. Interestingly, methanol fixation (at either −20°C or room temperature for 15 min) led to the complete preservation of the mitotically-bound HNF1β, either in fusion with the GFP or in cell lines that express HNF1β endogenously (Figure 2C).
Figure 2.

Effect of fixation on the mitotic localization of HNF1β. (A) Mitotic localization of Nter-HNF1β-GFP fusion protein detected through the fluorescence of the GFP moiety with confocal microscopy in live MDCK cells. (B) Effect of formaldehyde (4%) fixation on the mitotic localization of HNF1β. Shown above is the endogenously expressed HNF1β in IMCD3 cells detected with an HNF1β specific antibody. Shown below is the GFP fluorescence of the Nter-HNF1β-GFP fusion protein in MDCK cells. (C) Effect of methanol fixation. Shown above is the localization of the endogenously expressed HNF1β in IMCD3 cells detected with an HNF1β specific antibody. Shown below is the confocal GFP fluorescence of the Nter-HNF1β-GFP fusion protein in MDCK cells. DNA (red signal) was stained with DAPI (fixed cells) or Hoechst 33 342 (live cells). White arrows indicate the position of the mitotic cells. Scale bars, 10 μm.
All these results suggest that a minimal mitotic-module of HNF1β corresponding to the first 313 amino-acids in the N-terminal part contains a DNA-binding domain that is absolutely required for mitotic chromosomal binding. In addition, the endogenously expressed full-length protein (in renal cells) follows the same behaviour than that of GFP fusion constructs.
The impact of human MODY mutations on mitotic chromatin binding
One of the possible difficulties in the interpretation of results obtained with proteins carrying deletions is that these mutations may dramatically modify the global structure of the protein to an extent that could have prevented their ability to bind mitotic chromatin, independently from the loss of their specific DNA binding capacity. In order to take into account this consideration, we decided to assess mitotic binding of HNF1β protein by examining single amino-acid substitutions. The genes encoding for HNF1α and HNF1β (HNF1A and HNF1B, respectively) are frequently mutated in diabetic and renal cystic patients (12,14,25,26). The vast majority of these mutations is likely to encode for hypomorphic or null variants, since the phenotype of the patients carrying these mutations resembles that presented by mouse models carrying null mutations of Hnf1a and Hnf1b, respectively. Therefore, we may reasonably assume that the mutations observed in these patients might imply the impairment of some functions. A considerable number of these mutations (23,24,27,28) are localized in the minimal mitotic module described above, conferring chromosomal mitotic binding to GFP-fusion proteins. HNF1α and HNF1β share a particularly strong homology in this functional domain with 62% identity and 80% similarity. In addition, the three-dimensional structure of these two proteins, in this domain, is almost identical given the fact that the protein structures superimpose very well with a root-mean-square deviation of atomic positions in the backbone atoms in the order of only 1 Angström (23,24). Furthermore, in HNF1β-deficient embryonic stem cells, the forced expression of HNF1α rescues the function played by HNF1β (29), suggesting that the two transcription factors may share common specific binding sites on the genome. In fact, in EMSAs, both HNF1α and HNF1β bind to a common consensus motif with similar affinities (9). To further assess the functional similarity of the two proteins, we monitored the capacity of HNF1α to bind to mitotic chromatin. As previously described (30), we confirmed that when fused to the GFP, the corresponding minimal N-terminal functional mitotic binding module of HNF1α had an intrinsic defective nuclear localization in interphase cells (Supplementary Figure S2). In fact, in interphase cells, a considerable amount of the protein was localized in the cytoplasm. Nevertheless, upon nuclear envelope breakdown, we could not detect any significant increase in the intensity of the signal in the cytoplasm. When we inspected the possible association with chromosomal mitotic barrels, we could systematically see a clear association of HNF1α-GFP with mitotic chromatin indicating that the corresponding minimal mitotic-module of HNF1α shares with HNF1β the ability to remain associated to mitotic chromatin (Supplementary Figure S2).
Given the fact that a considerable number of MODY3 and MODY5 mutations map to this functional mitotic-module, it was interesting to investigate if some of them may have impaired mitotic binding. In order to test this possibility we selected, from the pool of known mutations that occur in MODY3 and MODY5 patients, amino-acid substitutions that involved residues particularly well conserved in both HNF1A and HNF1B and across different species throughout evolution, and that were not necessarily clearly involved in DNA binding. Then, we introduced by directed mutagenesis the amino-acid substitutions corresponding to the selected human mutations in the minimal mitotic module of Nter-HNF1β-GFP fusion that, per se, has a very robust basal interphase nuclear localization. We then assessed the effect of these mutations on the ability of Nter-HNF1β-GFP fusion protein to bind to mitotic chromatin. The first two mutations that we tested (G255S (28) and V233L (31)) were identified in MODY3 (HNF1α) patients and correspond to positions G287S and V265L in HNF1β, respectively. The third one (P256S) was found in an HNF1B patient presenting bilateral cortical renal cysts (14). Our results showed that the three mutant proteins were efficiently localized to the nucleus in interphase cells (Figure 3). During mitosis, the mutation G287S did not strongly impair mitotic localization whereas the other two (P256S and V265L) were completely excluded from mitotic chromatin, indicating that they drastically impaired mitotic binding (Figure 3).
Figure 3.
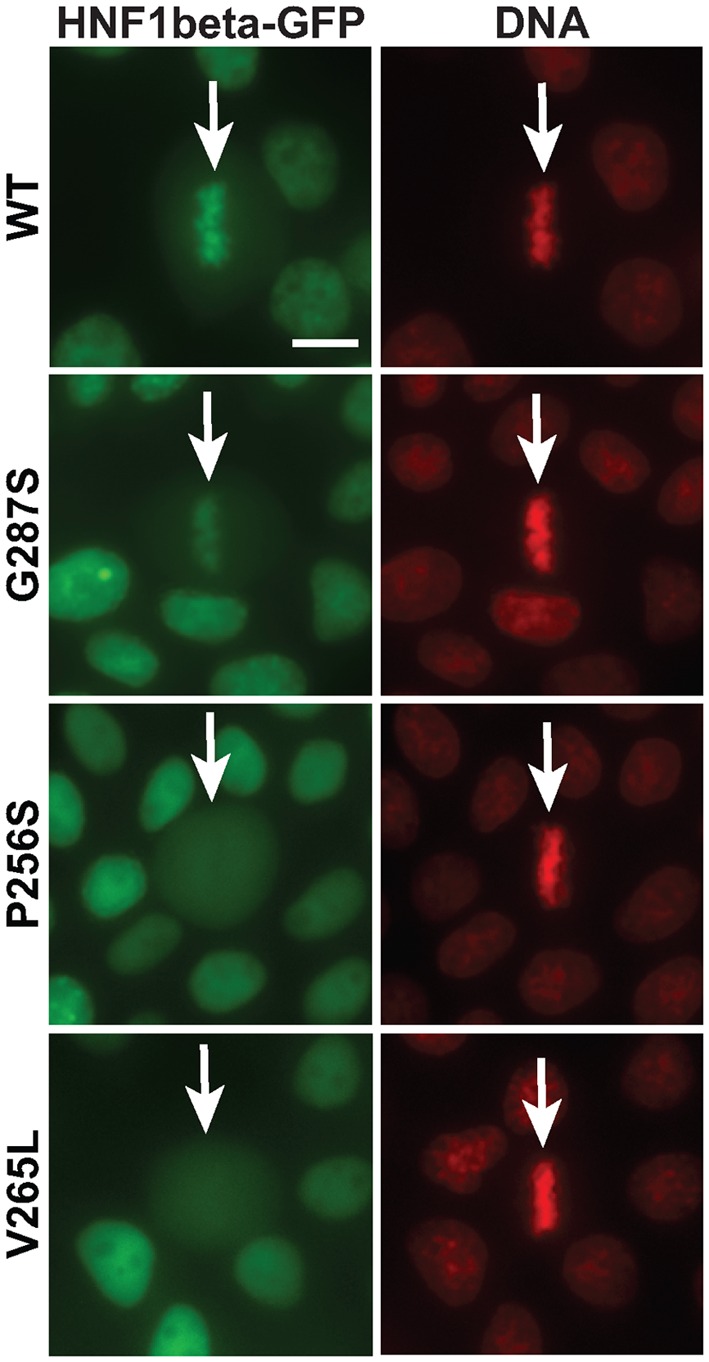
Mitotic chromatin localization defect of MODY mutants. Live imaging of MDCK cells expressing Nter-HNF1β-GFP (WT or MODY mutants) counterstained with Hoechst 33342 (red signal, DNA). White arrows indicate the position of the mitotic cells. Scale bar, 10 μm.
Thermosensitivity of MODY mutants
Strikingly, we discovered that the extent of mitotic chromatin binding of P256S and V265L mutant proteins was significantly affected by the temperature conditions in which the cells were set. The extent of mitotic chromatin retention of the wild-type Nter-HNF1β-GFP fusion protein and of the G287S mutant was essentially unaltered over a relatively wide range of temperatures with chromosomal barrels intensely decorated by the HNF1β fusion protein (Supplementary Figures S3 and 4). On the other hand, P256S and V265L mutant fusion proteins showed a marked decreased binding to mitotic chromatin with the increase in temperature. For example, the P256S mutant protein was completely dissociated from mitotic chromatin at temperatures above 30–32°C (restrictive temperature) (Figure 4A). When tested at temperatures below these critical values (permissive temperature), P256S mutant protein was associated with mitotic chromatin (Figure 4A). The V265L mutant followed the same transition, with a higher temperature switch around 36–37°C (Figure 4B). This striking effect of temperature transition allowed us to unmask an unpredicted high degree of rapid re-localization of mutant fusion proteins, upon abrupt temperature shifts. In fact, our live imaging experiments (with the use of in situ temperature sensors following the temperature changes induced by the addition of cold or hot medium) indicated that thermo-sensitive mutant proteins rapidly re-localized to mitotic chromatin after a sudden decrease of temperature (Figure 4A and B), or were dispersed in the cytoplasm after a rapid increase of temperature (Figure 4C and D). It is worth noting that when cells over-expressing these temperature-sensitive mutations were grown at restrictive temperature, the localization of the signal was perfectly nuclear (Figure 4A and B). With the use of the ‘Intensity Correlation Quotient’ (ICQ) method (32), we could measure the average extent of mitotic binding of these mutants. Our results showed that on average, both P256S and V265L mutants exhibited a significantly lower ICQ (at 37°C), in respect to wild-type Nter-HNF1β-GFP or the G287S mutant, confirming a drastic mitotic delocalization (Figure 4E). On the other hand, at room temperature (25°C) we could not see any significant difference of ICQs between WT and mutant Nter-HNF1β-GFP (Figure 4F). In order to validate the pertinence of these observations we decided to assess the mitotic binding ability of HNF1β directly in renal cells from a CAKUT patient carrying a heterozygous (P256S) mutation (14). To this end, we established primary urinary renal epithelial cell cultures (URECs) and assessed the mitotic localization of the endogenously expressed HNF1β protein, presumably composed by a mixture of heterodimers (WT:P256S) and homodimers (WT:WT and P256S:P256S). Immunofluorescence on mitotic patient cells cultured at restrictive temperature (37°C) showed a modest but significant and visible delocalization of HNF1β signal in the cytoplasm, compared to the healthy control cells (Figure 5A and C). The rather modest entity of the delocalization defect is probably related to the fact that this mutation is heterozygous. When these cells were incubated at permissive temperature (25°C) for 30 min before immunofluorescence, HNF1β signal was observed bound to mitotic chromosomes similarly to what is observed in the URECs from a healthy control (Figure 5B and D).
Figure 4.

MODY mutations are thermosensitive. (A–D) Effect of abrupt temperature variation on the mitotic chromatin localization of MODY mutations in Nter-HNF1β-GFP fusion protein (green signal) expressed in MDCK cells (red signal, DNA). The temperature in the medium was measured with three independent probes, as shown by the temperature curves. Black arrows indicate the moment in which the picture was taken. P256S and V265L mutants, when grown at restrictive temperature, were dissociated from mitotic chromatin. However, they rapidly re-associated to mitotic chromatin upon sudden temperature drop (panels A and B). Conversely, they rapidly dissociated from mitotic chromatin when temperature was rapidly raised (panels C and D). White arrows indicate the position of the mitotic cells. Scale bars, 10 μm. (E and F) Quantification of the ‘Intensity Correlation Quotient’ (ICQ) between Hoechst and GFP signals at 37°C (panel E) or at room temperature (panel F), in mitotic MDCK cells stably expressing Nter-HNF1β-GFP WT or mutants. WT 37°C, n = 34; G287S 37°C, n = 32; P256S 37°C, n = 32; V265L 37°C, n = 35; WT 25°C, n = 30; G287S 25°C, n = 34; P256S 25°C, n = 26; V265L 25°C, n = 31. Statistical significance was determined by Kruskal–Wallis test followed by Dunn's multiple comparisons test. ***P < 0.0001.
Figure 5.

Primary urinary renal epithelial cells (URECs) from a healthy subject (WT) and a patient carrying a heterozygous mutation of P256S in HNF1B gene. (A) Mitotic localization of endogenous HNF1β (green) in heterozygous (P256S) or WT URECs at 37°C. (B) Mitotic localization of endogenous HNF1β (green) in heterozygous P256S or WT URECs at low temperature (25°C). Scale bar, 10 μm. (C and D) Quantification of the ‘ICQ’ between DAPI and GFP (HNF1β) signals at 37°C (panel C) or at 25°C (panel D), in mitotic URECs. Statistical significance between WT (n = 35) and P256S (n = 24) at 37°C, and between WT (n = 14) and P256S (n = 16) at 25°C was determined by Kruskal–Wallis test followed by Dunn's multiple comparisons test. *P < 0.05.
Temperature shifts affect DNA binding ability
The profound effect of temperature transition on mitotic binding indicated that these mutations may induce temperature-dependent conformational changes that impair mitotic chromatin binding. In order to monitor if these putative conformational changes might have directly affected DNA binding, we evaluated the ability of mutant proteins to bind DNA by EMSA in temperature-controlled conditions. We optimized a protocol for EMSA to monitor DNA-binding saturation curves for Nter-HNF1β-GFP wild-type and mutants in nuclear extracts from stably transfected inducible MDCK cells. Our results showed that when the DNA–protein complexes were electrophoretically separated at low temperature (4°C), the sequence-specific DNA binding characteristics of P256S and V265L were comparable to that of the wild-type protein (Figure 6A). However, when the same extracts were incubated and electrophoretically separated at restrictive temperature conditions (38°C), a drastic decrease in specific shifts could be observed for these two mutants compared to the control wild-type protein (Figure 6A). These defects were confirmed by the comparison of saturation curves (Figure 6B). The observed drastically reduced binding was not due to degradation of the proteins since western blot experiments showed that all these extracts (heated or not) contained equivalent amount of HNF1β protein compared to controls (Figure 7A). Moreover, when nuclear extracts were temporarily incubated at 38°C for 20 min and then cooled down at 4°C, mutant proteins recovered their binding activity since they gave rise to shifts comparable to those obtained with the wild-type protein (Figure 7B). Interestingly, the peculiar reversible aspect of the binding of the MODY mutants to DNA was a dynamic and reversible phenomenon also in live cells. In fact, the fluorescence of the GFP fusion protein in live cells consistently dissociated from mitotic chromosomes upon a rise of the temperature and systematically re-associated to mitotic chromosomes when the temperature was again set to a lower level (Figure 7C and Supplementary Movie S4).
Figure 6.

Thermo-sensitivity of MODY mutants is linked to a switch in DNA binding ability. (A) Electrophoretic mobility shift assays (EMSA) experiments using 2 μg of nuclear extracts from MDCK cells stably expressing Nter-HNF1β-GFP WT or mutants, and variable amounts of double stranded oligonucleotide probe (from 0.4 to 100 nM), at the indicated temperature. Probe: migration of the radioactive probe in the absence of protein. Nc: non-specific competition, with a 30-fold excess of cold arbitrary and unrelated probe. Sc: specific competition, with a 30-fold excess of cold HNF1 probe. TOP: position of the well. S: specific band-shift. NS: non-specific band-shift. F: free probe. (B) Saturation curves established after signal measurement on ImageJ.
Figure 7.

The switch in DNA binding is a reversible phenomenon. (A) Western blot analysis of nuclear extracts from MDCK cells expressing Nter-HNF1β-GFP WT or mutants upon doxycycline induction. Protein extracts were incubated on ice, or at 38°C, for 20 min, then directly resuspended in Laemmli Buffer and boiled for 5 min. The negative control corresponds to nuclear extract of wild-type MDCK cells (NT). (B) EMSA using 3 μg of nuclear extracts, and different amounts of radioactive HNF1 probe (between 12.5 and 100 nM). Binding reactions were carried out at 38°C. Half of the binding reaction of the P256S was electrophoretically separated 38°C, while the second half was incubated on ice for 20 min and then separated at 4°C. (C) Live imaging of MDCK cells stably expressing Nter-HNF1β-GFP P256S (green signal) submitted to a first cold shock (COLD SHOCK 1), left to heat up to 37°C and submitted to a second cold shock (COLD SHOCK 2). White arrows indicate the position of the mitotic cells. Time (in minutes) is indicated for each frame at the bottom right corner. Scale bar: 10 μm.
All these results suggest that at restrictive temperature, P256S and the V265L mutant proteins undergo a reversible conformational change that prevents DNA binding. Similar to the phenotypic effect of ΔHom or ΔH3 deletion mutations, the loss of the ability to bind DNA leads to a sudden loss of mitotic chromatin binding. When the conformational change due to an increase of the temperature is induced, it takes only few seconds for the protein to be dispersed in the cytoplasm and, conversely, only few seconds to re-associate to mitotic chromatin when rapidly cooled down.
In order to verify if these mutants were able to bind chromatin, we performed ChIP assays on stably transfected inducible MDCK cells. We identified HNF1 binding sites that are highly conserved throughout evolution using a previously described in silico approach (33). Our results demonstrated that the mutant proteins had only a slightly decreased binding activity. In fact, in spite of the drastic defect of their DNA binding activity in EMSA, the two mutants were still able to bind the promoters of two of HNF1β's target genes, PKHD1 and HNF1B itself (Supplementary Figure S5).
HNF1β mitotic re-association is impaired by importazole
The thermosensitive nature of the phenotype observed with these MODY mutant proteins allowed us to characterize some critical aspects concerning the chromatin recruitment of HNF1β to mitotic chromatin. In particular, the extreme rapidity of the thermosensitive re-association of MODY mutant proteins suggested that such a translocation might be mediated by an active process. It is known that the Ran-GTP/Ran-GDP cycle plays an essential role in the cytoplasmic to nuclear transport by regulating the interactions between cargos and nuclear transport receptors including, for example, those of the importin-β family (34,35). The common principle underlying this function is the anisotropy of the distribution of the Ran-GTP gradient driven by the chromatin associated guanine nucleotide exchange factor RCC1. Interestingly, it has been shown that RCC1 and Ran-importin-β system also plays an important role in the spatio-temporal regulation of mitosis, in particular concerning mitotic-spindle assembly factors (36). We therefore inspected the effect of the inhibition of the importin-β system on the recruitment of HNF1β mutant fusion protein to mitotic chromatin with the use of importazole, a specific inhibitor of importin-β (37). In order to avoid the toxic effect of importazole (on nuclear compartmentalization and possibly on cell cycle), we treated our cells for a relatively short period of time (20 min) before observation, to be able to monitor cells that had already been engaged in mitosis. Our results showed that such a short treatment did not interfere with the nuclear localization of either wild-type or mutant HNF1β protein in interphase (Supplementary Figure S6). However, the mitotic chromatin re-association of the P256S mutant, after cold shock, was dramatically delayed and globally decreased in its extent, indicating that the importin-β system seems to be involved in the mitotic chromatin recruitment of our GFP fusion protein (Figure 8A). In fact, the statistical analysis of our results showed a very significantly decreased ICQ in importazole treated cells (Figure 8B).
Figure 8.

Importazole affects the dynamic relocalization of P256S mutant on mitotic chromatin. (A) Representative images of MDCK cells expressing the P256S mutant stained with Hoechst (red signal, DNA) submitted to a cold shock, in the presence or absence of importazole. (B) Quantification of the difference of the Intensity Correlation Quotient (ΔICQ) between Hoechst and GFP signal before and after cold shock, in cells treated or not with importazole. Images after cold-shock were taken 1 min after addition of cold medium. White arrows indicate mitotic cells. Scale bars: 10 μm. Results are represented by mean ± SEM. Statistical significance between ΔICQ for DMSO (n = 27) and Importazole (n = 30) treated cells was determined by a paired t-test. **P < 0.001.
DISCUSSION
In recent years, the study of the functions played by transcription factors has disclosed some novel crucial aspects on the epigenetic control of gene expression. Several transcription factors were shown to be involved in the mitotic transmission of epigenetic information. Mitosis is characterized by a dramatic reorganization of the nuclear structure. The typical progressive chromatin condensation is accompanied by the loss of nuclear envelope integrity. This leads to the cytoplasmic diffusion of many nuclear components, including most transcription factors that dissociate from the typically condensed mitotic chromatin. These phenomena lead to a genome wide silencing of gene expression. After mitosis, daughter cells must faithfully reactivate their genetic programs in order to ‘remember’ and preserve their identity. Interestingly, some transcription factors have the ability to remain bound to mitotic chromatin and play important roles in the epigenetic transmission of regulatory information through mitosis. This concept has been defined as ‘mitotic bookmarking’. However, relatively little is known about the way bookmarking factors vehicle this form of epigenetic transmission. In particular, the molecular mechanisms that endow bookmarking transcription factors their ability to remain associated to mitotic chromatin remain poorly understood. Our previous studies have shown that the transcription factor HNF1β acts as a bookmarking factor. Indeed, HNF1β is capable of binding to mitotic chromatin and reactivating the expression of specific target genes after mitosis (8).
In the present study, we have demonstrated that the binding of HNF1β to mitotic chromatin requires an efficient DNA binding ability. In fact, a series of deletions, known to prevent DNA binding, significantly impairs this function. Interestingly, specific point mutations leading to residue substitutions (P256S and V265L), responsible for loss of function phenotypes in MODY3 and MODY5 patients, abrogate DNA binding and as a consequence, mitotic chromatin binding. All these results indicate that the mitotic chromatin localization of HNF1β relies on its specific ability to bind to DNA. Interestingly, the analysed mutant proteins (P256S and V265L) are characterized by thermo-sensitive phenotypes. They bind to DNA only at temperatures below specific critical threshold. These observations indicate that these mutant proteins may exist in two alternative conformations with drastically different DNA binding capabilities that can rapidly interconvert one another according to the temperature.
In this respect, we were able to show that primary URECs from an HNF1B patient carrying a heterozygous P256S mutation displayed a slight but significant decrease of HNF1β mitotic localization. Moreover, this defect was rescued upon temperature decrease, further confirming the results we obtained with the mutant HNF1β GFP-fusion proteins.
Our ChIP assay experiments showed that HNF1B mutants were still able to bind the promoters of target genes although to a slightly lesser extent compared to the wild-type protein. In this regard, we cannot completely rule out that the adopted experimental conditions for ChIP were not able to prevent the conformational change that allows the mutant GFP-fusion proteins to bind DNA at lower temperatures. In fact, given the rapidity that characterizes this binding switch/relocalization phenomenon we cannot exclude that during the crosslinking step mutant proteins might have suddenly recovered their permissive conformation. In addition, it has also been recently reported that pioneer transcription factors can target nucleosomes by recognizing only partial DNA motifs (38). Thus, we cannot rule out that MODY mutants with decreased DNA binding abilities, as the ones described in this study, might still be able to associate to DNA in a chromatin context, at least in interphase chromatin.
The temperature sensitivity of our mutant proteins allowed us to probe and assess their in vivo behaviour upon sudden loss of DNA binding. Our results showed that mitotic chromatin binding during mitosis can be modulated in a very highly dynamic way. In fact, it takes <1 min to completely disperse a mutant protein in the cytoplasm if the temperature is suddenly raised above a critical threshold. This suggests that when DNA binding is suddenly compromised, HNF1β very rapidly diffuses in the surrounding cytoplasm. On the other hand, a rapid temperature drop, converting the mutant HNF1β into a DNA binding compatible conformation, leads to a surprising massive mitotic chromatin recruitment and localization in <1 min.
Such a prompt response in localization might rely on very efficient translocation machineries. Interestingly, our results have demonstrated that this translocation can be very efficiently inhibited, in already engaged mitotic cells, by a short-lived (20 min) treatment with importazole, a known inhibitor of the importin-β machinery. This observation indicates that mitotic chromatin localization of bookmarking factors is probably helped by the importin system known to be based on a Ran-GDP/Ran-GTP gradient that is maintained around mitotic chromatin during mitosis (36). It is worth to note that importin-β inhibition, per se, is not able to prevent wild-type HNF1β from binding mitotic chromatin, indicating that when the protein is solidly bound, importin-β inhibition is not sufficient to displace the protein from mitotic chromosomes. All these observations indicate that cells are genetically programmed with a system to ensure the transport of HNF1β to mitotic chromatin.
Recent studies have indicated that bookmarking factors can be associated to mitotic chromatin in two distinct ways. The first one, DNA-sequence-specific, seems to account for the association of transcription factors to precise locations on the genome. The second one, non-sequence specific, seems to ensure that bookmarking factors stay enriched in close proximity to mitotic chromatin throughout the whole cell division process (5). Our results seem to indicate that the action of bookmarking factors might be mediated by a two-step process (Figure 9). In the first step, an importin-β-dependent translocation would ensure that bookmarking factors are translocated into the proximity of mitotic chromatin. This enrichment is based on the indirect active transport/translocation dependent on the importin-β and on the typical RanGTP-RanGDP gradient/hydrolysis cycle that characterizes mitotic chromosomes (39). The second step, requiring the sequence specific DNA binding activity of bookmarking transcription factors, would allow the bookmarking of chromatin territories of specific target genes. This association, thermodynamically more stable, would explain the fact that once associated to DNA, HNF1β, for example, can remain associated to mitotic chromatin for relatively long time without requiring the importin-β translocation system (Supplementary Figure S6). On the other hand, when the DNA binding activity is suddenly inactivated, the very abundant inducible expression of the GFP-fusion mutant HNF1β proteins, at restrictive temperatures, might possibly rapidly dissipate the GTP/GDP gradient through a futile cycle of translocation/diffusion process that eventually would disperse the GFP fusion protein throughout the entire cytoplasm of mitotic cells. One of the possible consequences of this phenomenon is the fact that our inducible expression system is highly toxic since induced cells cannot survive the expression of the HNF1β-GFP fusion proteins more than 2–3 days.
Figure 9.

Model of association of HNF1β to mitotic chromatin. The localization of HNF1β on mitotic chromatin may be considered to be based on a two-step process. In the first step, RAN-GTP gradient and Importin-β system could bring HNF1β in close proximity to mitotic chromatin. In the second step, the DNA-binding ability of HNF1β could provide a more intimate and stable association of the protein with mitotic chromatin.
The discrepancy between the localization of HNF1β GFP-fusion proteins in live cells versus that observed in formaldehyde-fixed cells has disclosed the artefactual impact of this type of fixation in the study of subcellular localization of transcription factors during mitosis. It has already been suggested that formaldehyde crosslinking may not be necessarily appropriate for the study of chromatin binding of specific set of proteins (40). Concerns about formaldehyde crosslinking have also been previously raised in the case of chromatin crosslinking experiments (41). In the case of bookmarking factors, similar discrepancies have already been described. For example, GATA1, a bookmarking transcription factor involved in erythroid cell specification, shows retention on mitotic chromosomes in live cell-imaging (GFP fusion protein and ChIP experiment) whereas upon Paraformaldehyde (PFA) fixation the protein is globally dispersed in the cytoplasm during mitosis (6). In a similar way, FOXA1, a hepatocyte transcription factor with bookmarking activity, shows the same discrepancy: FOXA1 in fusion with GFP is perfectly localized on mitotic chromatin in live cells, whereas when fixed with formaldehyde, the endogenously expressed protein exhibits a diffused dispersion in indirect immunofluorescence experiments (5). The discovery of alternative fixation methodologies that can preserve mitotic localizations could bring novel insights in the study of binding of transcription factors to mitotic chromatin.
In conclusion, our results highlight the crucial role played by the DNA binding ability of HNF1β in the interaction with mitotic chromatin. In this context, the importin-β system is likely to be involved in the maintenance and collection of HNF1β protein to mitotic chromatin. More importantly, our studies have provided experimental evidence that defective mitotic chromatin binding may be responsible for human pathological conditions.
Supplementary Material
Acknowledgments
We thank Yasuyuki Fujita of University College (London, UK) for kindly providing us MDCK cells stably expressing the repressor pcDNA6/TR. We are grateful to Rémi Salomon for helpful suggestions and for providing clinical data on CAKUT patients. We also thank Moshe Yaniv, Fabiola Terzi and the members of the Pontoglio lab, for helpful discussions and critical reading of the manuscript. We are very grateful to the platform of ‘Imagerie Cellulaire’ of the Cochin Institute.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
‘Fondation pour la Recherche Médicale’ [équipe FRM DEQ2012032376]; Fondation Bettencourt-Schueller (Prix Coup d'Elan); European Community's Seventh Framework Programme FP7/2009 [241955, SYSCILIA]; Trancyst ITN; ‘Agence Nationale pour la Recherche’ and the ‘Who Am I?’ Laboratory of Excellence; ‘Ecole Normale Supérieure de Cachan’ Fellowship (to J.L.); ‘Ligue contre le Cancer’ Fellowship (to J.L.); ‘Fondation pour la Recherche Médicale’ Fellowship (to A.B.); ‘Association pour la Recherche sur le Cancer’ (ARC) Fellowship (to M.P.M.). Funding for open access charge: ‘Fondation pour la Recherche Médicale’.
Conflict of interest statement. None declared.
REFERENCES
- 1.Egli D., Birkhoff G., Eggan K. Mediators of reprogramming: transcription factors and transitions through mitosis. Nat. Rev. Mol. Cell Biol. 2008;9:505–516. doi: 10.1038/nrm2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vagnarelli P. Mitotic chromosome condensation in vertebrates. Exp. Cell Res. 2012;318:1435–1441. doi: 10.1016/j.yexcr.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 3.Zaidi S.K., Young D.W., Montecino M.A., Lian J.B., van Wijnen A.J., Stein J.L., Stein G.S. Mitotic bookmarking of genes: a novel dimension to epigenetic control. Nat. Rev. Genet. 2010;11:583–589. doi: 10.1038/nrg2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blobel G.A., Kadauke S., Wang E., Lau A.W., Zuber J., Chou M.M., Vakoc C.R. A reconfigured pattern of MLL occupancy within mitotic chromatin promotes rapid transcriptional reactivation following mitotic exit. Mol. Cell. 2009;36:970–983. doi: 10.1016/j.molcel.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caravaca J.M., Donahue G., Becker J.S., He X., Vinson C., Zaret K.S. Bookmarking by specific and nonspecific binding of FoxA1 pioneer factor to mitotic chromosomes. Genes Dev. 2013;27:251–260. doi: 10.1101/gad.206458.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kadauke S., Udugama M.I., Pawlicki J.M., Achtman J.C., Jain D.P., Cheng Y., Hardison R.C., Blobel G.A. Tissue-specific mitotic bookmarking by hematopoietic transcription factor GATA1. Cell. 2012;150:725–737. doi: 10.1016/j.cell.2012.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao R., Nakamura T., Fu Y., Lazar Z., Spector D.L. Gene bookmarking accelerates the kinetics of post-mitotic transcriptional re-activation. Nat. Cell Biol. 2011;13:1295–1304. doi: 10.1038/ncb2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verdeguer F., Le Corre S., Fischer E., Callens C., Garbay S., Doyen A., Igarashi P., Terzi F., Pontoglio M. A mitotic transcriptional switch in polycystic kidney disease. Nat. Med. 2010;16:106–110. doi: 10.1038/nm.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rey-Campos J., Chouard T., Yaniv M., Cereghini S. vHNF1 is a homeoprotein that activates transcription and forms heterodimers with HNF1. EMBO J. 1991;10:1445–1457. doi: 10.1002/j.1460-2075.1991.tb07665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Simone V., De Magistris L., Lazzaro D., Gerstner J., Monaci P., Nicosia A., Cortese R. LFB3, a heterodimer-forming homeoprotein of the LFB1 family, is expressed in specialized epithelia. EMBO J. 1991;10:1435–1443. doi: 10.1002/j.1460-2075.1991.tb07664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coffinier C., Thépot D., Babinet C., Yaniv M., Barra J. Essential role for the homeoprotein vHNF1/HNF1β in visceral endoderm differentiation. Development. 1999;126:4785–4794. doi: 10.1242/dev.126.21.4785. [DOI] [PubMed] [Google Scholar]
- 12.Horikawa Y., Iwasaki N., Hara M., Furuta H., Hinokio Y., Cockburn B.N., Lindner T., Yamagata K., Ogata M., Tomonaga O., et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat. Genet. 1997;17:384–385. doi: 10.1038/ng1297-384. [DOI] [PubMed] [Google Scholar]
- 13.Ulinski T., Lescure S., Beaufils S., Guigonis V., Decramer S., Morin D., Clauin S., Deschênes G., Bouissou F., Bensman A., et al. Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. J. Am. Soc. Nephrol. 2006;17:497–503. doi: 10.1681/ASN.2005101040. [DOI] [PubMed] [Google Scholar]
- 14.Heidet L., Decramer S., Pawtowski A., Morinière V., Bandin F., Knebelmann B., Lebre A.-S., Faguer S., Guigonis V., Antignac C., et al. Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin. J. Am. Soc. Nephrol. 2010;5:1079–1090. doi: 10.2215/CJN.06810909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gresh L., Fischer E., Reimann A., Tanguy M., Garbay S., Shao X., Hiesberger T., Fiette L., Igarashi P., Yaniv M., et al. A transcriptional network in polycystic kidney disease. EMBO J. 2004;23:1657–1668. doi: 10.1038/sj.emboj.7600160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ibraghimov-Beskrovnaya O., Bukanov N. Polycystic kidney diseases: from molecular discoveries to targeted therapeutic strategies. Cell. Mol. Life Sci. 2008;65:605–619. doi: 10.1007/s00018-007-7362-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hogan C., Dupré-Crochet S., Norman M., Kajita M., Zimmermann C., Pelling A.E., Piddini E., Baena-López L.A., Vincent J.-P., Itoh Y., et al. Characterization of the interface between normal and transformed epithelial cells. Nat. Cell Biol. 2009;11:460–467. doi: 10.1038/ncb1853. [DOI] [PubMed] [Google Scholar]
- 18.Bach I., Yaniv M. More potent transcriptional activators or a transdominant inhibitor of the HNF1 homeoprotein family are generated by alternative RNA processing. EMBO J. 1993;12:4229–4242. doi: 10.1002/j.1460-2075.1993.tb06107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schreiber E., Matthias P., Müller M.M., Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomei L., Cortese R., De Francesco R. A POU-A related region dictates DNA binding specificity of LFB1/HNF1 by orienting the two XL-homeodomains in the dimer. EMBO J. 1992;11:4119–4129. doi: 10.1002/j.1460-2075.1992.tb05505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou T., Benda C., Dunzinger S., Huang Y., Ho J.C., Yang J., Wang Y., Zhang Y., Zhuang Q., Li Y., et al. Generation of human induced pluripotent stem cells from urine samples. Nat. Protoc. 2012;7:2080–2089. doi: 10.1038/nprot.2012.115. [DOI] [PubMed] [Google Scholar]
- 22.Ajzenberg H., Slaats G.G., Stokman M.F., Arts H.H., Logister I., Kroes H.Y., Renkema K.Y., van Haelst M.M., Terhal P.A., van Rooij I.A., et al. Non-invasive sources of cells with primary cilia from pediatric and adult patients. Cilia. 2015;4:8. doi: 10.1186/s13630-015-0017-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu P., Rha G.B., Chi Y.-I. Structural basis of disease-causing mutations in hepatocyte nuclear factor 1beta. Biochemistry. 2007;46:12071–12080. doi: 10.1021/bi7010527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chi Y.-I., Frantz J.D., Oh B.-C., Hansen L., Dhe-Paganon S., Shoelson S.E. Diabetes mutations delineate an atypical POU domain in HNF-1alpha. Mol. Cell. 2002;10:1129–1137. doi: 10.1016/s1097-2765(02)00704-9. [DOI] [PubMed] [Google Scholar]
- 25.Yamagata K. Roles of HNF1α and HNF4α in pancreatic β-cells: lessons from a monogenic form of diabetes (MODY) Vitam. Horm. 2014;95:407–423. doi: 10.1016/B978-0-12-800174-5.00016-8. [DOI] [PubMed] [Google Scholar]
- 26.Yamagata K., Oda N., Kaisaki P.J., Menzel S., Furuta H., Vaxillaire M., Southam L., Cox R.D., Lathrop G.M., Boriraj V.V., et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3) Nature. 1996;384:455–458. doi: 10.1038/384455a0. [DOI] [PubMed] [Google Scholar]
- 27.Bellanné-Chantelot C., Clauin S., Chauveau D., Collin P., Daumont M., Douillard C., Dubois-Laforgue D., Dusselier L., Gautier J.-F., Jadoul M., et al. Large genomic rearrangements in the hepatocyte nuclear factor-1beta (TCF2) gene are the most frequent cause of maturity-onset diabetes of the young type 5. Diabetes. 2005;54:3126–3132. doi: 10.2337/diabetes.54.11.3126. [DOI] [PubMed] [Google Scholar]
- 28.Bellanné-Chantelot C., Carette C., Riveline J.-P., Valéro R., Gautier J.-F., Larger E., Reznik Y., Ducluzeau P.-H., Sola A., Hartemann-Heurtier A., et al. The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity-onset diabetes of the young (MODY)-3. Diabetes. 2008;57:503–508. doi: 10.2337/db07-0859. [DOI] [PubMed] [Google Scholar]
- 29.Haumaitre C., Reber M., Cereghini S. Functions of HNF1 family members in differentiation of the visceral endoderm cell lineage. J. Biol. Chem. 2003;278:40933–40942. doi: 10.1074/jbc.M304372200. [DOI] [PubMed] [Google Scholar]
- 30.Sourdive D.J., Chouard T., Yaniv M. The HNF1 C-terminal domain contributes to transcriptional activity and modulates nuclear localisation. C. R. Acad. Sci. III. 1993;316:385–394. [PubMed] [Google Scholar]
- 31.Tonooka N., Tomura H., Takahashi Y., Onigata K., Kikuchi N., Horikawa Y., Mori M., Takeda J. High frequency of mutations in the HNF-1alpha gene in non-obese patients with diabetes of youth in Japanese and identification of a case of digenic inheritance. Diabetologia. 2002;45:1709–1712. doi: 10.1007/s00125-002-0978-3. [DOI] [PubMed] [Google Scholar]
- 32.Li Q., Lau A., Morris T.J., Guo L., Fordyce C.B., Stanley E.F. A syntaxin 1, Galpha(o), and N-type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. J. Neurosci. 2004;24:4070–4081. doi: 10.1523/JNEUROSCI.0346-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vallania F., Schiavone D., Dewilde S., Pupo E., Garbay S., Calogero R., Pontoglio M., Provero P., Poli V. Genome-wide discovery of functional transcription factor binding sites by comparative genomics: the case of Stat3. Proc. Natl. Acad. Sci. U.S.A. 2009;106:5117–5122. doi: 10.1073/pnas.0900473106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melchior F., Paschal B., Evans J., Gerace L. Inhibition of nuclear protein import by nonhydrolyzable analogues of GTP and identification of the small GTPase Ran/TC4 as an essential transport factor. J. Cell Biol. 1993;123:1649–1659. doi: 10.1083/jcb.123.6.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moore M.S., Blobel G. The GTP-binding protein Ran/TC4 is required for protein import into the nucleus. Nature. 1993;365:661–663. doi: 10.1038/365661a0. [DOI] [PubMed] [Google Scholar]
- 36.Clarke P.R., Zhang C. Spatial and temporal coordination of mitosis by Ran GTPase. Nat. Rev. Mol. Cell Biol. 2008;9:464–477. doi: 10.1038/nrm2410. [DOI] [PubMed] [Google Scholar]
- 37.Soderholm J.F., Bird S.L., Kalab P., Sampathkumar Y., Hasegawa K., Uehara-Bingen M., Weis K., Heald R. Importazole, a small molecule inhibitor of the transport receptor importin-β. ACS Chem. Biol. 2011;6:700–708. doi: 10.1021/cb2000296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soufi A., Garcia M.F., Jaroszewicz A., Osman N., Pellegrini M., Zaret K.S. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell. 2015;161:555–568. doi: 10.1016/j.cell.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kalab P., Weis K., Heald R. Visualization of a Ran-GTP gradient in interphase and mitotic Xenopus egg extracts. Science. 2002;295:2452–2456. doi: 10.1126/science.1068798. [DOI] [PubMed] [Google Scholar]
- 40.Schmiedeberg L., Skene P., Deaton A., Bird A. A temporal threshold for formaldehyde crosslinking and fixation. PLoS One. 2009;4:e4636. doi: 10.1371/journal.pone.0004636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gavrilov A., Razin S.V., Cavalli G. In vivo formaldehyde cross-linking: it is time for black box analysis. Brief. Funct. Genomics. 2015;14:163–165. doi: 10.1093/bfgp/elu037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.