

Abstract
Here, we describe gene expression compositional assignment (GECA), a powerful, yet simple method based on compositional statistics that can validate the transfer of prior knowledge, such as gene lists, into independent data sets, platforms and technologies. Transcriptional profiling has been used to derive gene lists that stratify patients into prognostic molecular subgroups and assess biomarker performance in the pre-clinical setting. Archived public data sets are an invaluable resource for subsequent in silico validation, though their use can lead to data integration issues. We show that GECA can be used without the need for normalising expression levels between data sets and can outperform rank-based correlation methods. To validate GECA, we demonstrate its success in the cross-platform transfer of gene lists in different domains including: bladder cancer staging, tumour site of origin and mislabelled cell lines. We also show its effectiveness in transferring an epithelial ovarian cancer prognostic gene signature across technologies, from a microarray to a next-generation sequencing setting. In a final case study, we predict the tumour site of origin and histopathology of epithelial ovarian cancer cell lines. In particular, we identify and validate the commonly-used cell line OVCAR-5 as non-ovarian, being gastrointestinal in origin. GECA is available as an open-source R package.
INTRODUCTION
Gene expression profiling provides a downstream reflection of the system under study, capturing the effects of multiple drivers of disease behaviour, including gene methylation, mutation and copy number aberration. It can thus be used to characterise patient cohorts and model drug response, facilitating the discovery of novel molecular sub-group clinical tests (1,2). However, due to unrecognised systematic bias, under-powering and lack of validation, the generalisability and efficacy of many gene-list based stratification methods is questioned (3,4). In breast cancer, random gene-sets can be as effective in stratifying patients as published prognostic gene lists (5). Therefore, not surprisingly, only 0.07% of published biomarkers have made their way into routine clinical use (6), such as those with Section 510(k) clearances from the US Food and Drug Administration including MammaPrint, Prosigna (PAM50) and Pathwork Tissue of Origin Test (7–9).
With the advent of next generation sequencing (NGS), researchers are now faced with the challenge of validating gene-lists and multiple sub-groups derived from archived microarray-based transcriptome data. There is a risk of systematic bias if data sets are integrated, due to differences in the scale of intensity values (10,11). Tools to address cross-study effects do exist, but cannot be cannot be guaranteed to work as further biases may be introduced through cross-platform normalisation (10,12). In the pre-clinical setting, the integration of gene expression data sets is of particular importance when selecting the most appropriate cell lines to model newly-discovered molecular subgroups. Semi-supervised clustering with such gene lists has proved problematic if data sets have been profiled separately. Often the cell lines would simply cluster together in a distinct group separate from clinical samples (13,14).
An approach is therefore required that will enable the transfer of prior knowledge between different data sets, platforms and technologies in order to support translational discovery efforts and in silico validation.
In this paper, we demonstrate that data integration issues can be addressed by through the use of proportions (compositional ratios) rather than the actual values of expression levels. The comparability of gene expression compositional ratios is evaluated using two compositional data measures: Aitchison's (AD), a distance metric as used in geostatistics (15,16) and Kullback–Leibler divergence (KLD), a dissimilarity distance, which is derived from information theory (17). We evaluate both measures (a composite term for AD and KLD), in an approach termed gene expression compositional assignment (GECA), with comparison to a Spearman rank correlation (SRC)-based method (18). We hypothesised that compositional ratios would contain information on the inter-relationships between gene expression levels, thus having the potential to outperform SRC approaches. We applied GECA successfully to gene list transfer in data sets covering bladder cancer staging, tumour site of origin, epithelial ovarian cancer (EOC) prognosis and mislabelled cell lines, across different platforms and technologies. Finally, using EOC as a case study, we present how our approach can determine the tumour site of origin and histopathology of cell lines using random gene-sets and microarray transcriptional profiles of primary tumours and pathologically-reviewed EOC histopathology data sets.
MATERIALS AND METHODS
Definition of distance metric/dissimilarity distance
Data are in compositional form when its components sum up to a whole, e.g. the unit one or 100%. Both AD, a distance metric, and KLD, a dissimilarity distance, are suitable for comparing data in compositional form. Given two samples, S1 and S2, that are represented by two sets of gene expression profiles, geS1 and geS2, respectively, using gene lists of size n, such that:
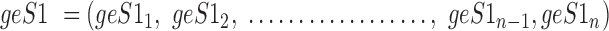 |
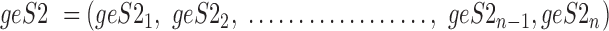 |
where the expression level of gene 1 in sample 1 is represented by geS11 and the expression level of the same gene in sample 2 is represented by geS21, then the compositional gene expression profiles are given by:
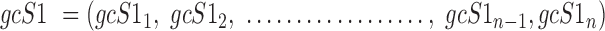 |
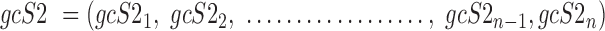 |
where 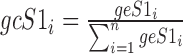 and
and 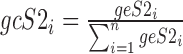 .
.
The AD metric is given by (15):
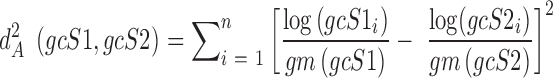 |
where 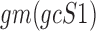 and
and 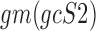 are the geometric means of compositional profiles
are the geometric means of compositional profiles 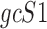 and
and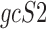 , respectively, and are defined as:
, respectively, and are defined as: 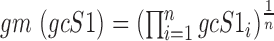 and
and 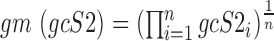 .
.
The KLD distance is given by (17):
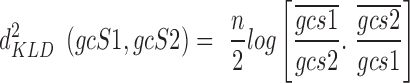 |
where: 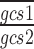 and
and 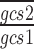 are the arithmetic means of the ratios
are the arithmetic means of the ratios 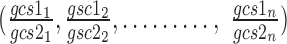 and
and 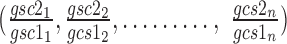 respectively.
respectively.
Compositional data analysis is based on two key principles: invariance to scale and compliance with subcompositional coherence (19). To be scale invariant 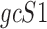 does not differ when
does not differ when 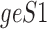 is transformed by a scalar factor. In subcompositional coherence, the relative ratios of components within
is transformed by a scalar factor. In subcompositional coherence, the relative ratios of components within 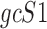 , are the same as the relative ratios of components within a subcomposition of
, are the same as the relative ratios of components within a subcomposition of 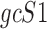 (19).
(19).
In the bladder staging, tumour of origin, prognostic EOC and mislabelled cell line data sets, the two compositional measures were compared against SRC (20). In the EOC case study, only the AD measure was used.
Gene expression compositional assignment using gene lists
The following analytical pipeline was followed for the bladder staging, tumour of origin and prognostic EOC data sets (Supplementary Figure S1).
Gene list analytical pipeline
Data processing
In studies with multiple platforms or technologies (bladder staging and epithelial ovarian cancer prognosis) genes with multiple probesets mappings were median summarised, otherwise the data were maintained at the probeset level. The gene list associated with the reference data set was then applied to both reference and query data sets. If the data were in log-transformed form, it was inverse transformed. Gene expression levels were then converted to gene expression compositional ratio format.
Leave-one-out cross-validation
Leave-one-out cross-validation (LOOCV) was carried out in both reference and query data sets using the reference data set gene list and observed group labels using GECA (AD and KLD) and SRC measures. For LOOCV in each data set, one sample was removed and compared to the remaining samples using the chosen measure. In a process similar to Dancik et al. (18), the measure scores for each group label were aggregated and the average measure calculated. For GECA (AD and KLD), the lowest score was taken as the assigned group. When using SRC, the highest average score was used to assign the group of the unknown sample. Against the observed group labels of each data set, the accuracy metric (number of samples with correctly assigned group labels/total number of samples) was used. For each accuracy metric, 95% confidence intervals were devised using the mean squared error.
Reference data set group label assignment
We evaluated the assignment of group labels to query data sets using a reference data set. The same gene list as in the LOOCV section was used. Each sample in the query data set was compared to all the samples in the reference data set. As in the LOOCV stage the average of each measure was calculated and the assigned group allocated and the accuracy metric calculated against observed groups provided with each data set.
Gene list permutation
We considered the effect of the removal of genes, from a gene list, on the performance of AD, KLD and SRC to assign group labels. In a two-class context, using reference and query data sets with a common gene list, an overall measure was calculated for each query sample with respect to each reference group. Taking each query sample, genes were removed, without replacement, in decreasing order of percentage composition. After the removal of each gene, the measure was re-calculated and presented as a fraction of the original score using the full gene list. The mean group measure scores (averaged over all query data set samples) were plotted against the number of genes removed.
Bootstrapping
For robustness, a bootstrapping procedure was carried out. In this procedure, the observed ratio of subgroup samples was maintained in the reference data set, while samples were selected with replacement. This was repeated 10 000 times, the number of times (divided by 10 000) each group was assigned was taken as a P-value. This P-value together with the observed and assigned groups was used to calculate the bootstrapped area under the curve (AUC) score.
Random gene-set permutation
Random gene-sets (n = 10 000) were used to evaluate the confidence in each of the gene lists. Group assignment using random gene-sets was repeated 10 000 times, the number of times (divided by 10 000) each group was assigned was taken as a P-value.
Application to data sets with gene lists
Bladder staging
To demonstrate the ability of GECA (AD and KLD) to work across microarray platforms we first selected four bladder cancer data sets (21–24) as used in Lauss et al.'s study (25), together with a bladder staging gene list (21). The Sanchez–Carbayo et al. (21) bladder staging gene list, consisting of 249 probesets, was selected as the Lauss et al. (25) study concluded that lists of at least 150 genes in length were the most accurate in stage and grade classification. To use this gene list, bladder cancer stages Ta/T1 were classed as 1 (non-muscle invasive (NMI)), with stages T2 and above labelled as 2 (muscle invasive (MI)). Staging information was available for all four data sets and was used as observed group labels. All four data sets were processed as described in Lauss et al. (25) with the exception of cross-platform normalisation, merging the sample sets, quantile normalisation and gene-centring. Additional data set compositional details can be found in Supplementary Table S1. LOOCV, reference data set group label assignment, signature permutation, bootstrapping and random gene-set permutation were carried out as previously described.
Tumour site of origin
To demonstrate the ability of GECA (AD and KLD) to work in a setting with multiple group labels, a tumour site of origin data set was used [http://biogps.org/downloads/] (26). In the original study a training set of 100 and a test set of 75 tumour site of origin samples were used (one of the test samples was unavailable). The training set was used as a reference data set in this study, while the test set was used as a query data set. The reference data set consisted of primary tumour samples from: prostate, bladder, breast, colorectal, pancreas, gastroesophageal, kidney, liver, ovarian and lung. The query data set comprised both primary tumours and metastatic samples. The provided tumour site of origin data provided with each sample was used as observed group labels. The original study (26) had developed a 216 gene (probeset level) list to distinguish between the 11 different tumour sites of origin within the reference data set. Additional data set compositional details can be found in Supplementary Table S2. LOOCV, reference data set group label assignment, bootstrapping and random gene-set permutation were carried out as previously described, with the exception of the AUC score, a version suitable for multi-class predictions was used instead (27). Results were presented for both the full query data set and ten metastatic samples as these were treated as ‘tumour of unknown origin’ being derived from secondary tumours where the primary site was known.
Epithelial ovarian cancer prognosis
To demonstrate the ability of GECA (AD and KLD) to work in a cross-technology setting we selected a data set comprising 215 EOC samples (Stage II-IV) which had had been used to develop a prognostic signature of 193 genes in The Cancer Genome Atlas (TCGA) EOC study (28). The signature classified patients (using overall survival right-censored at 60 months) based on a t-statistic comparing defined ‘good’ and ‘poor’ prognosis genes. The data for the 215 samples was available in gene-centric normalised form, based on the integration of gene expression from Affymetrix (Affy) U133A, Agilent and Affy HuEx platforms (28,29). Next 294 samples from the TCGA ovarian data set were identified which had been profiled on both an NGS and on an Affy-based microarray platform. Additional data set compositional details can be found in Supplementary Table S3. Using the gene-centric data and the classification method as described (29), good or poor prognosis group labels were assigned to the 294 patient samples. As 94 samples had been used in the development of the prognostic signature in the original study, these were retained for use as a reference set, while the 200 samples were used as a query set (which was available in two versions – Affy U133A and Illumina HiSeq). LOOCV, reference data set group label assignment, bootstrapping and random gene-set permutation were carried out as previously described. Survival plots (overall survival, right censored at 60 months) and hazard ratios (Kaplan–Meier estimation, log-rank test) with P-values were calculated for the reference set (Supplementary Figure S3A) and the query set (Supplementary Figure S3B).
Gene expression compositional assignment (gene-sets)
The following analytical pipeline was followed for the mislabelled cell lines and EOC cell line studies (Supplementary Figure S4).
Random gene-set analytical pipeline
Data processing
If available, data sets were downloaded and used in their processed format (NCI-60: GSE5846), otherwise CEL files were provided and samples processed and normalised using the Robust Multi-array Average (RMA) algorithm. In studies with multiple platforms or technologies genes with multiple probesets mappings were median summarised, otherwise the data were maintained at the probeset level. All data sets were inverse-transformed.
Random gene-sets
Random gene-sets were selected (n = 10 000). For each random gene-set, the data were converted to a gene expression compositional ratio format. GECA (AD and KLD) and SRC were used to compare each member of the query data sets to the reference data set (AD only in the EOC tumour site of origin/histopathology study). The number of gene-sets for which a sample from the reference data set produced the best measure for a sample from the query data set was combined to result in an aggregate assignment. Initially in the mislabelled cell lines study, we examined the effect of varying lengths of random gene-sets (20, 30, 40, 50, 60, 70, 80, 90,100,125,150,175, 200, 225, 250, 275, 300, 350, 400, 450, 500, 550, 600, 650, 700 and 750) with respect to aggregate assignments using a data set as both query and reference. In the cross-platform/cross-data set section of the mislabelled cell lines study, we compared random gene-set lengths of 100, 250 and 500, using the latter length only in the EOC tumour site of origin/histopathology study.
Random allocation of assignments
The robustness of observed aggregate assignments was tested by random allocation of assignments. A sample in the reference data set was chosen at random as producing the best measure, this was repeated 10 000 times, resulting in a random aggregate assignment. The random aggregate assignment was replicated 10 000 times and compared against the observed aggregate assignment matrix to produce a confidence value. Results were treated as P-values and adjusted by Benjamini and Hochberg false discovery rate (FDR) method. Aggregate assignments with an FDR of 0.01 were used. Percentage aggregate assignments were calculated from the remaining significant assignments. In the cell line data sets in which replicates were available, each replicate was treated as an individual sample. After checking for concordance with respect to individual data (all replicates demonstrated a high level of agreement), the median assignment was taken and aggregate assignment calculated as before, followed by random aggregate assignments.
Application to data sets using random gene-sets
Mislabelled cell lines
To demonstrate the ability of GECA (AD and KLD) to work with random gene-sets in the absence of a gene list we identified a group of potentially mislabelled cell lines, where concerns have previously been expressed regarding cell line authenticity or tissue identity. Two versions of the National Cancer Institute 60 cell line data set (NCI-60) were used, accession number GSE5846 (30,31), was obtained from the GEO database, the second, Cell-Miner, from the National Cancer Institute's (NCI) online database (32). An internal EOC cell line data set, (AL-OV), as described later (GEO accession number GSE73638 (31)), was also used. The NCI-60 data set comprised breast, central nervous system, colorectal, leukaemia, melanoma, lung, ovarian, prostate and renal cancers. The lung cell line, NCI-H23, was missing from the Cell-Miner version of the NCI-60 data sets. The seven mislabelled cell lines, (i) MDA-MB-435, MDA-N and M14, (ii) SNB-19 and U251 and (iii) OVCAR-8 and NCI-ADR-RES were selected for investigation into their alignment with other cell lines (33–36). All seven cell lines were present in the two versions of the NCI-60 data set, only OVCAR-8 and NCI-ADR-RES were present in the AL-OV data set. Additional data set compositional details can be found in Supplementary Tables S4, S5 and S6.
The GSE5846 data set was first used as both a query and a reference data set, in a LOOCV-style comparison and tested the effect of varying random gene-set length (from 20 to 750) on percentage assignment, performing the random allocation of assignments on three key lengths, 100, 250, 500. Next, to demonstrate the validity of GECA in assigning cell lines to their equivalent across data sets, on the same platform, the seven cell lines in the GSE5846 data set (together with the lung cancer cell line NCI-H23) were used as a query data set against the Cell-Miner version, as a reference data set (32). Then, the two cell lines OVCAR-8 and NCI-ADR-RES, profiled on the OV-DSA platform from the AL-OV data set were used as the query data set, with the GSE5846 NCI-60 data set used as reference.
Tumour site of origin and histopathology of epithelial ovarian cancer cell lines
Three query data sets were used in the EOC cell line case study: Cell-Miner (32); CCLE (37), and an internal EOC cell line data set (AL-OV). In total, 58 cell lines were represented in at least one query data set.
The largest EOC cell line data set was obtained from the Broad Institute's Cancer Cell Line Encyclopedia (CCLE) (37), comprising 52 cell line samples. The NCI-60 data set was obtained from Cell-Miner, the NCI's online database (32), comprising seven cell lines, including three replicates per cell line. Both were profiled on the Affy U133 Plus 2 platform. The 18 cell lines in the AL-OV were prepared and processed as described in Supplementary Methods. The final data set comprised 18 cell lines, including three replicates (n = 17) and one replicate (n = 1). The data have been deposited in GEO (31) and is accessible through GEO Series accession number GSE73638. Additional data set compositional details can be found in Supplementary Table S7. Two reference data sets were used: expO and an internal EOC histopathology data set, AL-OVH (GEO (31) accession number GSE73638). The tumour site of origin data set was obtained from the International Genomics Consortium's Expression Project for Oncology (expO) (data accessible from the GEO database (31), accession number GSE2109). The data set comprised 2158 samples profiled on the Affy U133 Plus 2 platform. The data set was then curated by primary tumour site of origin. Additional data set compositional details can be found in Supplementary Tables S8 and S9. The EOC histopathology data set (AL-OVH) comprised 50 samples obtained from a retrospective data set collected in Edinburgh from 2000 to 2010 (Ethics reference number: 07/S1102/33). All patients had received platinum-based chemotherapy. The EOC histopathology data set was reviewed by two pathologists (ARW and WGM) and comprised the five main ovarian sub-types: high-grade serous (n = 13), low-grade serous (n = 7), clear cell (n = 12), mucinous (n = 9) and endometrioid (n = 9). The data set was profiled on the OV-DSA .The data have been deposited in GEO (31) and is accessible through GEO Series accession number GSE73638. All 58 cell lines in three query data sets were first compared to the tumour site of origin data set. Cell lines with highest-ranked percentage grouped aggregate assignments to ovarian, fallopian tube and endometrium tumour site of origin group labels were retained. The remaining cell lines were next compared to the internal EOC histopathology data set. Compound aggregate assignments, with respect to EOC sub-type labels, i.e. clear cell, endometrioid, mucinous, serous (high-grade) and serous (low-grade) were calculated as with the tumour site of origin reference data set. For percentage compound aggregate assignments, a minimum threshold of 50% was selected by which to classify a majority histology.
RESULTS
Gene lists
To apply GECA, the user selects reference (with group labels) and query gene expression data sets (Figure 1A). In both, a user-defined set of genes is selected and their expression levels converted into compositional form (Figure 1B). Using pairwise comparisons, the similarity between each query and reference sample is calculated using a measure (Figure 1C). As the value of the measure decreases, the relative similarity between samples increases (Figure 1D). The label of the reference group with the lowest average measure is assigned to the query sample. In the absence of a pre-defined gene list, the second approach utilises random gene-sets (a list of genes selected without replacement randomly from all available genes) from the full list of intersecting genes. Reference assignments can be viewed at the individual sample level or at group label aggregate form. To demonstrate the utility of GECA, we selected three studies in which a gene list and observed group labels were available for both query and reference data sets.
Figure 1.

Schema of gene expression compositional assignment (A) Heatmaps of hypothetical query and reference data sets using normalised, log-transformed gene expression levels. The query samples are to be assigned reference group labels. (B) Bar charts of gene expression compositional ratios in query and reference samples. Gene expression levels are inverse-log-transformed and converted to gene expression compositional ratio format (values summing to 1). (C) A query sample is compared to each reference sample using GECA. (D) In the network, shaded nodes represent reference samples, coloured according to group labels. The white node represents a query sample. The relative position of reference nodes corresponds inversely to the GECA score against query samples. The query sample is assigned the reference group label with the lowest relative measure score (or mean score). Bootstrapping or random assignment tests the robustness of observed assignments. Non-significant assignments are faded, significant assignments are opaque.
Bladder cancer staging
First, the effectiveness of GECA was tested in a binary class, multi-microarray platform setting. We used a gene list and data sets (21–24) from a validation study of bladder cancer staging (25). LOOCV results using GECA were comparable to results obtained by SRC and the results published in the original study (25) (Supplementary Table S10A). Importantly, this performance was achieved without the original study's data merging and cross-platform normalisation steps. In assigning reference group labels to the three query data sets, GECA produced similar AUC results to SRC in two, out-performing SRC in the third (this data set was profiled on a non-standard custom-DNA microarray platform) (Supplementary Tables S10B, S11A–C). This demonstrated GECA's ability to work across data sets and platforms. To investigate the effect of the sequential removal of genes, in decreasing order of compositional contribution, from each query data set gene list, we considered the measures for the reference group labels as a fraction of the measure using the full gene list. For GECA (AD and KLD) this resulted in a linear-like decrease (Supplementary Figure S5A, B, D, E, G and H), this contrasted with the non-linear behaviour of SRC (Supplementary Figure S5C, F and I). This shows that GECA (AD and KLD) is robust to the underlying input, as compared to SRC, under depletion of relevant genes.
Tumour site of origin
We next evaluated GECA in transferring the discriminatory power of a gene list in a multi-class setting, comprising 11 different tumour sites of origin (26). LOOCV accuracy using GECA and SRC was comparable to the original study's results (Supplementary Table S12A). GECA and SRC produced similar AUC scores and accuracy result, the latter being comparable to the original paper's classification results (26) (Supplementary Tables S12B, C and S13). This showed GECA's effectiveness in both multi- and binary-class contexts.
Epithelial ovarian cancer prognosis
We then tested GECA's ability to transfer a prognostic gene signature from one technology, microarray, to another, NGS, using EOC data sets (28). Samples in both query (two versions, NGS and microarray) and reference (microarray-only) data sets were assigned ‘good’ or ‘poor’ prognosis labels as in the original study (28,29) (Supplementary Figure S3A and B). In particular, GECA (AD) maintained the same AUC in both microarray and NGS query data sets, respectively (Supplementary Tables S14A–C, S15A, B and Supplementary Figures S3C–H). We have therefore demonstrated GECA's capacity to transfer prior knowledge from one technology to another.
Random Gene-Sets
In some cases, a discriminatory gene list is either not available or appropriate. We therefore assessed the performance of GECA using random gene-sets, focusing on the effectiveness of different set lengths (n = 100, 250 and 500).
Mislabelled cell lines
We tested GECA's ability to identify mislabelled or cross-contaminated cancer cell lines between different data sets and platforms. Seven cell lines, with revised classifications, e.g. breast to melanoma, were selected from a National Cancer Institute (NCI-60) reference data set (30). The data set contained both the original cell lines and their experimentally-determined equivalent. Within the NCI-60 data set, using a random gene-set version of LOOCV, varying the random gene-set length between n = 20 and 750 showed that the association with GECA or SRC's performance (without significance adjustment) varied by cell line, with all but one cell line's assignment level stabilising by n = 500 (Supplementary Figure S5B). In this case, GECA (AD) and SRC produced comparable results between n = 300 and 700, with SRC's performance dropping thereafter. Also GECA (AD) produced improved assignments over GECA (KLD) at shorter gene-set lengths (n = 100 to n = 500) (Supplementary Figure S5C and D) and between n = 100 to n = 300 (Supplementary Figure S5B). Examining GECA's and SRC's performance, this time adjusted for significance, at three gene-set sizes (n = 100, 250, 500) confirmed improved assignments with increasing gene-set length, in particular using GECA (AD) (Figure 2A and Supplementary Tables S16A and B). One ovarian cell line, OVCAR-8, produced a 100% assignment with a lung cancer cell line (NCI-H23), suggesting a mislabelling error (Supplementary Table S16C). Next, we tested GECA's performance across data sets, using an independent NCI-60 reference data set (32). All seven cell lines had 100% assignments, regardless of method, with their independent replicate (Supplementary Table S17). The lung cancer cell line was again assigned to the same ovarian cancer cell line, appearing to be identical, transcriptionally. In a second independent data set (AL-OV, deposited in the National Center for Biotechnology Information's (NCBI) Gene Expression Omnibus (GEO) (31), accession number GSE73638), which included two of the seven cell lines, increased gene-set size (n = 500) also produced improved assignments, in particular with GECA (KLD) (Supplementary Tables S18 and Supplementary Figures S6A–C). We thus demonstrated GECA's ability to transfer prior knowledge across data sets and platforms without a defined gene list.
Figure 2.

Cell lines by gene expression compositional aggregate assignment (A) Heatmaps showing percentage assignment of cell lines BR: MDA-MB-435, BR: MDA-N, ME: M14, CNS: SNB-19, CNS: U251, OV: OVCAR-8, UN: NCI-ADR-RES, within a query/reference data set (30) by GECA (AD) for three different gene-set lengths: (i) 500 (ii) 250 and (iii) 100. (B) Tumour site of origin aggregate assignment, using GECA (AD) for 58 cell lines across three query data sets (32,37) against a primary tumour site of origin reference data set. Assignments are presented as a spring-embedded network in which the size of nodes (circular = tumour samples, square = cell line) are proportional to the number of aggregate assignments attributed. Width and transparency and relative positioning of connections between nodes are representative of the aggregate assignment. The robustness of observed aggregate assignments was tested by random allocation of assignments. Results were treated as P-values and adjusted by the Benjamini and Hochberg false discovery rate (FDR) method. Assignments with an FDR of <0.01 were used. (C) Photomicrograph of the OVCAR-5 cell line showing diffusely positive staining with CA19.9 indicating upper gastrointestinal origin.
Epithelial ovarian cancer cell lines: tumour site of origin
As a final proof of principle, we tested GECA in a study to determine the tumour site of origin and histopathology of EOC cell lines. We chose EOC as some tumours may arise from non-ovarian sites such as the gastrointestinal (GI) tract (38). We used random gene-sets of length 500 with GECA (AD), as this combination produced the most consistent assignments. We first filtered the cell by tumour site of origin, referencing a publicly available data set of solid tumours (expO) (Supplementary Table S2). This was refined by a second stage of histopathological assignments, using a reference data set of expert-pathologically reviewed EOC samples, each allocated to one of five histopathologies (AL-OVH, deposited in NCBI's GEO (31), accession number GSE73638). We queried these reference data sets using EOC cell lines from three cross-platform data sets (Supplementary Table S7) (32,37). Endometrioid EOC and endometrial cancer are similar at the molecular and morphological levels (39), therefore we filtered cell lines whose majority aggregate assignments were to EOC, fallopian tube (FT) or endometrium tumour sites of origin (Supplementary Figure S7). All but eight of the 58 cell lines showed assignments to either EOC, FT or endometrial tumour sites of origin (Figure 2B, Supplementary Figure S7, Supplementary Table S19). We selected OVCAR-5, assigned by GECA to GI tumour sites of origin, for review by an expert pathologist (WGM). In pathological review, the immunophenotype indicated a tumour of upper GI origin (Figure 2C), consistent with the GECA assignment.
Epithelial ovarian cancer cell lines: histopathology
In the next stage, we were able to obtain majority histopathology assignments for 26 (out of 50) cell lines (Supplementary Figure S8, Supplementary Table S20). One cell line, OVCAR-3 was assigned the same histopathology (high-grade serous) in two out of three query data sets, the non-consensus version of the cell line being from a different source. We took forward OVCAR-3 for pathological review (WGM), which confirmed the immunophenotype as characteristic of an ovarian or tubal serous carcinoma. Our assignment results were next compared to classifications from two recent studies both of which had used mutation and copy number analyses to characterise EOC cell lines (40,41) (Supplementary Table S21). We were able to achieve expert pathology confirmation of our GECA (AD) assignments in examples of where we disagreed (OVCAR-5) and agreed (OVCAR-3) with both studies.
DISCUSSION
The biology represented by gene lists and signatures must have the capacity to be validated across platforms or technologies. We have described an approach that circumvents data integration and cross-platform normalisation limitations through the use of gene expressional compositional ratios. Using our method, GECA (AD and KLD), we demonstrated at least comparable accuracies with a rank-based correlation method in multi-platform and technology data sets, outperforming the latter in certain cases, e.g. bladder cancer staging and EOC prognosis. We have also shown that GECA can identify mislabelled cell lines, including the NCI-H23 lung cancer cell line which has previously been shown, through short-tandem-repeat (STR) profiling, to have a common donor origin with HCC60, an ovarian cell line (42). We established a further use for GECA in the identification of the tumour site of origin and histopathology of EOC cell lines. Authentication testing of cell lines is now possible through short tandem repeat (STR) profiling (43). The use of cell line data from archived gene expression data sets, without access to the original samples, is particularly problematic. As STR-profiling is both simple and inexpensive, the use of a gene expression-based bioinformatics method should, in theory, be redundant. However, the EOC cell line, OVCAR-5, that GECA identified as GI (from two data sets), and was confirmed as such by an expert pathologist, was included in two STR-based studies (44,45), neither of which suggested a non-ovarian source. STR profiling looks at donor origin, aiming to exclude cross-contamination by cells from a different donor, which is an important problem in cell line research (44). Assessment of tissue origin can complement authentication testing methods. Problems with donor or tissue identity indicate the need to explore the cell line's origin further, to ensure that the cell line is an appropriate model for future work.
In using GECA, a number of factors need to be considered. The measures used provide relative assignments, so thresholding cannot be applied such as with SRC. However LOOCV, bootstrapping and random assignment testing can determine the robustness of the reference data set and subsequent assignments. As with any classification-type approach, an appropriately sampled reference data set is also required to produce results with translational research utility. In using random gene-sets, aggregate group assignments can be adjusted for unbalanced groupings within the reference data set, using propensity-based (normalised probability) scores. In conclusion, we have demonstrated that GECA is able to transfer prior knowledge across data sets, platforms and technologies and can make effective use of publicly available archived microarray data in conjunction with those derived from newer technologies available today.
AVAILABILITY
GECA, including AD, KLD and SRC, is available as an open-source R package. Gene and probe-set level versions of expO are also available for download, as are example R scripts for pre-processing data [https://sourceforge.net/projects/geca/].
Supplementary Material
Acknowledgments
The authors would like to thank Andrew Tikhonov (European Bioinformatics Institute) for his technical advice.
Footnotes
Present address: Caroline Greenan, Clovis Oncology, Cambridge, CB3 0AX, UK.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Invest Northern Ireland. Funding for open access charge: Invest Northern Ireland.
Conflict of interest statement. T.D., N.M., S.W., C.G., T.D., K.K., D.P.H. and R.D.K. are employees of Almac Diagnostics. R.D.K. receives payment as the medical director for Almac Diagnostics.
REFERENCES
- 1.Hornberger J., Alvarado M.D., Rebecca C., Gutierrez H.R., Yu T.M., Gradishar W.J. Clinical validity/utility, change in practice patterns, and economic implications of risk stratifiers to predict outcomes for early-stage breast cancer: a systematic review. J. Natl. Cancer Inst. 2012;104:1068–1079. doi: 10.1093/jnci/djs261. [DOI] [PubMed] [Google Scholar]
- 2.Lu A.T., Salpeter S.R., Reeve A.E., Eschrich S., Johnston P.G., Barrier A.J, Bertucci F., Buckley N.S., Salpeter E.E., Lin A.Y. Gene expression profiles as predictors of poor outcomes in stage II colorectal cancer: A systematic review and meta-analysis. Clin. Colorectal Cancer. 2009;8:207–214. doi: 10.3816/CCC.2009.n.035. [DOI] [PubMed] [Google Scholar]
- 3.Dupuy A., Simon R.M. Critical review of published microarray studies for cancer outcome and guidelines on statistical analysis and reporting. J. Natl. Cancer Inst. 2007;99:147–157. doi: 10.1093/jnci/djk018. [DOI] [PubMed] [Google Scholar]
- 4.Simon R. Roadmap for developing and validating therapeutically relevant genomic classifiers. J. Clin. Oncol. 2005;23:7332–7341. doi: 10.1200/JCO.2005.02.8712. [DOI] [PubMed] [Google Scholar]
- 5.Venet D., Dumont J.E., Detours V. Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput. Biol. 2011;7:e1002240. doi: 10.1371/journal.pcbi.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poste G. Bring on the biomarkers. Nature. 2011;469:156–157. doi: 10.1038/469156a. [DOI] [PubMed] [Google Scholar]
- 7.Nielsen T., Wallden B., Schaper C., Ferree S., Liu S., Gao D., Barry G., Dowidar N., Maysuria M., Storhoff J. Analytical validation of the PAM50-based Prosigna Breast Cancer Prognostic Gene Signature Assay and nCounter Analysis System using formalin-fixed paraffin-embedded breast tumor specimens. BMC Cancer. 2014;14:177. doi: 10.1186/1471-2407-14-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glas A.M., Floore A., Delahaye L.J., Witteveen A.T., Pover R.C., Bakx N., Lahti-Domenici J.S., Bruinsma T.J., Warmoes M.O., Bernards R., et al. Converting a breast cancer microarray signature into a high-throughput diagnostic test. BMC Genomics. 2006;7:278. doi: 10.1186/1471-2164-7-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dumur C.I., Fuller C.E., Blevins T.L., Schaum J.C., Wilkinson D.S., Garrett C.T., Powers C.N. Clinical verification of the performance of the Pathwork Tissue of Origin Test utility and limitations. Am. J. Clin. Pathol. 2011;136:924–933. doi: 10.1309/AJCPDQPFO73SSNFR. [DOI] [PubMed] [Google Scholar]
- 10.Rung J., Brazma A. Reuse of public genome-wide gene expression data. Nat. Rev. Genet. 2013;14:89–99. doi: 10.1038/nrg3394. [DOI] [PubMed] [Google Scholar]
- 11.Fan X., Lobenhofer E.K., Chen M., Shi W., Huang J., Luo J., Zhang J., Walker S.J., Chu T.M., Li L., et al. Consistency of predictive signature genes and classifiers generated using different microarray platforms. Pharmacogenomics J. 2010;10:247–257. doi: 10.1038/tpj.2010.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tseng G.C., Ghosh D., Feingold E. Comprehensive literature review and statistical considerations for microarray meta-analysis. Nucleic Acids Res. 2012;40:3785–3799. doi: 10.1093/nar/gkr1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neve R.M., Chin K., Fridlyand J., Yeh J., Baehner F.L., Fevr T., Clark L., Bayani N., Coppe J.P., Tong F., et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Auman J.T., McLeod H.L. Colorectal cancer cell lines lack the molecular heterogeneity of clinical colorectal tumors. Clin. Colorectal Cancer. 2010;9:40–47. doi: 10.3816/CCC.2010.n.005. [DOI] [PubMed] [Google Scholar]
- 15.Aitchison J. The statistical analysis of compositional data. 2nd edn. Caldwell: The Blackburn Press; 2003. [Google Scholar]
- 16.Lovell D., Müller W., Taylor J., Zwart A., Helliwell C.C. Proportions, percentages, PPM: Do the molecular biosciences treat compositional data right? In: Pawlowsky-Glahn V, Bucciant A, editors. Compositional data analysis: Theory and applications. Chichester: Wiley and Sons; 2011. pp. 193–207. [Google Scholar]
- 17.Martín-Fernández J.A., Bren M. Proceedings of 2001 Annual Conference of the International Association for Mathematical Geology. Cancun: 2001. Some practical aspects on multidimensional scaling of compositional data. September 6-12, 2001. [Google Scholar]
- 18.Dancik G.M., Ru Y., Owens C.R., Theodorescu D. A framework to select clinically relevant cancer cell lines for investigation by establishing their molecular similarity with primary human cancers. Cancer Res. 2011;71:7398–7409. doi: 10.1158/0008-5472.CAN-11-2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aitchison J., Egozcue J.J. Compositional data analysis: where are we and where should we be heading? Math. Geol. 2005;37:829–850. [Google Scholar]
- 20.Spearman C. The proof and measurement of association between two things. Am. J. Psychol. 1904;15:72–101. [PubMed] [Google Scholar]
- 21.Sanchez-Carbayo M., Socci N.D., Lozano J., Saint F., Cordon-Cardo C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J. Clin. Oncol. 2006;24:778–789. doi: 10.1200/JCO.2005.03.2375. [DOI] [PubMed] [Google Scholar]
- 22.Dyrskjøt L., Zieger K., Real F.X., Malats N., Carrato A., Hurst C., Kotwal S., Knowles M., Malmström P.U., de la Torre M., et al. Gene expression signatures predict outcome in non-muscle-invasive bladder carcinoma: a multicenter validation study. Clin. Cancer Res. 2007;13:3545–3551. doi: 10.1158/1078-0432.CCR-06-2940. [DOI] [PubMed] [Google Scholar]
- 23.Stransky N., Vallot C., Reyal F., Bernard-Pierrot I., de Medina S.G., Segraves R., de Rycke Y., Elvin P., Cassidy A., Spraggon C., et al. Regional copy number-independent deregulation of transcription in cancer. Nat. Genet. 2006;38:1386–1396. doi: 10.1038/ng1923. [DOI] [PubMed] [Google Scholar]
- 24.Blaveri E., Simko J.P., Korkola J.E., Brewer J.L., Baehner F., Mehta K., Devries S., Koppie T., Pejavar S., Carroll P., et al. Bladder cancer outcome and subtype classification by gene expression. Clin. Cancer Res. 2005;11:4044–4055. doi: 10.1158/1078-0432.CCR-04-2409. [DOI] [PubMed] [Google Scholar]
- 25.Lauss M., Ringnér M., Höglund M. Prediction of stage, grade, and survival in bladder cancer using genome-wide expression data: A validation study. Clin. Cancer Res. 2010;16:4421–4433. doi: 10.1158/1078-0432.CCR-10-0606. [DOI] [PubMed] [Google Scholar]
- 26.Su A.I., Welsh J.B., Sapinoso L.M., Kern S.G., Dimitrov P., Lapp H., Schultz P.G., Powell S.M., Moskaluk C.A., Frierson H.F., Jr, et al. Molecular classification of human carcinomas by use of gene expression signatures. Cancer Res. 2001;61:7388–7393. [PubMed] [Google Scholar]
- 27.Hand D.J., Till R.J. A simple generalisation of the area under the ROC curve for multiple class classification problems. Mach. Learn. 2001;45:171–186. [Google Scholar]
- 28.The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–661. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X.V., Verhaak R.G.W., Purdom E., Spellman P.T., Speed T.P. Unifying gene expression measures from multiple platforms using factor analysis. PLoS One. 2011;6:e1769. doi: 10.1371/journal.pone.0017691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee J.K., Havaleshko D.M., Cho H., Weinstein J.N., Kaldjian E.P., Karpovich J., Grimshaw A., Theodorescu D. A strategy for predicting the chemosensitivity of human cancers and its application to drug discovery. Proc. Natl. Acad. Sci. U.S.A. 2007;10:13086–13091. doi: 10.1073/pnas.0610292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reinhold W.C., Sunshine M., Liu H., Varma S., Kohn K.W., Morris J., Doroshow J., Pommier Y. CellMiner: A web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res. 2011;72:3499–3411. doi: 10.1158/0008-5472.CAN-12-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rae J.M., Ramus S.J., Waltham M., Armes J.E., Campbell I.G., Clarke R., Barndt R.J., Johnson M.D., Thompson E.W. Common origins of MDA-MB-435 cells from various sources with those shown to have melanoma properties. Clin. Exp. Metastasis. 2004;21:543–552. doi: 10.1007/s10585-004-3759-1. [DOI] [PubMed] [Google Scholar]
- 34.Rae J.M., Creighton C.J., Meck J.M., Haddad B.R., Johnson M.D. MDA-MB-435 cells are derived from M14 melanoma cells–a loss for breast cancer, but a boon for melanoma research. Breast Cancer Res.Treat. 2007;104:13–19. doi: 10.1007/s10549-006-9392-8. [DOI] [PubMed] [Google Scholar]
- 35.Azari S., Ahmadi N., Tehrani M.J., Shokri F. Profiling and authentication of human cell lines using short tandem repeat (STR) loci: Report from the National Cell Bank of Iran. Biologicals. 2007;35:195–202. doi: 10.1016/j.biologicals.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Liscovitch M., Ravid D. A case study in misidentification of cancer cell lines: MCF-7/AdrR cells (re-designated NCI/ADR-RES) are derived from OVCAR-8 human ovarian carcinoma cells. Cancer Lett. 2007;245:350–352. doi: 10.1016/j.canlet.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 37.Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D., et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurman R.J., Shih I.M. The origin and pathogenesis of epithelial ovarian cancer- a proposed unifying theory. Am. J. Surg. Pathol. 2010;34:433–443. doi: 10.1097/PAS.0b013e3181cf3d79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mangili G., Bergamini A., Taccagni G., Gentile C., Panina P., Viganò P., Candiani M. Unraveling the two entities of endometrioid ovarian cancer: A single center clinical experience. Gynecol. Oncol. 2012;126:403–407. doi: 10.1016/j.ygyno.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 40.Anglesio M.S., Wiegand K.C., Melnyk N., Chow C., Salamanca C., Prentice L.M., Senz J., Yang W., Spillman M.A., Cochrane D.R., et al. Type-specific cell line models for type-specific ovarian cancer research. PLoS One. 2013;8:e72162. doi: 10.1371/journal.pone.0072162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Domcke S., Sinha R., Levine D.A., Sander C., Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013;4:2126. doi: 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu M., Selvaraj S.K., Liang-Chu M.M.Y., Aghajani S., Busse M., Yuan J., Lee G., Peale F., Klijn C., Bourgon R., et al. A resource for cell line authentication, annotation and quality control. Nature. 2015;520:307–312. doi: 10.1038/nature14397. [DOI] [PubMed] [Google Scholar]
- 43.Dirks W., MacLeod R.A., Jäger K., Milch H., Drexler H.G. First searchable database for DNA profiles of human cell lines: sequential use of fingerprint techniques for authentication. Cell Mol. Biol. 1999;45:841–853. [PubMed] [Google Scholar]
- 44.Korch C., Spillman M.A., Jackson T.A., Jacobsen B.M., Murphy S.K., Lessey B.A., Jordan V.G., Bradford A.P. DNA profiling analysis of endometrial and ovarian cell lines reveals misidentification, redundancy and contamination. Gynecol. Oncol. 2012;127:241–248. doi: 10.1016/j.ygyno.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lorenzi P.L., Reinhold W.C., Varma S., Hutchinson A.A., Pommier Y., Chanock S.J., Weinstein J.N. DNA fingerprinting of the NCI-60 cell line panel. Mol. Cancer Ther. 2009;8:713–724. doi: 10.1158/1535-7163.MCT-08-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.