


Abstract
Topoisomerase 1 (Top1) is essential for removing the DNA supercoiling generated during replication and transcription. Anticancer drugs like camptothecin (CPT) and its clinical derivatives exert their cytotoxicity by reversibly trapping Top1 in covalent complexes on the DNA (Top1cc). Poly(ADP-ribose) polymerase (PARP) catalyses the addition of ADP-ribose polymers (PAR) onto itself and Top1. PARP inhibitors enhance the cytotoxicity of CPT in the clinical trials. However, the molecular mechanism by which PARylation regulates Top1 nuclear dynamics is not fully understood. Using live-cell imaging of enhanced green fluorescence tagged-human Top1, we show that PARP inhibitors (Veliparib, ABT-888) delocalize Top1 from the nucleolus to the nucleoplasm, which is independent of Top1–PARP1 interaction. Using fluorescence recovery after photobleaching and subsequent fitting of the data employing kinetic modelling we demonstrate that ABT-888 markedly increase CPT-induced bound/immobile fraction of Top1 (Top1cc) across the nuclear genome, suggesting Top1-PARylation counteracts CPT-induced stabilization of Top1cc. We further show Trp205 and Asn722 of Top1 are critical for subnuclear dynamics. Top1 mutant (N722S) was restricted to the nucleolus in the presence of CPT due to its deficiency in the accumulation of CPT-induced Top1-PARylation and Top1cc formation. This work identifies ADP-ribose polymers as key determinant for regulating Top1 subnuclear dynamics.
INTRODUCTION
Topoisomerase I (Top1) is a ubiquitous enzyme essential for the relaxation of DNA supercoiling inside cells during the process of replication, transcription and chromosomal recombination (1,2). The mechanism by which Top1 alters the DNA supercoiling involves three major steps: (i) nucleophilic attack by the hydroxyl group of the active site tyrosine (Tyr723 for human Top1) on the scissile phosphate resulting in the covalent attachment of Top1 to the 3′ end of the broken strand (i.e. transient Top1–DNA cleavage complex; Top1cc), (ii) DNA relaxation involving controlled free rotation and (iii) religation of the DNA strand and release of the enzyme (1,2).
Top1 religation rate is much faster than cleavage rate, thus, the covalent enzyme–DNA complexes (Top1cc) are fleeting catalytic intermediates and normally not detectable. In contrast, the array of conditions that significantly enhance the frequency of trapped Top1cc inside the cells are: Top1 poisons, such as camptothecin (CPT) and its clinically used derivatives (irinotecan and topotecan), as well as several non-CPT Top1 inhibitors including the indenoisoquinolines and the indolocarbazoles (3). Endogenous and carcinogenic DNA lesions can also trap Top1cc (3). Top1cc catalytic intermediates can be converted into irreversible Top1–DNA cleavage complexes by colliding replication fork or transcription machinery (3,4), which trigger DNA double strand breaks (DSBs) and cell death (3).
Point mutations that confer CPT-resistance are distributed in the different domains of Top1 and are conserved among species (2,5–8). The Asn722Ser mutation in the C-terminal domain of Top1 is widely available among CPT-resistant cancer cell lines and is also conserved in CPT producing plants (7–9). Though Asn722 is next to the catalytic Tyr723 (Figure 1A), still Top1-Asn722Ser mutation was found to be catalytically active in vitro but resistant to CPT (8). Recent studies also highlight the importance of the N-terminal domain (191–206 aa) of Top1 related to in vivo activity, interaction with cofactors and CPT sensitivity (10–12). Precisely, Trp205 residue was suggested to control Top1 dynamics through tryptophan anchoring together with Trp203 and Trp206 on the DNA (13,14); (see the domain structure, Figure 1A), and stabilization of CPT-induced Top1cc (6,15). However, the impact of these point mutations on Top1 nuclear dynamics together with its cofactors and post-translational modifications (PTMs) have not been elucidated in live cells.
Figure 1.

Differential nuclear dynamics of enhanced green fluorescence (EGFP)–Top1 variants in live cell. (A) Schematic representation of human Top1 organized into four domains: N-terminal domain (1–214 aa), core (215–635 aa), linker (636–712 aa) and C-terminal domain (713–765 aa) based on the crystal structure(2,13). N-terminal domain harbours tryptophan anchor (W203, 205, 206), C-terminal domain harbours catalytic active site (Y723) and CPT interacting site (N722) are also shown. (B) Expression of EGFP–Top1 was unaffected with indicated mutations. Immunoblotting of HCT116 cells expressing ectopic EGFP–Top1WT (lane 1), EGFP–Top1Y723F (lane 2), EGFP–Top1N722S (lane 3), EGFP–Top1W205G (lane 4) and EGFP empty vector [VC] (lane 5). Blots were probed with antibodies against GFP (top) or Actin as loading control (below) (C) EGFP-linked and endogenous Top1 are indicated on the extreme right probed with human Top1 antibody. (D) Representative images showing colocalization of the ectopic EGFP–Top1WT (green) with the nucleoli (red). EGFP–Top1 WT construct was expressed in HCT116 cells in the absence (Ctr) or presence of CPT (10 μM for 30 min). Immunofluorescence staining of EGFP–Top1 with anti-GFP antibody (green) and anti-nucleolin antibody (red) was done. Cells were counterstained with DAPI to visualize nuclei. (E) Representative images showing differential nucleolar localization of EGFP–Top1 variants in the presence of camptothecin (CPT). All the EGFP–Top1 constructs were separately expressed in HCT116 cells and were imaged 24 h post-transfection under live cell confocal microscopy. Cells were treated with CPT (10 μM for 30 min) as indicated. Nuclei were stained with Hoechst 33342 (blue). (F) Densitometry analysis of CPT-induced delocalization of EGFP–Top1 variants (shown in panel E) from the nucleolus to the nucleoplasm. The percentage of cells displaying nucleolar fluorescence was determined from at least 60–70 cells expressing individual EGFP–Top1 constructs (WT, Y723F, N722S, W205G) in the presence and absence of indicated drugs. Error bars represent mean ± S.E. (n = 70).
The nucleolus is often considered as the stress sensing subnuclear compartment and functions as a site of ribosome biogenesis (16). Several DNA processing enzymes including Top1 are predominantly nucleolar protein and are required to relax DNA supercoils generated during rRNA synthesis (17,18). CPT-induced Top1cc has been shown to activate accumulation of antisense RNA polymerase (RNAP) II transcripts, which causes an interim block of RNAP II at the promoter and a transitory increase of R-loops particularly at the highly transcribed regions (clusters of ribosomal genes in the nucleolus) leading to transcription-dependent DSBs and genome instability (19–22). CPT causes delocalization of nucleolar Top1 to the nucleoplasm (23). The complex coordination of Top1 between nucleolus and nucleoplasm was previously attributed to mRNA synthesis (18), while others alternatively proposed that CPT-mediated modification of Top1 with small ubiquitin-like modifiers (SUMOs) may induce translocation of Top1 from the nucleoli to the nucleoplasm (24,25). Poly(ADP-ribose) polymerase-1 (PARP1) interacts with Top1 and shows colocalization in the nucleolus and nucleoplasm throughout the cell cycle (26–28). PARP1 is a ubiquitous chromatin-associated enzyme that catalyses the nicotinamide adenine dinucleotide (NAD+)-dependent addition of ADP-ribose polymers (PAR) onto itself (90% of PAR is found on PARP1) and several chromatin proteins including Top1 (29,30). Top1cc accumulates PAR polymers in CPT treated cells (31–33), which play vital role in Top1cc repair by directly regulating the recruitment of DNA repair proteins including Tyrosyl–DNA phosphodiesterase 1 (TDP1), X-ray repair cross-complementing protein 1 (XRCC1) and Ligase III (31–35). PARP1 also helps in releasing Top1 from stalled replication complexes by promoting the restart of replication forks reversed by Top1cc (36–38). In vitro studies suggest that PARP1 favours faster Top1 religation activity in the presence of CPT either through its direct interaction with Top1 or by the formation of PARylated Top1 (39,40).
DNA damaging anti-cancer agents constitute the mainstay of treatment for most solid tumours. PARP inhibitors generate considerable interest as anti-tumour agents especially for tumours defective in BRCA1 or BRCA2 (41–43), or in combination with Top1 inhibitors (CPT, irinotecan and topotecan) or with other DNA damaging agents (41,44,45). PARP inhibitors enhance the activity of CPT and its clinical derivatives both in cell culture, in xenograft model and in patients under clinical trials (31–33,44,46). Though Top1 is known to be PARylated for quite a long time (27,39,40), however the molecular mechanism by which PAR polymers regulate Top1 nuclear kinetics and localization has not been elucidated.
In the present study, we took the advantage of live-cell imaging and real-time monitoring of enhanced green fluorescence (EGFP) tagged-human Top1 variant combined with fluorescence recovery after photobleaching (FRAP) technology (47) to get new insights into Top1 dynamics such as association and dissociation reactions that are mediated by each of the functional amino acid residues. The FRAP kinetic modelling (48,49) indicates that Trp205 at the N-terminal and Asn722 at the C-terminal domain of Top1 are critical for subnuclear dynamics associated with CPT. Our study provides new evidence suggesting that ADP-ribose polymers are key molecular determinant for the regulation of Top1 mobility between subnuclear compartments and contributes new rationale for the combination of PARP inhibitors with Top1 inhibitors in cancer chemotherapy.
MATERIALS AND METHODS
Drug and antibodies
CPT was purchased from Sigma (St Louis, MO, USA). PARP inhibitor Veliparib (ABT-888) was kindly gifted by Dr Yves Pommier (NIH/NCI). Mouse monoclonal anti-human Top1 (C21) and anti-GFP (B-2) antibodies were from Santa Cruz Biotechnology (Santa Cruz, USA). Rabbit polyclonal anti-GFP (A-11122) antibody was from Invitrogen and anti-actin (ACTN05) antibody was from Neo Markers (USA). Rabbit polyclonal anti-nucleolin antibody (ab22758) was from Abcam (USA). The anti-PAR polymer mouse monoclonal (10H) and rabbit polyclonal antibodies were from Trevigen (USA). Rabbit polyclonal PARP1 antibody and secondary antibodies: horseradish peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG were obtained from Santa Cruz Biotechnology (Santa Cruz, USA).
Expression constructs and site-directed mutagenesis
The EGFP-tagged human Top1 fusion construct (24,50) was a kind gift from Dr William T. Beck (University of Illinois, IL, USA). The following point mutations: EGFP–Top1Y723F, EGFP–Top1N722S and EGFP–Top1W205G were constructed using the ‘QuikChange’ protocol (Stratagene, La Jolla, CA, USA). All constructs were confirmed by DNA sequencing.
Cell culture, treatment and transfections
The colon carcinoma cell line (HCT116) was obtained from the Developmental Therapeutics Program (NCI, NIH/USA). Cell cultures were maintained at 37°C under 5% CO2 in Dulbecco's modified Eagle's medium containing 10% fetal calf serum (Life Technologies). Cells were either treated with different concentrations of CPT or PARP inhibitor Veliparib (ABT-888) or combination of both as described (32,51). For Confocal microscopy experiments, cells (60–70% confluent) were grown on glass bottom dish where CPT or ABT-888 was added to a final concentration of 1–10 μM as indicated. Plasmid DNAs were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.
Cell extracts, immunoblotting and immunoprecipitation
Preparation of whole cell extracts, immunoprecipitation and immunoblotting were carried out as described (32,51,52). Briefly, HCT116 cells ectopically expressing EGFP–Top1WT, EGFP–Top1Y723F, EGFP–Top1N722S and EGFP–Top1W205G were separately lysed in a lysis buffer (10 mM Tris–HCl (pH 8), 150 mM NaCl, 0.1% sodium dodecyl sulphate, 1% NP40, 0.5% Na-deoxycholate supplemented with complete protease inhibitors) and phosphatase inhibitors (Phosphatase Inhibitor Cocktail 1 from Sigma). After thorough mixing and incubation at 4°C for 2 h, lysates were then centrifuged at 12 000 g for 20 min at 4°C. Supernatants were collected and stored in aliquotes at −80°C.
For immunoprecipitation, cells were lysed in a lysis buffer (50 mM Tris–HCl (pH 7.4), 300 mM NaCl, 0.4% NP-40, 10 mM MgCl2, 0.5 mM dithiothreitol supplemented with protease and phosphatase inhibitors). Supernatants of cell lysates were obtained by centrifugation at 15 000 g for 20 min at 4°C and pre-cleared with 50 μl of protein A/G-PLUS agarose beads (Santa Cruz, CA, USA). About 3–5 mg of pre-cleared lysate was incubated overnight at 4°C with indicated antibodies (2–5 mg/ml) and 50 μl of protein A/G-PLUS agarose beads. Isolated immunocomplexes were recovered by centrifugation, washed thrice with lysis buffer and were subjected to electrophoresis on 10% Tris-glycine gels and immunoblot analysis. Immunoblottings were carried out following standard procedures and immunoreactivity was detected using ECL chemiluminescence reaction (Amersham) under ChemiDoc™ MP System (Bio-Rad, USA).
In vitro topoisomerase I relaxation assay
Type I DNA topoisomerases are assayed by decreased mobility of the relaxed isomers of supercoiled pBS (SK+) DNA in agarose gel. The relaxation assays were carried out as described (53–55), briefly HCT116 cells ectopically expressing EGFP–Top1 variants (EGFP–Top1WT, EGFP–EGFP–Top1N722S or EGFP–Top1W205G) were immunoprecipitated (IP) with anti-GFP antibody and the immune complexes were used as source of Top1 (EGFP–Top1WT, EGFP–Top1Y723F, EGFP–Top1N722S or EGFP–Top1W205G) for the time course DNA relaxation experiments. The immune complexes with anti-IgG antibody served as control. The relaxation assays were performed in relaxation buffer (25 mM Tris–HCl, pH 7.5, 5% glycerol,0.5 mM Dithiothreitol (DTT), 10 mM MgCl2, 50 mM KCl, 25 mM ethylenediaminetetraacetic acid and 150 mg/ml bovine serum albumin) and supercoiled plasmid pBS (SK+) DNA (85–95% were negatively supercoiled, with remainder being nicked circles). For all kinetic studies, the reaction mixtures containing the buffer and DNA were heated to 37°C before addition of the enzyme. The reactions were rapidly quenched using stop solution and kept on ice. The amount of supercoiled monomer DNA band florescence after ethidium bromide (EtBr) (0.5 mg/ml) staining was quantitated by using Bio-Rad ChemiDoc™ MP system under UV illumination (Bio-Rad Quantity One software).
Live-cell confocal microscopy and immunocytochemistry
Live-cell imaging was carried out as described previously (51,52), using confocal laser-scanning microscope (Leica TCS SP8) with a UV-laser and 63X/1.4 NA oil objective equipped with a heated environmental chamber set to 37°C with optimal CO2 facility. Fluorophores were excited using a 488/514 nm argon laser line. All the EGFP–Top1 constructs were separately transfected in HCT116 cells cultured on cover glass bottom dish (Genetix, Biotech Asia Pvt. Ltd.) and were imaged for 24 h post-transfection under live cell confocal microscopy. Cells were treated with CPT, ABT-888 or combination of CPT + ABT-888 for 30 min with indicated concentrations. Nuclei were stained with Hoechst 33342 (Blue) (Sigma).
Immunofluorescence staining and confocal microscopy were performed as described previously (51,52). Briefly, HCT116 cells ectopically expressing EGFP–Top1WT treated with or without CPT were fixed with 4% paraformaldehyde for 10 min at room temperature. Primary antibodies against nucleolin and green fluorescent protein (GFP) were detected with anti-rabbit or anti-mouse IgG secondary antibodies labelled with Alexa 488/568 (Invitrogen). Cells were mounted in anti-fade solution with DAPI (Vector Laboratories, USA) and examined using a laser scanning confocal microscope. Images were collected and processed using the Leica software and sized in Adobe Photoshop 7.0. The percentage of cells displaying nucleolar fluorescence was determined with Adobe Photoshop 7.0 from at least 60–70 cells expressing individual EGFP–Top1 constructs.
Photobleaching experiments
Photobleaching experiments were carried out as described previously (32,51), using Andor Spining disc inverted confocal laser-scanning microscope equipped with a 60X/1.42 NA oil-immersion objective (Olympus). Fluorophores were excited using a 488 nm laser line. The microscope was equipped with a CO2-controlled on-stage heated environmental chamber set to 37°C. FRAP analyses were carried out with living HCT116 cells grown on chamber cover glass (Genetix, Biotech Asia Pvt. Ltd.). Cells were transfected with EGFP fusion proteins and mounted on an incubation chamber filled with medium 24 h after transfection. Cells were treated with different concentrations of CPT, PARP inhibitor (ABT-888) or combination of CPT + ABT-888 as indicated.
For FRAP analysis, a subnuclear spot was bleached for 30 ms by solid state laser line (488 nm for EGFP) adapted to the fluorescent protein of interest. For imaging, the laser power was attenuated to 0.1% of the bleach intensity. Subsequently, the recovery of fluorescence in the spot was continuously monitored for ∼90 s at 3 ms intervals. Relative fluorescence intensities of the bleached region were corrected for background. To show the FRAP curves, the fluorescence signal measured in a region of interest (ROI) was individually normalized to the pre-bleach signal in the ROI according to the following equation: ROI = (It-Ibg)/(Io-Ibg) × 100, where Io is the intensity in the ROI during pre-bleach, It is the intensity in the ROI at time point t and Ibg is the background signal determined in a region outside of the cell nucleus.
FRAP kinetic modelling and fitting
Analysis of FRAP data were performed by fitting them according to the general kinetic model of FRAP recovery that incorporates both free diffusion of the protein and binding of protein to other cellular components. In our present study the protein is Top1 (EGFP-tagged) and the binding partner is DNA. During DNA relaxation, Top1 binds DNA non covalently (during Top1 anchoring on the DNA) followed by transient covalent binding (nucleophilic attack by the hydroxyl group of the active site tyrosine on the scissile phosphate resulting in covalent attachment of Top1 to the 3′ end of the broken strand i.e. Top1–DNA cleavage complexes; Top1cc) and finally religation of the DNA strand and release of the enzyme (2,13). The dissociation timescale of strongly bound complex (irreversible Top1cc) in the presence of specific inhibitors (CPT) is generally very high and does not contribute to the recovery process. Therefore, we only consider the weak binding reaction and free diffusion of Top1 are the only two factors contributing towards recovery. In this situation, the equation for time dependent normalized FRAP can be written as (48,49),
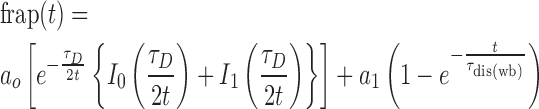 |
(1) |
Where, frap(t) represents normalized intensity of ROI or bleached point after photobleaching at time t. ao is the diffusion fraction, a1 is the fraction of weakly bound molecule. τD is the diffusion time and τdis(wb) is the dissociation time for weak bound molecule (koff = 1/τdis). Io and I1 are the basal function. (1-ao-a1) is the bound (immobile) fraction which corresponds to the fraction of strongly bound Top1 which does not contribute to the recovery. Using the value of 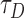 (diffusion time), we estimated the diffusion coefficient of the free Top1 as,
(diffusion time), we estimated the diffusion coefficient of the free Top1 as,
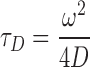 |
where, ω2 is the ROI area and D is diffusion coefficient.
RESULTS
CPT resistant mutant Top1 are defective in nucleolar delocalization
To investigate the impact of CPT on the nuclear dynamics of human Top1 (see the domain structure, Figure 1A) in live cells, we ectopically expressed wild-type (WT) EGFP-tagged nuclear Top1 (EGFP–Top1WT) and its mutant variants such as catalytically inactive mutant (EGFP–Top1Y723F), CPT-resistant mutant (EGFP–Top1N722S) in the C-terminal domain and N-terminal mutant (EGFP–Top1W205G) in HCT116 cells. We confirmed the expression of all the chimeric EGFP–Top1 variants by western blotting using anti-GFP antibody (Figure 1B). Top1 specific antibodies (Figure 1C), detect EGFP–Top1 variants as an additional band of slower migration (117 kDa) and with similar intensity as compared with endogenous Top1 (91 kDa), suggesting that the ectopic Top1 variants were not over expressed. Cells transfected with EGFP empty vector failed to show the additional band (Figure 1C, lane 5).
We confirmed the colocalization of the ectopic EGFP–Top1 with the nucleoli by double-immunostaining with an antibody specific against nucleolin (17,18,24). Figure 1D shows that in absence of CPT, EGFP–Top1WT was predominantly confined to the nucleolus, while CPT treatment markedly accumulate Top1 in the nucleoplasm (Figure 1D, panel CPT). Next, we followed the subnuclear distribution of the ectopic EGFP–Top1 variants using live cell confocal microscopy. CPT treatment delocalized Top1 (EGFP–Top1WT) from the nucleolus to the nucleoplasm in 85–90% of cells (Figure 1E, panel EGFP–Top1WT + CPT, see the quantification in Figure 1F), which is consistent with the earlier reports (18,23,24,56). Interestingly, under similar conditions, CPT failed to delocalize EGFP–Top1Y723F in 65–75% of cells (Figure 1E, panel EGFP–Top1Y723F + CPT, and the quantification in Figure 1F) and EGFP–Top1N722S in 70–85% of cells from the nucleoli to nucleoplasm (Figure 1E, panel EGFP–Top1N722S + CPT, and the quantification in Figure 1F). However, EGFP–Top1W205G showed CPT-induced partial re-distribution in the nucleoplasm (Figure 1E, panel EGFP–Top1W205G+ CPT, see the quantification in Figure 1F), suggesting that Top1 subnuclear rearrangements depend on CPT-sensitive Top1 activity.
To elucidate the mechanistic link between nuclear dynamics, catalytic activity and CPT sensitivity of Top1 variants, we tested them in plasmid DNA relaxation assays (53–55). We IP ectopic EGFP–Top1 and its mutants using anti-GFP antibody (Figure 2A) and the immune complexes were used as source of Top1 (EGFP–Top1WT, EGFP–Top1Y723F, EGFP–Top1N722S or EGFP–Top1W205G) for the time course DNA relaxation experiments (Figure 2B–F) in the presence or absence of CPT. As expected, no plasmid relaxation activity was observed with the IgG-control beads (Figure 2B) or catalytically inactive Top1 (EGFP–Top1Y723F) (Figure 2D). In the absence of CPT, EGFP–Top1WT relaxes supercoiled DNA at a similar rate as that of EGFP–Top1N722S and EGFP–Top1W205G (Figure 2C, E and F, panel (−) CPT). Under similar condition, the rate of plasmid DNA relaxation by EGFP–Top1WT was inhibited by CPT in a time dependent manner (see Figure 2C, panel (+) CPT and quantification in Figure 2G), whereas the drug has a reduced effect (∼2-fold) on the rate of relaxation by EGFP–Top1W205G (compare CPT-induced DNA relaxation inhibition (%) by WT versus W205G with time in Figure 2G), which may account for the abrogation of CPT-induced nucleolar delocalization in live cell microscopy (Figure 1E, panel CPT + EGFP–Top1W205G). Under similar conditions CPT failed to inhibit the DNA relaxation activity by EGFP–Top1N722S (Figure 2E and G). We therefore conclude that the molecular interactions of CPT with Top1–DNA cleavage complexes (Top1cc) drive or limit the mobility of Top1 in the nuclear compartments.
Figure 2.

Effect of CPT on the plasmid DNA relaxation activity of EGFP–Top1 variants. (A) EGFP-tagged wild-type (WT) and mutant (Y723F, N722S, W205G) Top1 constructs were ectopically expressed in HCT116 cells and were immunoprecipitated (IP) using anti-GFP antibody. The immune complexes were blotted with the anti-GFP antibody showing similar level of Top1 pull-down. Control immunoprecipitation with anti-IgG antibody demonstrates the specificity of the reactions. Protein molecular weight markers (kDa) are indicated at right. (B) Time kinetics plasmid DNA relaxation activity was performed with purified immune complexes of EGFP–Top1 variants (EGFP–Top1WT, EGFP–Top1Y723F, EGFP–Top1N722S or EGFP–Top1W205G) or anti-IgG antibody (as described in panel A) in the presence or absence of CPT. Representative gel showing relaxation of pBS (SK+) plasmid DNA (300 ng) by immune complexes of EGFP–Top1 variants (each reaction volume contains 0.5 μg protein) as source of Top1. Lanes 1 and 8, pBS (SK+) DNA; lanes 2–7, same as lane 1, but DNA was added with control IgG immune complex (B) with indicated time points at 37°C; lanes 9–14, same as lane 2–7, but in the presence of 10 μM CPT at 37°C for the indicated time periods. (C–F) Same as (B), but time kinetics plasmid DNA relaxation assays were performed with EGFP–Top1WT (C), EGFP–Top1Y723F (D), EGFP–Top1N722S (E) and EGFP–Top1W205G(F). All reactions were stopped by the addition of sodium dodecyl sulphate to a final concentration of 0.5% (w/v) and were electrophoresed in 1% agarose gel. Positions of supercoiled monomer (SM), relaxed and nicked monomer (RM/NM) are indicated. (G) Quantitative representation of CPT-induced Top1 (WT, W205G) relaxation inhibition (%) plotted as a function of time. Inhibition of DNA relaxation was calculated as the percentage of relaxed and nicked monomer (RM/NM) converted to supercoiled monomer (SM) as a function of time (C and F). Error bars represent mean ± S.E. (n = 3).
FRAP kinetic modelling shows accumulation of CPT-induced immobile Top1 in the nucleus
The FRAP techniques offer an effective tool to study the in vivo mobility of cellular proteins (47). To examine the FRAP kinetic properties of Top1 (EGFP–Top1WT) and the impact of CPT in living cells, we ectopically expressed EGFP–Top1 variants and analysed the same under a spinning disc confocal laser microscope (Figure 3A). We further utilized mathematical modelling (48,49) to estimate the quantitative parameters, such as the diffusion coefficient and immobile fractions of Top1 induced by CPT (Table 1).
Figure 3.

CPT accumulates immobile/bound Top1 in the nucleus. (A) Cartoon representing live cells Top1 dynamics in the presence and absence of CPT. Fluorescence tagged-human Top1 was evaluated with fluorescence recovery after photobleaching (FRAP) technology. Cells exhibit mainly two types of fluorescent molecules: mobile (unbound) and immobile (bound) (as indicated in the cartoon). After photobleaching at a region of interest (ROI) cells exhibit bleached molecules (grey). Exchanges occur between the mobile parts of two compartments. Top1 cleaves one strand of duplex DNA via the nucleophilic attack of its active site tyrosine (Y723) on the DNA phosphodiester backbone to yield a 3′-phosphotyrosyl bond. The short-lived covalent Top1–DNA cleavage complex (reversible Top1cc) is readily reversed by a second transesterification reaction in which the 5′-hydroxyl end acts as a nucleophile to religate the DNA and to free Top1 (No CPT). Top1 poisons i.e. CPT binds in the interface of Top1–DNA complex, stabilizes Top1cc (Top1 bound state/immobile) and inhibits the Top1-religation reaction. Bold arrow indicates the shift in the cleavage/religation equilibrium (3), with increasing population of bound/immobile fraction, fluorescence exchange rate is reduced in FRAP recoveries. (B) Representative images showing the FRAP of WT Top1 (EGFP–Top1WT) transiently expressed in HCT116 cells and their response to indicated CPT concentrations (1–10 μM). Cells expressing the ectopic proteins were kept untreated or treated with CPT (indicated) for 10 min and were analysed by live cell spinning disk confocal microscopy and photobleaching. A sub‐nuclear spot (ROI) indicated by a circle was bleached (BLH) for 30 ms and photographed at regular intervals of 3 ms thereafter. Successive images taken for ∼90 s after bleaching illustrate fluorescence return into the bleached areas. (C) Quantification of FRAP data showing mean curves of Top1 variants in the presence and absence of CPT. Error bars represent mean ± S.E. (n = 15).
Table 1. Estimation of diffusion constants and bound/immobile fraction of EGFP-tagged wild-type (WT) and mutant (Y723F, N722S, W205G) Top1 from FRAP recovery studies.
| TOP1 variants | Treatment | Bound(immobile) fraction | Diffusion constant (μm2/s) |
|---|---|---|---|
| WT | No drug | 0.18 | 1.66 |
| CPT | 0.35 | 1.16 | |
| ABT | 0.22 | 0.79 | |
| ABT + CPT | 0.64 | 1.75 | |
| Y723F | No drug | 0.1 | 1.02 |
| CPT | 0.11 | 1.35 | |
| ABT | 0.1 | 1.31 | |
| ABT + CPT | 0.14 | 1.09 | |
| N722S | No drug | 0.18 | 1.21 |
| CPT | 0.17 | 1.37 | |
| ABT | 0.19 | 1.29 | |
| ABT + CPT | 0.19 | 1.46 | |
| W205G | No drug | 0.14 | 1.23 |
| CPT | 0.2 | 1.16 | |
| ABT | 0.22 | 1.12 | |
| ABT + CPT | 0.27 | 1.95 |
Normalized fluorescence intensities after photobleaching were fitted into the equation Equation 1 (see ‘Materials and Methods’ section for details) to quantitate bound/immobile fraction and diffusion constant of Top1 variants in the absence or presence Top1 poison (CPT), PARP inhibitor (ABT-888) and combination of ABT-888 + CPT. Quantification are based on mean FRAP data (n = 15).
In absence of CPT, the FRAP recovery curves revealed two populations of EGFP–Top1WT: a mobile population representing ∼80–85% of the total and a smaller (∼15–20%) immobile population (see Figure 3B, and the quantification in 3C; No CPT), indicating that Top1 is mobile in the nucleus under steady state and were exchanged promptly between nuclear compartments. However, EGFP–Top1 shows slower recovery than EGFP alone (Supplementary Figure S1A, showing free diffusion of EGFP), suggesting that the Top1 is not freely diffusible. During DNA relaxation, Top1 binds DNA non-covalently followed by transient covalent binding (reversible Top1cc; Figure 3A) and finally religation of the DNA strand and release of the enzyme (2,13), which plausibly contributes to the (∼15–20%) immobile Top1 population in the FRAP recovery curves (Figure 3B). However in the presence of CPT (1 μM), Top1 is covalently trapped on the DNA (Figure 3A; CPT), which significantly blocks FRAP recovery by increasing ∼35–40% of Top1 immobile population in the nucleoplasm (Figure 3B, and the quantification in 3C; (+) CPT) as well as in the nucleolus (Supplementary Figure S1B). Fluorescence recovery of Top1 was further retarded by ∼40–55% with increasing dose of CPT (Figure 3B and C). The dissociation timescale of covalently bound complexes (irreversible Top1cc) in the presence of CPT are normally high and does not contribute to the FRAP recovery process. Therefore, we assume that CPT-induced immobile/bound fraction is exclusively due to the formation of Top1cc in the live cell nucleus (Figure 3A; CPT).
To further test this possibility, we performed the fluorescence recovery of Top1 inactive mutant (EGFP–Top1Y723F), which shows no significant variation in the mobile fraction of Top1Y723F either in the presence or absence of CPT (Supplementary Figure S1C, and the quantification in supplementary Figure 1D).
Next, kinetic modelling was applied to fit the complete set of Top1–FRAP data that best approximate the experimental dynamics (Supplementary Figures S2 and 3) and this yields an estimated CPT-induced bound/immobile fraction, mobile fraction and the diffusion rate of Top1. The diffusion coefficient for all the Top1 variants were comparable, suggesting no gross change in the diffusion rate of Top1 due to its point mutations (Table 1). However, we observed a marked accumulation (∼2-fold increase) of CPT-induced in vivo Top1cc (Table 1, WT), absent in the inactive mutant Top1Y723F (Table 1, Y723F), which is in agreement with the CPT mediated inhibition of the in vitro plasmid DNA relaxation activity (Figure 2C, panel WT (+) CPT). Together these results provide evidence that CPT impairs Top1 mobility and induce Top1cc (Figure 3) in the nucleus.
Top1-mutants (W205G and N722S) abrogate CPT-induced Top1 immobile fraction
Because Top1-mutants (N722S and W205G) were completely or partially resistant to CPT in the plasmid DNA relaxation assays (see Figure 2E and F, panel (+) CPT), we further investigated the FRAP kinetics of both EGFP–Top1N722S and EGFP–Top1W205G in the live cell's nucleus. As expected, we observed no significant variation in the fluorescence recovery of EGFP–Top1N722S in the presence or absence of CPT (Figure 4A and B). This is in complete agreement with the estimated bound fraction of Top1N722S in the presence or absence of CPT (Table 1, N722S; fittings of the FRAP data shown in Supplementary Figure S4), and accounts for resistance to CPT-induced dynamic mobility of Top1N722S in the nuclear compartments (Figure 1E and F).
Figure 4.

Top1 W205 and N722 residues are critical for CPT-induced nuclear dynamics. Representative images showing the FRAP of Top1mutants (A) EGFP–Top1N722S or (C) EGFP–Top1W205G transiently expressed in HCT116 cells. Cells were treated with CPT (indicated CPT concentration) for 10 min and were analysed by live cell spinning disk confocal microscopy and photobleaching (FRAP analysis). A sub-nuclear spot (ROI) indicated by a circle was bleached (BLH) for 30 ms and photographed at regular intervals of 3 ms thereafter. Successive images taken for ∼90 s after bleaching illustrate fluorescence return into the bleached areas. Right panels (B and D) quantification of FRAP data showing mean curves of Top1 variants in the presence and absence of CPT. Error bars represent mean ± S.E. (n = 15).
Interestingly, in absence of CPT, Top1W205G mutant shows faster rate of fluorescence recovery (∼85–90% of the total within 30 s, Figure 4D) indicating a significant increase in the mobile population when compared with the fluorescence recovery of the EGFP–Top1WT (70–80% at 30 s) (compare Figure 4D with 3C). However, CPT fails to trap Top1W205G on DNA with an estimated reduction of ∼1.5–2-fold in the CPT-induced bound/immobile Top1W205G compared to Top1WT (Table 1, compare WT versus W205G; fittings of Top1W205G-FRAP data shown in Supplementary Figure S5), which may account for CPT-induced partial nucleolar delocalization (Figure 1E and F). Therefore our live cell analyses provide new evidence suggesting Trp205 and Asn722 regulate CPT-induced nuclear dynamics of Top1.
(ADP)ribose polymers regulate Top1 subnuclear distribution
Top1 is heavily PARylated when trapped on DNA in the presence of CPT or due to DNA breaks associated with endogenous DNA damage, chromosomal transactions or DNA lesions (26,30,57). To determine the functional relationship between PAR polymers and Top1 dynamics, we tested the impact of the PARP inhibitor (veliparib, ABT-888) in the absence or presence of CPT under live cells microscopy. Top1 dynamics was determined under conditions that blocked cellular PAR formation (31,32). Figure 5A shows that in absence of ABT-888, EGFP–Top1WT was predominantly confined to the nucleolus as shown in Figure 1D. ABT-888 only, delocalized Top1 from the nucleolus to the nucleoplasm in 55–65% of cells (Figure 5A, panel WT + ABT-888 and the quantification in Figure 5B). However, ABT-888 failed to delocalize Top1Y723F and Top1N722S in 70–80% of cells (Figure 5A, panel Y723F or N722S + ABT-888 and the quantification in Figure 5B) like CPT (Figure 1E). Co-treatment of ABT-888 + CPT, shows similar impact on nuclear localization of Top1Y723F and EGFP–Top1N722S as observed independently with CPT (Figure 5A, panels Y723F and N722S; quantification in Figure 5B). In contrast, ABT-888 or co-treatment with ABT-888 + CPT induces significant delocalization of Top1 mutants (EGFP–Top1W205G) from the nucleoli to nucleoplasm (Figure 5A; panel W205G and the quantification in Figure 5B), suggesting PAR polymers recruit Top1 in the nucleolus (Figure 1).
Figure 5.

(ADP)ribose polymers regulate Top1 subnuclear distribution. (A) Representative live cell images showing nucleolar delocalization of ectopic EGFP–Top1WT when treated with PARP inhibitor (ABT-888; 10 μM for 30 min) or combination of CPT + ABT-888 (10 μM each for 30 min) in HCT116 cells. EGFP–Top1 mutant variants: EGFP–Top1Y723F, EGFP–Top1N722S, EGFP–Top1W205G show differential nuclear dynamics in presence of PARP inhibitor (ABT-888) or combination of CPT + ABT-888 under live cell confocal microscopy. Nuclei were stained with Hoechst 33342 (blue). (B) Densitometry analysis of delocalization of EGFP–Top1 variants (shown in panel A) from the nucleolus to the nucleoplasm in the presence and absence of indicated drug treatment. The percentage of cells displaying nucleolar fluorescence was determined from at least 60–70 cells expressing individual EGFP–Top1 constructs (WT, Y723F, N722S, W205G) in the absence and presence of drugs. Error bars represent mean ± S.E. (n = 60). (C) Detection of CPT-induced PARylation of ectopic Top1. EGFP-tagged WT and mutant (Y723F, N722S, W205G) Top1 constructs were ectopically expressed in HCT116 cells. Following treatment with 10 μM CPT for 2 h, EGFP–Top1 was IP using anti-GFP antibody and the immune complexes were probed with anti-PAR and anti-PARP1 antibody. Blots were subsequently stripped and probed with anti-GFP antibody to detect equal loading of ectopic EGFP-tagged Top1 variants. Aliquots (10%) of the input show the uniform presence of PARP1 prior to immunoprecipitation. Protein molecular weight markers (kDa) are indicated at right. Control immunoprecipitation with anti-IgG antibody demonstrates the specificity of the reactions. Note: PAR signal was observed with a prominent band ∼120 kDa, consistent with PARylation of ectopic EGFP–Top1 as indicated.
Because Top1 is a part of PARP1 complexes (26,27) and ABT-888 induces delocalization of Top1 to the nucleoplasm (Figure 5A and B), therefore, the next question was whether the differential nuclear dynamics of EGFP–Top1N722S is due to its deficiency in the formation of PARP1–Top1 complex or PARylation of Top1N722S. To further examine these possibilities, we co-IP ectopic EGFP–Top1 variants in cells treated with CPT. PARP1 was readily detected in all the immunoprecipitates independent of Top1 mutations (Figure 5C). Under these conditions, PAR signal was observed with a prominent band of ∼120 kDa, consistent with PARylation of ectopic EGFP–Top1 (Figure 5C). However, under similar conditions we detected limited PAR signal in the co-immunoprecipitates of EGFP–Top1N722S (Figure 5C, top panel, fifth lane) compared to other Top1 variants (Top1WT or Top1W205G) as measured by western blotting. Therefore, the attenuation in the subnuclear rearrangement of the Top1N722S mutant in response to CPT + ABT-888 is not due to its inability to form PARP1–Top1 complexes but due to its deficiency in accumulation of PAR polymers (Figure 5C, N722S), which is in agreement with the decreased PAR signal on the catalytically inactive Top1 (Top1Y723F) (Figure 5C, top panel, third lane). Taken together, these results suggest that PAR polymers regulate Top1 dynamics in the nucleus.
PARP inhibitor (ABT-888) enhance CPT-induced Top1 trapping on the DNA
Because PARP inhibitors (ABT-888) efflux Top1 from the nucleolus to the nucleoplasm (Figure 5A), we investigated the potential involvement of PAR polymers on Top1 trapping on DNA using FRAP experiments in cells treated with PARP inhibitors and/or in combination with CPT (Figure 6). FRAP kinetics of EGFP–Top1WT in the presence of ABT-888 (Figure 6A and B) was determined under conditions that blocked cellular PAR formation (31,32). In the presence of ABT-888, Top1 shows faster rate of fluorescence recovery similar to untreated condition (70–80%) at early times (30 s), however between 50 and 90 s we observed small but significant reduction (∼5–8%) in the ABT-888 induced mobile population of Top1 (Figure 6B). The impact was more dramatic with combination of CPT + ABT-888 compared to CPT alone (Figure 6A and B) as evidenced by marked reduction (25–30%) in the mobile fraction of EGFP–Top1WT (Figure 6C). FRAP kinetic modelling shows CPT + ABT-888 markedly increased (∼2-fold) the bound/immobile fraction of Top1 (Top1cc) compared to CPT alone (fittings of FRAP data are shown in Supplementary Figure S2 and estimated values are shown in Table 1; WT).
Figure 6.

PARP inhibitor (ABT-888) promotes CPT-induced prolong Top1 trapping across nuclear genome. (A) Representative images showing the FRAP of WT Top1 (EGFP–Top1WT) transiently expressed in HCT116 cells and their response to CPT or ABT-888 or combination of ABT-888+ CPT at indicated concentrations for 10 min. A sub-nuclear spot (ROI) indicated by a circle was bleached (BLH) for 30 ms and photographed at regular intervals of 3 ms thereafter. Successive images taken for ∼90 s after bleaching illustrate fluorescence return into the bleached areas. (B) Quantification of FRAP data showing mean curves of EGFP–Top1WT separately treated with CPT, ABT-888 or combination of ABT-888 + CPT. Error bars represent mean ± S.E. (n = 15). (C) Quantification of bound Top1cc calculated from the FRAP of ectopic EGFP-tagged WT and mutant (Y723F, N722S, W205G) Top1 constructs in the presence of indicated drugs. Percentage of FRAP recovery was estimated after 60 s normalized with FRAP recovery with no treatment. Bound fraction of all Top1 variants were plotted as function of drug treatment [CPT (1 μM) or ABT-888 (1 μM) or combination of CPT + ABT-888 (1 μM each) for 10 min]. Error bars represent the standard error of the mean.
The impact of ABT-888 + CPT combination on prolonged Top1 trapping was further rationalized by the deficiencies in FRAP recoveries for Top1 mutants (EGFP–Top1Y723F and EGFP–Top1N722S), which failed to show the accumulation of Top1 bound fractions (see the FRAP quantitation in Figure 6C) in the presence of ABT-888 + CPT (fittings of FRAP data are shown in Supplementary Figure S3 and 4 and the estimated values are shown in Table 1; Y723F and N722S). Consistent with the resistance of EGFP–Top1W205G to form CPT-induced Top1–DNA covalent complexes in the plasmid relaxation assays (Figure 2F) and FRAP kinetic modelling (Supplementary Figure S5 and estimated values are shown in Table 1; W205G), ABT-888 failed to markedly increase the CPT-induced bound/immobile fraction of EGFP–Top1W205G in FRAP recoveries (Figure 6C). Taken together, these results indicate that PAR polymers indeed counteract CPT-induced stabilization of Top1cc in the live cell nucleus.
DISCUSSION
Poly(ADP)ribose polymerases are DNA nick sensors and have been proposed to play a critical role in the early detection and repair of Top1cc-induced DNA breaks (29–35,57–59). Top1 is an acceptor of PAR polymers (26,30), we describe here a new role for PAR polymers on Top1 nuclear dynamics, which is independent from Top1–PARP1 interaction but dependent on PARP catalytic activity. We examine the effects of PARP inhibitors and/or in combination with CPT on EGFP-tagged-human Top1 subnuclear dynamics in living cells. Using a combination of live cell microscopy and FRAP kinetic modelling, we establish that orally bioactive PARP inhibitors (Veliparib, ABT-888) efflux Top1 from the nucleolus to the nucleoplasm. FRAP data reveals that combination of ABT-888 with CPT markedly increased the bound/immobile fraction of Top1 (Top1cc) across the nuclear genome compared to CPT alone. In an unprecedented finding, we conclusively demonstrate that EGFP–Top1N722S are restricted to the nucleolus due to its deficiency in accumulation of CPT-induced Top1–PARylation and Top1cc formation.
The regulation of cellular Top1 mobility is only partially understood. Top1 is a dynamic protein in the nucleus and there is continuous efflux between the nucleolus and nucleoplasm (23,24,56). The CPT induced nuclear redistribution of Top1 was suggested to be p53-dependent (56) or associated with SUMOs, which may induce translocation of Top1 from the nucleoli to the nucleoplasm (25,60). The nucleolus is the subnuclear compartment that requires high rate of replication and transcription for ribosomal RNA synthesis (16). Therefore, Top1 is essential for the maintenance of rDNA supecoiling (61). Hence, we observed EGFP–Top1 was predominantly confined to the nucleolus (Figure 1D and E), which is consistent with the previous reports (23,24,56). Other than Top1, a high density of RNA Polymerase I (RNA Pol I) and a large number of chromatin associated proteins like PARP1, XRCC1, BRCA1, RNF8, Werner syndrome helicase, Bloom syndrome helicase and NBS1 are recruited to the nucleolus for the mRNA synthesis and maintenance of the rDNA (18,28,62,63). Consistent with the requirement of Top1 catalytic activity in rRNA synthesis, selective inhibition of RNA polymerase I with actinomycin D or inhibition of transcription with DRB (5,6-dichlorobenzimidazole 1-β-D-ribofuranoside) results in a rapid redistribution of EGFP–Top1 from nucleolus to the nucleoplasm (18,56).
The N-terminal region of Top1 (1–215 aa) is highly charged, and harbours the nucleolar targeting regions as it forms complexes with nucleolin (64). Although this domain is dispensable for catalytic activity of Top1 in vitro, however is important for in vivo activity, interactions with cofactors and CPT sensitivity (10,15,53,56). Our live cell imaging with EGFP–Top1 single point mutation (W205G) in the N-terminal region shows resistance to CPT-induced redistribution in the nucleoplasm (Figure 1E and F) as well as CPT-mediated inhibition of the plasmid DNA relaxation activity (Figure 2F). Top1W205G mutant shows faster rate of fluorescence recovery compared to EGFP–Top1WT (Figure 4C) indicating a significant increase in the mobile population (compare Figure 3C with Figure 4D) due to its weak association with DNA (10,12,15). Thus our live cell data provide evidence in favour of the conclusion that Trp205 helps in anchoring of Top1 on DNA (14,15). However, CPT was significantly deficient in trapping Top1W205G on DNA compared to Top1WT (Table 1; compare CPT-induced immobile fraction for WT versus W205G) as well as in the in vitro plasmid DNA relaxation assays (Figure 2G), suggesting Trp205 is critical for in vivo molecular interaction of CPT with Top1–DNA cleavage complexes.
PARP1 is responsible for the majority of cellular PAR formation (29–31). PARylation is a key PTM orchestrating a variety of cellular processes including DNA repair, transcription, ribosome biogenesis, mitotic segregation, telomere homeostasis and genome maintenance (29,65). Poly(ADP)ribosylation of ribosomal proteins is essential for nucleolar structure, ribosome biogenesis (66,67), rRNA synthesis (65,68) and epigenetic upkeep of the rDNA (63). PARP1 plays a crucial role in DNA damage response by controlling the nuclear localization and biological activities of DNA repair complexes and the conservation and stability of ribosomal genes (29–32,65). PARP1 is a key regulator of the single-strand break repair and base excision repair pathway and are backup pathway for the non-homologous end joining double-strand break repair pathway (29,30,32). Like Top1, PARP1 is also dynamic in the nucleus, unmodified PARP1 protein molecules bind to chromatin and accumulate in the nucleoli, upon automodification with poly(ADP-ribose), PARP1 dissociates from the chromatin template and facilitates the recruitment of other chromatin factors (29,30). Consistent with the role of PARP1 activity in rRNA transcription, inhibition of transcription or RNA polymerase I activity delocalize PARP1 from the nucleolus to the nucleoplasm (69). Cells exposed to CPT results in delocalization of both Top1 (Figure 1) and PARP1 (26,28) from the nucleolus to nucleoplasm. Because CPT failed to delocalize the Top1-mutants (W205G, N722S and Y723F) from the nucleolus (Figure 1E and F) and ABT-888 markedly accumulates Top1 in the nucleoplasm (Figure 5A and B), therefore the phenomenon of Top1 nucleolar delocalization is coupled with PARP catalytic activity (Figure 5A). However, our data suggests that PARP1–Top1 interactions (Figure 5C) do not play a role in the delocalization of Top1 from the nucleolus. This is in keeping with the previous report indicating CPT-induced nucleolar delocalization kinetics of Top1 and PARP1 are not concurrent (26,28). Thus it is plausible that activated PARP1 catalyse Top1–PARylation (27,39) (Figures 5C and 7D), which facilitates Top1 catalytic activity (39,40) and turnover of the rDNA synthesis, as PARP1 and Top1 complexes are readily detectable in cells (Figure 5C). Consistent with this conclusion, our FRAP data indicates that both CPT and ABT-888 failed to show accumulation of Top1N722S-bound fractions (see the FRAP quantitation in Figure 6C and Table 1; N722S) as well as redistribution from the nucleolus to the nucleoplasm (Figure 5A) due to limited CPT-induced Top1–PARylation (Figure 5C). Thus, we conclude that the PARylation of Top1 not only counteracts CPT-induced stabilization of Top1cc but also recruits Top1 to the active site of rDNA and rRNA synthesis.
Figure 7.

Schematic representation of our findings regarding the Top1 dynamics in live cells and response to combination of PARP inhibitor (ABT-888) plus CPT as established by FRAP. (A) Cartoon representing a cell expressing fluorescent molecules (EGFP–Top1). The selected circular ROI of the cell is bleached by laser and imaged before and after photobleaching. The fluorescent intensity recovery of ROI by the surrounding molecule into the bleached area is monitored over time. Cells exhibit mainly two types of fluorescent molecule: mobile (unbound) and immobile (bound) (as indicated in the cartoon). After photobleaching cells exhibit bleached molecules. Exchanges occur between the mobile parts of two compartments. (B) Top1 poison (CPT) induces bound Top1cc formation. With increasing population of bound/immobile fraction, fluorescence exchange rate is reduced in FRAP recoveries. (C) PARP inhibitor (ABT-888) increased Top1 density in the nucleoplasm and enhanced the activity of CPT by trappingTop1cc (immobile fraction) across the genome. (D) Schematic representation of PARP1–Top1 coupling for regulation of Top1 catalytic activity. PARylation of Top1 helps in the religation of CPT-induced Top1 cleavage complex. ABT-888 prevents the Top1-PARylation and induces prolong trapping of CPT-induced Top1cc.
PARP inhibitors generate considerable interest in clinical trials in combination with wide variety of DNA damaging drugs which includes alkylating agents (temozolomide), Top1 inhibitors (CPT and its clinical derivatives topotecan and irinotecan) and others (30,41,46). The potential ability of PARP inhibitors for trapping PARP–DNA complexes in addition to their NAD+-competitive catalytic inhibitory mechanism accounts for the cytotoxicity in the proliferative cells (44). PARP trapping is critical when combined with temozolomide (45), while combination with Top1 poisons (CPT) is synergistic in killing cells due to inhibition of PARP catalytic activity (32,41,44,45). The present study provides new mechanistic insights into the action of PARP inhibitors in combination with Top1 inhibitors relevant for cancer chemotherapy. Using live cell microscopy in the presence of ABT-888 [PARP inhibitor with least potential in trapping PARP–DNA complex; (41,44,45)], we show delocalization of Top1 from the nucleolus which results in increased Top1 density in the nucleoplasm. PARylation of Top1 counteracts CPT-induced stabilization of Top1cc. Therefore ABT-888 markedly increased CPT-induced trapping of Top1 across the nuclear genome (Figure 7), which is associated with increased cytotoxicity in the proliferating cells exposed to the combination of PARP inhibitor with Top1 inhibitor (31–33,41,44–46). Indeed, (ADP)ribose polymerase appears to act as a key molecular determinant both for Top1 nuclear dynamics (present study) as well as Top1cc repair by recruitment of PARylated–TDP1–PARP1 complex at Top1cc damage sites (32,35,57–59). Thus, this study provides rationale for the combination of PARP inhibitors with Top1 inhibitors in cancer treatment, which is highly relevant for the ongoing clinical trials.
Supplementary Material
Acknowledgments
The authors thank Dr Yves Pommier, NIH/NCI, USA and Dr Hemanta K Majumder, CSIR-Indian Institute of Chemical Biology, India for their suggestions and helpful discussions during the study. We are also thankful to Dr Suvendra Nath Bhattacharyya, CSIR-Indian Institute of Chemical Biology for giving us the opportunity to use his Andor Spining disc confocal laser-scanning microscope facility.
Footnotes
Present address: Souvik Sengupta, Institute of Life Sciences, Ahmedabad University, Navrangpura, Ahmedabad, Gujarat-380009, India.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
WellcomeTrust/DBT India Alliance Intermediate Fellowship [IA/I/13/1/500888 to B.B.D. team]; Indian Association for the Cultivation of Science, Department of Science and Technology, Government of India startup fund (to B.B.D. team); CSIR/NET Senior Research Fellowship (to AG). Funding for open access charge: WellcomeTrust/DBT India Alliance Intermediate Fellowship [IA/I/13/1/500888 to B.B.D.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Wang J.C. Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 2.Champoux J.J. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 3.Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem. Biol. 2013;8:82–95. doi: 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sordet O., Redon C.E., Guirouilh‐Barbat J., Smith S., Solier S., Douarre C., Conti C., Nakamura A.J., Das B.B., Nicolas E. Ataxia telangiectasia mutated activation by transcription‐and topoisomerase I‐induced DNA double‐strand breaks. EMBO Rep. 2009;10:887–893. doi: 10.1038/embor.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pommier Y., Pourquier P., Urasaki Y., Wu J., Laco G.S. Topoisomerase I inhibitors: selectivity and cellular resistance. Drug Resist. Updat. 1999;2:307–318. doi: 10.1054/drup.1999.0102. [DOI] [PubMed] [Google Scholar]
- 6.Pommier Y., Marchand C. Interfacial inhibitors: targeting macromolecular complexes. Nat. Rev. Drug Discov. 2012;11:25–36. doi: 10.1038/nrd3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rasheed Z.A., Rubin E.H. Mechanisms of resistance to topoisomerase I-targeting drugs. Oncogene. 2003;22:7296–7304. doi: 10.1038/sj.onc.1206935. [DOI] [PubMed] [Google Scholar]
- 8.Fujimori A., Harker W.G., Kohlhagen G., Hoki Y., Pommier Y. Mutation at the catalytic site of topoisomerase I in CEM/C2, a human leukemia cell line resistant to camptothecin. Cancer Res. 1995;55:1339–1346. [PubMed] [Google Scholar]
- 9.Sirikantaramas S., Yamazaki M., Saito K. Mutations in topoisomerase I as a self-resistance mechanism coevolved with the production of the anticancer alkaloid camptothecin in plants. Proc. Natl. Acad. Sci. U.S.A. 2008;105:6782–6786. doi: 10.1073/pnas.0801038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christensen M.O., Barthelmes H.U., Boege F., Mielke C. Residues 190–210 of human topoisomerase I are required for enzyme activity in vivo but not in vitro. Nucleic Acids Res. 2003;31:7255–7263. doi: 10.1093/nar/gkg927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lisby M., Olesen J.R., Skouboe C., Krogh B.O., Straub T., Boege F., Velmurugan S., Martensen P.M., Andersen A.H., Jayaram M. Residues within the N-terminal domain of human topoisomerase I play a direct role in relaxation*. J. Biol. Chem. 2001;276:20220–20227. doi: 10.1074/jbc.M010991200. [DOI] [PubMed] [Google Scholar]
- 12.Frøhlich R.F., Andersen F.F., Westergaard O., Andersen A.H., Knudsen B.R. Regions within the N-terminal domain of human topoisomerase I exert important functions during strand rotation and DNA binding. J. Mol. Biol. 2004;336:93–103. doi: 10.1016/j.jmb.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 13.Redinbo M.R., Stewart L., Kuhn P., Champoux J.J., Hol W.G. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science. 1998;279:1504–1513. doi: 10.1126/science.279.5356.1504. [DOI] [PubMed] [Google Scholar]
- 14.Laco G., Pommier Y. Role of a tryptophan anchor in human topoisomerase I structure, function and inhibition. Biochem. J. 2008;411:523–530. doi: 10.1042/BJ20071436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frøhlich R.F., Veigaard C., Andersen F.F., McClendon A.K., Gentry A.C., Andersen A.H., Osheroff N., Stevnsner T., Knudsen B.R. Tryptophane-205 of human topoisomerase I is essential for camptothecin inhibition of negative but not positive supercoil removal. Nucleic Acids Res. 2007;35:6170–6180. doi: 10.1093/nar/gkm669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lam Y.W., Trinkle-Mulcahy L., Lamond A.I. The nucleolus. J. Cell Sci. 2005;118:1335–1337. doi: 10.1242/jcs.01736. [DOI] [PubMed] [Google Scholar]
- 17.Mao Y., Mehl I.R., Muller M.T. Subnuclear distribution of topoisomerase I is linked to ongoing transcription and p53 status. Proc. Natl. Acad. Sci. U.S.A. 2002;99:1235–1240. doi: 10.1073/pnas.022631899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christensen M.O., Krokowski R.M., Barthelmes H.U., Hock R., Boege F., Mielke C. Distinct effects of topoisomerase I and RNA polymerase I inhibitors suggest a dual mechanism of nucleolar/nucleoplasmic partitioning of topoisomerase I. J. Biol. Chem. 2004;279:21873–21882. doi: 10.1074/jbc.M400498200. [DOI] [PubMed] [Google Scholar]
- 19.Bertozzi D., Iurlaro R., Sordet O., Marinello J., Zaffaroni N., Capranico G. Characterization of novel antisense HIF-1alpha transcripts in human cancers. Cell Cycle. 2011;10:3189–3197. doi: 10.4161/cc.10.18.17183. [DOI] [PubMed] [Google Scholar]
- 20.Marinello J., Chillemi G., Bueno S., Manzo S.G., Capranico G. Antisense transcripts enhanced by camptothecin at divergent CpG-island promoters associated with bursts of topoisomerase I-DNA cleavage complex and R-loop formation. Nucleic Acids Res. 2013;41:10110–10123. doi: 10.1093/nar/gkt778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marinello J., Bertoncini S., Aloisi I., Cristini A., Malagoli Tagliazucchi G., Forcato M., Sordet O., Capranico G. Dynamic Effects of Topoisomerase I Inhibition on R-Loops and Short Transcripts at Active Promoters. PLoS One. 2016;11:e0147053. doi: 10.1371/journal.pone.0147053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cristini A., Park J.H., Capranico G., Legube G., Favre G., Sordet O. DNA-PK triggers histone ubiquitination and signaling in response to DNA double-strand breaks produced during the repair of transcription-blocking topoisomerase I lesions. Nucleic Acids Res. 2016;44:1161–1178. doi: 10.1093/nar/gkv1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christensen M.O., Barthelmes H.U., Feineis S., Knudsen B.R., Andersen A.H., Boege F., Mielke C. Changes in mobility account for camptothecin-induced subnuclear relocation of topoisomerase I. J. Biol. Chem. 2002;277:15661–15665. doi: 10.1074/jbc.C200066200. [DOI] [PubMed] [Google Scholar]
- 24.Mo Y.-Y., Yu Y., Shen Z., Beck W.T. Nucleolar delocalization of human topoisomerase I in response to topotecan correlates with sumoylation of the protein. J. Biol. Chem. 2002;277:2958–2964. doi: 10.1074/jbc.M108263200. [DOI] [PubMed] [Google Scholar]
- 25.Rallabhandi P., Hashimoto K., Mo Y.-Y., Beck W.T., Moitra P.K., D'Arpa P. Sumoylation of topoisomerase I is involved in its partitioning between nucleoli and nucleoplasm and its clearing from nucleoli in response to camptothecin. J. Biol. Chem. 2002;277:40020–40026. doi: 10.1074/jbc.M200388200. [DOI] [PubMed] [Google Scholar]
- 26.Yung T.M., Sato S., Satoh M.S. Poly (ADP-ribosyl) ation as a DNA damage-induced post-translational modification regulating poly (ADP-ribose) polymerase-1-topoisomerase I interaction. J. Biol. Chem. 2004;279:39686–39696. doi: 10.1074/jbc.M402729200. [DOI] [PubMed] [Google Scholar]
- 27.Bauer P.I., Chen H.-J., Kenesi E., Kenessey I., Buki K.G., Kirsten E., Hakam A., Hwang J.I., Kun E. Molecular interactions between poly (ADP-ribose) polymerase (PARP I) and topoisomerase I (Topo I): identification of topology of binding. FEBS Lett. 2001;506:239–242. doi: 10.1016/s0014-5793(01)02919-2. [DOI] [PubMed] [Google Scholar]
- 28.Rancourt A., Satoh M.S. Delocalization of nucleolar poly (ADP-ribose) polymerase-1 to the nucleoplasm and its novel link to cellular sensitivity to DNA damage. DNA Repair. 2009;8:286–297. doi: 10.1016/j.dnarep.2008.11.018. [DOI] [PubMed] [Google Scholar]
- 29.Schreiber V., Dantzer F., Ame J.-C., De Murcia G. Poly (ADP-ribose): novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 30.Rouleau M., Patel A., Hendzel M.J., Kaufmann S.H., Poirier G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y.-W., Regairaz M., Seiler J.A., Agama K.K., Doroshow J.H., Pommier Y. Poly (ADP-ribose) polymerase and XPF–ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res. 2011;39:3607–3620. doi: 10.1093/nar/gkq1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Das B.B., Shar-yin N.H., Murai J., Rehman I., Amé J.-C., Sengupta S., Das S.K., Majumdar P., Zhang H., Biard D. PARP1–TDP1 coupling for the repair of topoisomerase I–induced DNA damage. Nucleic Acids Res. 2014;42:4435–4449. doi: 10.1093/nar/gku088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patel A.G., Flatten K.S., Schneider P.A., Dai N.T., McDonald J.S., Poirier G.G., Kaufmann S.H. Enhanced killing of cancer cells by poly (ADP-ribose) polymerase inhibitors and topoisomerase I inhibitors reflects poisoning of both enzymes. J. Biol. Chem. 2012;287:4198–4210. doi: 10.1074/jbc.M111.296475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.El‐Khamisy S.F., Masutani M., Suzuki H., Caldecott K.W. A requirement for PARP‐1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003;31:5526–5533. doi: 10.1093/nar/gkg761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pommier Y., Shar-yin N.H., Gao R., Das B.B., Murai J., Marchand C. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2) DNA Repair. 2014;19:114–129. doi: 10.1016/j.dnarep.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugimura K., Takebayashi S.-I., Taguchi H., Takeda S., Okumura K. PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J. Cell Biol. 2008;183:1203–1212. doi: 10.1083/jcb.200806068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berti M., Chaudhuri A.R., Thangavel S., Gomathinayagam S., Kenig S., Vujanovic M., Odreman F., Glatter T., Graziano S., Mendoza-Maldonado R. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013;20:347–354. doi: 10.1038/nsmb.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chaudhuri A.R., Hashimoto Y., Herrador R., Neelsen K.J., Fachinetti D., Bermejo R., Cocito A., Costanzo V., Lopes M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012;19:417–423. doi: 10.1038/nsmb.2258. [DOI] [PubMed] [Google Scholar]
- 39.Malanga M., Althaus F.R. Poly (ADP-ribose) reactivates stalled DNA topoisomerase I and induces DNA strand break resealing. J. Biol. Chem. 2004;279:5244–5248. doi: 10.1074/jbc.C300437200. [DOI] [PubMed] [Google Scholar]
- 40.Park S.-Y., Cheng Y.-C. Poly (ADP-ribose) polymerase-1 could facilitate the religation of topoisomerase I-linked DNA inhibited by camptothecin. Cancer Res. 2005;65:3894–3902. doi: 10.1158/0008-5472.CAN-04-4014. [DOI] [PubMed] [Google Scholar]
- 41.Lord C.J., Tutt A.N., Ashworth A. Synthetic lethality and cancer therapy: lessons learned from the development of PARP inhibitors. Annu. Rev. Med. 2015;66:455–470. doi: 10.1146/annurev-med-050913-022545. [DOI] [PubMed] [Google Scholar]
- 42.Bryant H.E., Schultz N., Thomas H.D., Parker K.M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N.J., Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly (ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 43.Farmer H., McCabe N., Lord C.J., Tutt A.N., Johnson D.A., Richardson T.B., Santarosa M., Dillon K.J., Hickson I., Knights C. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 44.Murai J., Shar-yin N.H., Das B.B., Renaud A., Zhang Y., Doroshow J.H., Ji J., Takeda S., Pommier Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murai J., Zhang Y., Morris J., Ji J., Takeda S., Doroshow J.H., Pommier Y. Rationale for poly (ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J. Pharmacol. Exp. Therap. 2014;349:408–416. doi: 10.1124/jpet.113.210146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kummar S., Chen A., Ji J., Zhang Y., Reid J.M., Ames M., Jia L., Weil M., Speranza G., Murgo A.J. Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res. 2011;71:5626–5634. doi: 10.1158/0008-5472.CAN-11-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reits E.A., Neefjes J.J. From fixed to FRAP: measuring protein mobility and activity in living cells. Nat. Cell Biol. 2001;3:E145–E147. doi: 10.1038/35078615. [DOI] [PubMed] [Google Scholar]
- 48.Sprague B.L., Pego R.L., Stavreva D.A., McNally J.G. Analysis of binding reactions by fluorescence recovery after photobleaching. Biophys. J. 2004;86:3473–3495. doi: 10.1529/biophysj.103.026765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mueller F., Mazza D., Stasevich T.J., McNally J.G. FRAP and kinetic modeling in the analysis of nuclear protein dynamics: what do we really know. Curr. Opin. Cell Biology. 2010;22:403–411. doi: 10.1016/j.ceb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mo Y.-Y., Wang P., Beck W.T. Functional expression of human DNA topoisomerase I and its subcellular localization in HeLa cells. Exp. Cell Res. 2000;256:480–490. doi: 10.1006/excr.2000.4864. [DOI] [PubMed] [Google Scholar]
- 51.Das B.B., Antony S., Gupta S., Dexheimer T.S., Redon C.E., Garfield S., Shiloh Y., Pommier Y. Optimal function of the DNA repair enzyme TDP1 requires its phosphorylation by ATM and/or DNA‐PK. EMBO J. 2009;28:3667–3680. doi: 10.1038/emboj.2009.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Das B.B., Dexheimer T.S., Maddali K., Pommier Y. Role of tyrosyl-DNA phosphodiesterase (TDP1) in mitochondria. Proc. Natl. Acad. Sci. U.S.A. 2010;107:19790–19795. doi: 10.1073/pnas.1009814107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Das B.B., Sen N., Dasgupta S.B., Ganguly A., Majumder H.K. N-terminal region of the large subunit of Leishmania donovani bisubunit topoisomerase I is involved in DNA relaxation and interaction with the smaller subunit. J. Biol. Chem. 2005;280:16335–16344. doi: 10.1074/jbc.M412417200. [DOI] [PubMed] [Google Scholar]
- 54.Das B.B., Sen N., Ganguly A., Majumder H.K. Reconstitution and functional characterization of the unusual bi-subunit type I DNA topoisomerase from Leishmania donovani. FEBS Lett. 2004;565:81–88. doi: 10.1016/j.febslet.2004.03.078. [DOI] [PubMed] [Google Scholar]
- 55.Das B.B., Sen N., Roy A., Dasgupta S.B., Ganguly A., Mohanta B.C., Dinda B., Majumder H.K. Differential induction of Leishmania donovani bi-subunit topoisomerase I–DNA cleavage complex by selected flavones and camptothecin: activity of flavones against camptothecin-resistant topoisomerase I. Nucleic Acids Res. 2006;34:1121–1132. doi: 10.1093/nar/gkj502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mao Y., Okada S., Chang L.-S., Muller M.T. p53 dependence of topoisomerase I recruitment in vivo. Cancer Res. 2000;60:4538–4543. [PubMed] [Google Scholar]
- 57.de Murcia G., de Murcia J.M. Poly (ADP-ribose) polymerase: a molecular nick-sensor. Trends Biochem. Sci. 1994;19:172–176. doi: 10.1016/0968-0004(94)90280-1. [DOI] [PubMed] [Google Scholar]
- 58.Ashour M.E., Atteya R., El-Khamisy S.F. Topoisomerase-mediated chromosomal break repair: an emerging player in many games. Nat. Rev. Cancer. 2015;15:137–151. doi: 10.1038/nrc3892. [DOI] [PubMed] [Google Scholar]
- 59.Pommier Y., Barcelo J.M., Rao V.A., Sordet O., Jobson A.G., Thibaut L., Miao Z.H., Seiler J.A., Zhang H., Marchand C. Repair of topoisomerase I‐mediated DNA damage. Prog. Nucleic Acid Res. Mol. Biol. 2006;81:179–229. doi: 10.1016/S0079-6603(06)81005-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mao Y., Sun M., Desai S.D., Liu L.F. SUMO-1 conjugation to topoisomerase I: a possible repair response to topoisomerase-mediated DNA damage. Proc. Natl. Acad. Sci. U.S.A. 2000;97:4046–4051. doi: 10.1073/pnas.080536597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang H., Wang J.C., LIu L.F. Involvement of DNA topoisomerase I in transcription of human ribosomal RNA genes. Proc Natl. Acad. Sci. U.S.A. 1988;85:1060–1064. doi: 10.1073/pnas.85.4.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Larsen D.H., Stucki M. Nucleolar responses to DNA double-strand breaks. Nucleic Acids Res. 2016;44:538–544. doi: 10.1093/nar/gkv1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guetg C., Scheifele F., Rosenthal F., Hottiger M.O., Santoro R. Inheritance of silent rDNA chromatin is mediated by PARP1 via noncoding RNA. Mol. Cell. 2012;45:790–800. doi: 10.1016/j.molcel.2012.01.024. [DOI] [PubMed] [Google Scholar]
- 64.Bharti A.K., Olson M.O., Kufe D.W., Rubin E.H. Identification of a nucleolin binding site in human topoisomerase I. J. Biol. Chem. 1996;271:1993–1997. doi: 10.1074/jbc.271.4.1993. [DOI] [PubMed] [Google Scholar]
- 65.Meder V.S., Boeglin M., de Murcia G., Schreiber V. PARP-1 and PARP-2 interact with nucleophosmin/B23 and accumulate in transcriptionally active nucleoli. J. Cell Sci. 2005;118:211–222. doi: 10.1242/jcs.01606. [DOI] [PubMed] [Google Scholar]
- 66.Boamah E.K., Kotova E., Garabedian M., Jarnik M., Tulin A.V. Poly(ADP-Ribose) polymerase 1 (PARP-1) regulates ribosomal biogenesis in Drosophila nucleoli. PLoS Genet. 2012;8:e1002442. doi: 10.1371/journal.pgen.1002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tulin A., Stewart D., Spradling A.C. The Drosophila heterochromatic gene encoding poly(ADP-ribose) polymerase (PARP) is required to modulate chromatin structure during development. Genes Dev. 2002;16:2108–2119. doi: 10.1101/gad.1003902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tulin A., Spradling A. Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puff loci. Science. 2003;299:560–562. doi: 10.1126/science.1078764. [DOI] [PubMed] [Google Scholar]
- 69.Desnoyers S., Kaufmann S.H., Poirier G.G. Alteration of the nucleolar localization of poly(ADP-ribose) polymerase upon treatment with transcription inhibitors. Exp. Cell Res. 1996;227:146–153. doi: 10.1006/excr.1996.0259. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.