

Abstract
Aim: Cytosolic NADH-NAD+ redox state is central to cellular metabolism and a valuable indicator of glucose and lactate metabolism in living cells. Here we sought to quantitatively determine NADH-NAD+ redox in live cells and brain tissue using a fluorescence lifetime imaging of the genetically-encoded single-fluorophore biosensor Peredox. Results: We show that Peredox exhibits a substantial change in its fluorescence lifetime over its sensing range of NADH-NAD+ ratio. This allows changes in cytosolic NADH redox to be visualized in living cells using a two-photon scanning microscope with fluorescence lifetime imaging capabilities (2p-FLIM), using time-correlated single photon counting. Innovation: Because the lifetime readout is absolutely calibrated (in nanoseconds) and is independent of sensor concentration, we demonstrate that quantitative assessment of NADH redox is possible using a single fluorophore biosensor. Conclusion: Imaging of the sensor in mouse hippocampal brain slices reveals that astrocytes are typically much more reduced (with higher NADH:NAD+ ratio) than neurons under basal conditions, consistent with the hypothesis that astrocytes are more glycolytic than neurons. Antioxid. Redox Signal. 25, 553–563.
Introduction
Nicotinamide adenine dinucleotide (NAD+) and the corresponding reduced molecule NADH are key biological electron carriers. In eukaryotic cells, they are used both in the cytosol and mitochondria mainly for catabolic reactions, while the closely related NADP/NADPH couple is involved mainly in biosynthetic electron transfer. NADH and NADPH are both weakly fluorescent, while the oxidized forms are not, and this fluorescence difference has enabled fluorimetric assays of redox state, as well as numerous enzyme-coupled assays for specific metabolites. However, in the complicated context of a living cell, the indistinguishability of NADH and NADPH fluorescence makes the resultant signal (usually referred to as “NAD(P)H fluorescence”) ambiguous, particularly because the two redox couples are typically in very different redox states.
Innovation.
We show that cytosolic NADH-NAD+ redox state can be visualized in brain tissue using two-photon fluorescence lifetime imaging microscopy, using the genetically encoded biosensor Peredox. The sensor is completely specific to the NADH-NAD+ couple, without interference from NADPH-NADP. The lifetime images provide a direct calibrated measurement of the NADH redox state that is independent of sensor expression level.
This ambiguity between NADH and NADPH can be resolved using genetically encoded fluorescent biosensors that are completely specific for NADH over NADPH (12, 29, 30). These sensors couple a conformational change in a bacterial NADH binding protein to a fluorescence change in a linked fluorescent protein (FP). Not only are the sensors very chemically specific, they also can be targeted to specific compartments (cytosol or mitochondria) that may have very different redox states. In some cases, competition for binding sites between NADH and NAD+ causes the sensors to detect the NAD+/NADH ratio (effectively, the redox state), approximately independent of the total size of the NADH + NAD+ pool (12, 29).
When using such fluorescent biosensors in living cells, a key challenge is distinguishing whether cell-to-cell differences in fluorescence correspond to different redox states or to different levels of biosensor expression. To date, this has generally been done by ratiometric imaging: normalizing the sensor fluorescence to the fluorescence of a second, attached FP of a different color (12), or normalizing the biosensor fluorescence at one excitation wavelength to that at another (29, 30). Either ratiometric approach can work reliably, but it still can be difficult to make quantitative comparisons between cells measured in different instruments, because most fluorescence measurements are made in arbitrary units that vary with the spectral filters and excitation powers. Additional problems with ratiometric quantitation can be encountered when using two-photon excitation (28).
An alternative method for quantitative biosensor imaging is to measure fluorescence lifetime. Fluorescence lifetime is the dwell time of a fluorescent molecule in the excited state, measured as the distribution of waiting times between absorption of an incident photon and emission of a photon (2, 16). For some fluorescent sensors, changes in fluorescence intensity correspond to changes in fluorescence lifetime. Because a change in the number of sensor molecules does not lead to any change in the measured fluorescence lifetime, no normalization or correction for sensor protein concentration is needed. Furthermore, fluorescence lifetime is measured in absolute units of time (usually nanoseconds), so that different experiments on different instruments can be compared with confidence.
Only certain biosensors exhibit changes in fluorescence lifetime. Most sensors that are used by excitation ratio measurement do not have a substantial change in fluorescence lifetime [e.g., (13, 25)], as the fluorescence intensity change is predominantly a change between different absorbance states that produce the same, or similar excited states. Most sensors designed to exploit Förster resonance energy transfer (FRET) between two different FPs do show a change in fluorescence lifetime of the “donor” FP, as FRET provides an additional nonradiative pathway out of the excited state and thus speeds its decay [e.g., (10, 14, 15)].
Here we show that a single-FP sensor of NADH:NAD+ redox, Peredox, exhibits a large change in fluorescence lifetime upon conversion between the NAD+-bound and NADH-bound states. This change can be imaged using a two-photon microscope equipped for fluorescence lifetime imaging microscopy (2p-FLIM), which exploits the fact that all two-photon microscopy is performed with femtosecond pulsed lasers whose pulses provide a “time zero” for measuring fluorescence lifetime. The 2p-FLIM images of Peredox expressed in brain tissue provide a direct calibrated readout of the cytosolic NADH:NAD+ redox in both neurons and astrocytes of an acutely prepared tissue slice from the mouse hippocampus.
Results
The NAD+- and NADH-loaded states of Peredox have substantially different fluorescence lifetime
To assess whether the Peredox sensor could be used in the fluorescence lifetime mode, we first imaged the affinity-purified sensor protein in the 2p-FLIM microscope in the fully NAD+- or fully NADH-loaded states. Bacterially expressed Peredox protein with an N-terminal his6 tag was purified by its binding to Ni2+-chelate beads. The purified protein was mixed with purified NAD+ or NADH and imaged in sealed-tip micropipettes, placed in a flowing temperature-controlled stream of water in the microscope chamber. As a test of the 2p-FLIM lifetime calibration, we determined the fluorescence lifetime of two dyes, fluorescein and rhodamine B; the results were in excellent agreement with the published values (Table 1).
Table 1.
Fluorescence Lifetime Properties of Peredoxa and Dye Standards
| τ1 | f1 | τ2 | f2 | 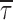 |
τEmpirical | |
|---|---|---|---|---|---|---|
| Peredox NADH, 25°C | 1.38 ± 0.15 | 0.18 ± 0.03 | 3.22 ± 0.05 | 0.82 ± 0.03 | 2.63 ± 0.01 | 2.63 ± 0.01 |
| Peredox NAD+, 25°C | 0.99 ± 0.02 | 0.58 ± 0.04 | 2.49 ± 0.06 | 0.42 ± 0.04 | 1.87 ± 0.02 | 1.87 ± 0.02 |
| Peredox NADH, 35°C | 1.00 ± 0.09 | 0.29 ± 0.06 | 3.19 ± 0.11 | 0.71 ± 0.06 | 2.53 ± 0.01 | 2.53 ± 0.01 |
| Peredox NAD+, 35°C | 0.84 ± 0.02 | 0.65 ± 0.03 | 2.28 ± 0.09 | 0.35 ± 0.03 | 1.65 ± 0.01 | 1.66 ± 0.01 |
| Fluoresceinb | 3.95 ± 0.05 | 1 | 3.08 ± 0.01 | 3.08 ± 0.004 | ||
| Rhodamine Bc | 2.45 ± 0.02 | 1 | 2.27 ± 0.01 | 2.32 ± 0.005 |
All lifetime values in ns. The values are derived from two-exponential fits of the form f1 e−t/τ1 + f2 e−t/τ2, convolved with a Gaussian of FWHM ≈140 ps. The summary values for the lifetime are as follows:
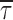 , the average lifetime value of the fit function over the measurement range (a window of ∼10 ns); τEmpirical, the average arrival time of the detected photons, over the measurement range. Protein was in MOPS buffer at pH 7.3, at 25°C. Excitation wavelength was 800 nm, and emission light collected from 500 to 550 nm. Expressed as mean ± SD, n = 4 for all measurements.
, the average lifetime value of the fit function over the measurement range (a window of ∼10 ns); τEmpirical, the average arrival time of the detected photons, over the measurement range. Protein was in MOPS buffer at pH 7.3, at 25°C. Excitation wavelength was 800 nm, and emission light collected from 500 to 550 nm. Expressed as mean ± SD, n = 4 for all measurements.
In 0.1 M sodium hydroxide, 5 μM. Excitation wavelength 800 nm, emission light collected from 500 to 550 nm. Expressed as mean ± SD, n = 3. Compare to reference fluorescein fluorescence lifetime value of 4.1 ± 0.1 ns (18, 21).
In methanol, 5 μM. Excitation wavelength 800 nm, emission light collected from 603 to 678 nm. Expressed as mean ± SD, n = 3. Compare to reference rhodamine B fluorescence lifetime value of 2.5 ± 0.1 (5).
The 2p-FLIM microscope uses an 80 MHz mode-locked Ti-Sapphire laser for excitation; the ∼75 fs excitation pulses occur approximately every 12.5 ns. The arrival times of individual photons are measured relative to the laser pulse and accumulated in an arrival time histogram that describes the decay behavior of the excited state (Fig. 1). Because of the substantial nanosecond scale delays in both the optical and electronic paths of the microscope and its detection system, the zero arrival time is determined from fitting the actual decay (roughly the peak of the decay). The photon detectors and the detection electronics have a response function that scatters the arrival times slightly into several bins; this instrument response function (IRF) is approximately Gaussian in shape (Fig. 1, inset).
FIG. 1.

Peredox fluorescence lifetime decay curves. Time-resolved single photon arrival distribution for purified Peredox protein fluorescence in both the NADH-bound (black dots) and NAD+-bound (gray dots) states. Corresponding double-exponential decay fits (solid lines) are shown overlaying data points. IRF also shown in gray (inset) with Gaussian fit. The inset has a linear vertical scale (scale bar corresponds to 200 photon counts) and the same horizontal scale as the main figure. Peredox protein was excited at 800 nm, and the IRF was obtained by eliciting second harmonic generation from mouse tail collagen using 1010 nm excitation and 525/50 emission. The IRF fit shown is a pure Gaussian with FWHM of ∼220 ps; the fit is improved by assuming a small amount of short lifetime fluorescence from the collagen, which then gives a Gaussian with FWHM of ∼140 ps (a typical value for the free fits of the sensor lifetime data). IRF, instrument response function.
The decay histograms are fitted with the sum of two exponentials; before comparison to the data, the fit function is convolved with a Gaussian function (for the IRF), offset by a fitted tpeak value, and “wrapped around” so that the small late tail (after ∼12.5 ns) is summed with the response for the first 10–12 ns.
Figure 1 illustrates the lifetime decay functions for the Peredox sensor, fully loaded with either NAD+ or NADH, at 25°C. In the presence of NADH, the decay is notably slower than in the presence of NAD+ (2.63 ± 0.01 ns vs. 1.87 ± 0.02 ns, mean ± SD, n = 4). The two exponential fit values are shown in Table 1, for both 25°C and 35°C. We have not attempted to learn the photophysical details of the decay; instead, we simply use the measured lifetime as a parametric indication of the state of the sensor. For the practical reasons described previously, our decay measurements are limited to a time window of about 10 ns, slightly shorter than the laser repetition time. We, therefore, use the “empirical mean lifetime”—the mean photon arrival time for all photons arriving within this window—for our sensor measurement.
These measurements (τEmpirical) agree well with the definite integral of the fit function over this same time window (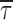 ). All of the subsequent values reported in this article are of τEmpirical, and the only adjustments needed to correct for the properties of a particular 2p-FLIM microscope are to recalculate the definite integrals over the measurement window for that microscope (as well as correcting for the IRF and time offset, as typically required for FLIM).
). All of the subsequent values reported in this article are of τEmpirical, and the only adjustments needed to correct for the properties of a particular 2p-FLIM microscope are to recalculate the definite integrals over the measurement window for that microscope (as well as correcting for the IRF and time offset, as typically required for FLIM).
We measured this change of lifetime over a wide range of excitation wavelengths, from 730 to 970 nm, at 25°C (Fig. 2A, B). Excitation efficiency falls steeply above ∼830 nm, but at wavelengths in the range 770–860 nm, the lifetime difference between the fully NAD+- and fully NADH-loaded states is roughly a constant 0.77 ns.
FIG. 2.

Peredox fluorescence lifetime characteristics. (A) Peredox protein fluorescence lifetime two-photon excitation spectra. Average fluorescence lifetime of purified Peredox protein in the NADH-bound (black) and NAD+-bound (gray) state (n = 5 independent protein samples). Two-photon excitation wavelength varied between 730 and 970 nm, in 10 nm increments. Error bars indicate standard deviation. All measurements performed at 25°C and pH 7.3. (B) Change in Peredox fluorescence lifetime with excitation wavelength. Average magnitude of purified Peredox protein fluorescence lifetime change between NADH-bound and NAD+-bound states (n = 5 independent protein samples). Error bars indicate standard deviation. (C) Peredox protein two-photon action cross section. Average fluorescence two-photon action cross section for purified Peredox protein in the NADH-bound (black) and NAD+-bound (gray) state (n = 3 independent protein samples). Dashed line indicates reference two-photon cross section for Sapphire fluorescent protein (32). (D) Fold-change in Peredox protein two-photon action cross section. Average magnitude of purified Peredox protein fluorescence two-photon action cross section change between NADH-bound and NAD+-bound states (n = 3 independent protein samples).
We also measured the two-photon action cross section (the product of the absorbance cross section and quantum yield) of the two states (Fig. 2C), calibrated by comparing parallel measurements for fluorescein to its published cross section. The cross sections of the two states are comparable to that previously reported for the Sapphire FP (the closest reported relative of the circularly permuted TSapphire FP used in the Peredox sensor); the NADH-loaded state is a bit higher and the NAD+-loaded state a bit lower than the Sapphire cross section. Over the middle range of excitation wavelengths, the change in 2p action cross section between the two states (Fig. 2D) corresponds well to the ∼2.5-fold change observed for Peredox fluorescence intensity using 1p excitation (12).
Lifetime calibration of the NADH-NAD+ redox response for the purified sensor protein in solution
To measure the “dose–response” of the sensor to changes in NADH-NAD+ redox, we used purified lactate dehydrogenase (LDH) enzyme, together with known amounts of lactate and pyruvate, to set the NAD+/NADH ratio. We found this method to be more reproducible than setting the NADH and NAD+ concentrations directly; we attribute this partly to reagent purity and partly to variable copurification of a small amount of NADH with the sensor protein.
The empirical lifetime response of Peredox to variation of the lactate/pyruvate ratio, in the presence of LDH, is shown in Figure 3, at both 25°C and 35°C. The graphs show the primary x-axis as the lactate/pyruvate ratio; for each temperature, there is a secondary x-axis showing the corresponding NAD+/NADH ratios calculated from the published equilibrium constants for LDH at these two temperatures (9, 26). The half-maximal change in lifetime occurs at an NAD+/NADH value of ∼529 at 25°C and ∼255 at 35°C. Note also that both the upper and lower lifetime limits are shorter at the higher temperature, perhaps because increased movement of the protein permits more collisional quenching of the fluorophore. Lifetime measurements of the sensor are very pH resistant (Fig. 3C) within a wide range of cytosolic pH values, as seen previously for the intensity response (12).
FIG. 3.

Peredox protein fluorescence lifetime NAD+/NADH dose–response curves. (A) Peredox protein fluorescence lifetime dose–response curve at 25°C. Purified Peredox protein fluorescence lifetime response to increasing ratios of lactate/pyruvate (bottom axis) and corresponding levels of NAD+/NADH (top axis). Error bars indicate standard deviation, n = 4 independent protein samples (black circles), and the Hill fit to the grouped average is shown in black. Fits of the individual experiment data sets yielded an average KR lactate/pyruvate value (ratio giving a half-maximal response) of 20.7 ± 2.0, which corresponds to a KR NAD+/NADH of 529 ± 50. (B) Peredox protein fluorescence lifetime dose–response curve at 35°C. Purified Peredox protein fluorescence lifetime response to increasing ratios of lactate/pyruvate (bottom axis) and corresponding levels of NAD+/NADH (top axis). Error bars indicate standard deviation, n = 3 independent protein samples (black circles), and the Hill fit to the grouped average is shown in black. The average KR lactate/pyruvate value for all experiments was 23.5 ± 4.2, corresponding to a KR NAD+/NADH of 255 ± 45. At elevated temperature, both the dynamic range and the KR of Peredox increase. (C) pH dependence of the fully NADH and NAD+ loaded states of the sensor (purified protein at 34°C, mean ± range, n = 2).
Calibration of the Peredox lifetime response in living cells
We performed a similar calibration of the Peredox lifetime response in HEK293 cells transfected with the sensor. Rapid transmembrane transport of lactate and pyruvate and the high cellular activity of LDH allow cytosolic NADH-NAD+ redox to be clamped (approximately) by changing extracellular lactate and pyruvate concentrations (12). Figure 4 shows the outcome of these calibrations. There is generally good agreement between the response of the sensor in cells and that of the purified protein, although the lowest lifetime values (at low NADH) are slightly higher in cells. We suspect that these cells continue to produce cytosolic NADH at a low level, despite having zero supplied glucose and strongly active LDH.
FIG. 4.

Peredox fluorescence lifetime dose–response curve in live cells. Average Peredox fluorescence lifetime response (black filled circles) to increasing ratios of lactate/pyruvate applied externally by perfusion of HEK293 cells at 35°C. Data are fit to a Hill equation (black trace). Error bars indicate standard deviation between responses of N = 4 independent groups of cells, with each group consisting of between 24 and 80 cells (n = 148 total cells). Individual fits to the data from each group of cells provided an average KR of 23.8 ± 3.2 lactate/pyruvate, corresponding to an NAD+/NADH KR of 246 ± 34. For comparison, the 35°C protein data of Figure 3B and the 35°C HEK293 cell dose–response using fluorescence intensity [from (12)] are also shown.
A typical 2p-FLIM calibration experiment on HEK293 cells is illustrated in Figure 5A–C. Note that at the start of the experiment, cells vary substantially in sensor lifetime (Fig. 5A). However, once glucose metabolism is inhibited and the various lactate/pyruvate mixtures are washed onto the cells, the lifetimes of all the cells become quite comparable (Fig. 5B), irrespective of the different expression levels of the sensor (Fig. 5C).
FIG. 5.

Peredox calibration and baseline values in live cells. (A) Peredox dose–response calibration in live cells. Fluorescence lifetime images of HEK293 cells expressing Peredox sensor show varying levels of NADH:NAD+ redox when perfused with glucose (panel 1). The same cells perfused with 10 mM lactate quickly and uniformly increase NADH levels in all cells, regardless of initial state (panel 2). Peredox fluorescence lifetime imaging indicates cytosolic NADH:NAD+ redox varies according to changes in lactate/pyruvate ratios of 30 (panel 3) and 0.1 (panel 4). Scale bar indicates 10 μm. (B) Time course of Peredox calibration in live cells. Representative traces from five cells shown in (A) quantifying the Peredox lifetime over the course of the calibration experiment. Although Peredox fluorescence lifetime in individual cells varies initially in the presence of glucose, lifetime values become uniform between cells after exposure to varying levels of pyruvate and lactate. Arrowheads indicate time of images shown in (A). (C) Peredox lifetime values are independent of intensity. Peredox fluorescence intensity as measured by photon counting (right panel), compared with the fluorescence lifetime of the same photons (left panel). Image of cells shown in (A). Scale bar indicates 10 μm. (D) Peredox fluorescence lifetime baseline values in hippocampal neurons. The three principal regions of the mouse hippocampus express Peredox protein after injection with a custom AAV8 virus, in both astrocytes and neurons. Granule neurons in the DG have uniform and low Peredox fluorescence lifetime values (top left panel), regardless of fluorescence intensity (top right panel). Similarly, pyramidal neurons of the CA3 (middle panels) and CA1 regions (lower panels) also showed fairly uniform and low Peredox fluorescence lifetime values, indicating oxidized NADH:NAD+ redox. Scale bars indicate 10 μm. (E) Peredox fluorescence lifetime baseline values in hippocampal astrocytes. Astrocytes expressing Peredox sensor were also found (although less frequently) in the three principal regions of the hippocampus. Astrocytes were recognized by live staining with SR101 (right panels of top, middle, and bottom). Peredox fluorescence lifetime values were markedly higher in astrocytes than in neurons. A single astrocyte in the cell layer of the dentate gyrus (top panel) has much more reduced NADH:NAD+ redox than its neighboring neurons. Astrocytes found in the molecular layer of the dentate gyrus (middle panel) were also more reduced compared to neurons and neuropil. Astrocytes near CA3 pyramidal cells (bottom panel) were similarly reduced compared to local neurons. Scale bars indicate 10 μm.
Peredox imaging with 2p-FLIM reveals the resting metabolic differences between neurons and astrocytes in a living mouse hippocampal brain slice
The great value of 2p-FLIM imaging of the Peredox sensor is that 2p-FLIM can be readily used in a highly light-scattering tissue like a brain slice, while ratiometric imaging can be costly, difficult, and inaccurate (28). Figure 5D and E show representative 2p-FLIM images of Peredox expressed in neurons and astrocytes of an acutely prepared mouse hippocampal slice. A viral vector to express the Peredox protein was injected intracranially, at precise coordinates, in mice at postnatal day 1 or 2, and mice were euthanized between postnatal day 15 and 22. Brain slices were prepared using standard methods, and the Peredox sensor was visualized on the 2p-FLIM microscope while the slices were perfused with oxygenated artificial CSF. These are typical conditions for electrophysiological and imaging studies of hippocampal slice, although they may differ from the actual conditions in vivo.
Fluorescence intensity and lifetime images are shown for principal neurons in three areas of the hippocampal slice: the dentate gyrus, the CA3 region, and the CA1 region (Fig. 5D). Neurons from all three regions have similar, low lifetime values, corresponding to relatively oxidized levels of NAD+/NADH (i.e., very low levels of NADH). Again, the lifetime values were quite uniform, despite marked differences in total sensor expression that are apparent from the monochrome intensity images.
In contrast, NADH:NAD+ redox in astrocytes was generally more reduced (i.e., higher NADH; Fig. 5E). The astrocytes were identified not only by their morphology but also by the strong staining by the dye sulforhodamine 101 (19) (red monochrome images at the right of Fig. 5E). Note how the higher lifetime (more reduced) astrocytes and their processes stand out against the neighboring more oxidized neurons or neuropil. The red sulforhodamine 101 dye does not influence the measurements in the green Peredox emission channel. This is apparent, for instance, from several astrocytes in Figure 5E, middle, which are brightly stained with SR101, but appear dark in the green channel, because they had not been transduced by the vector for Peredox expression.
Figure 6 summarizes the Peredox lifetime measurement values for hippocampal neurons as well as local astrocytes. In each area of the hippocampus, we found astrocytes were significantly more reduced than neurons. Overall, the mean NAD+/NADH value in hippocampal neurons was ∼660 and in hippocampal astrocytes was ∼270 (based on Fig. 6; different with p < 0.0001, Mann–Whitney), indicating that NADH levels are approximately 2.4-fold higher in astrocytes than neurons.
FIG. 6.

Peredox baseline lifetime values in brain slice. Peredox fluorescence lifetime in the somata of both astrocytes and neurons throughout the DG, CA1, and CA3 regions of the mouse hippocampus. Cells in a single slice were grouped and averaged by type (astrocyte vs. neuron) and location (DG, CA3, CA1). Slice averages (open circles) are shown next to the corresponding overall average (black filled circle) for each category. Standard deviation bars indicate variation between slices, N = slices and n = cells. Dashed lines indicate both the ceiling and floor values expected for Peredox lifetime based on results of purified protein at 35°C (Fig. 3B). The right-hand axis ticks are also calculated from the purified protein data.
Discussion
Use of Peredox as a lifetime sensor for monitoring NADH-NAD+ redox
The genetically encoded Peredox sensor reports NADH-NAD+ redox over a physiologically relevant range, with excellent resistance to interference from pH changes or from changes in other nucleotides (12). Compared with autofluorescence imaging of signals from NAD(P)H, Peredox gives a much brighter fluorescence signal that specifically reports free NADH:NAD+ ratio (and not NADPH) in the cytosolic compartment. Here we have shown that this sensor also has a large change in fluorescence lifetime over its sensing range, allowing the use of 2p-FLIM for direct and simple measurement of NADH-NAD+ redox in living cells in scattering tissue such as a brain slice, or potentially in vivo.
Because fluorescence lifetime is independent of the number of fluorophores or their brightness, no adjustment or normalization is necessary to correct for differences between cells in the level of sensor expression. This lifetime signal can be used for quantitative monitoring of the NADH-NAD+ redox either in the steady state (Figs. 5D, E and 6) or for time-dependent changes (Fig. 5B), although the time resolution is ultimately limited by the response time of the sensor [time constant of about 16 s at 35°C (12)].
Two excitation ratiometric sensors for NADH, which also use the bacterial Rex protein as a scaffold, have been reported by Yang and colleagues: Frex (30) and SoNar (29). We have not tested either of these sensors for a lifetime response, but based on our experience with our own ratiometric sensors, we would expect a relatively small or absent lifetime response. Our original Rex-based NADH sensor was ratiometric and had no detectable lifetime response; when we replaced the Venus FP with the low-pKa Sapphire FP, there was only a very tiny fluorescence change of any kind. Only after rescreening linker libraries to improve the response did we again see a substantial fluorescence change, accompanied by the large lifetime change reported here.
We suspect that the mechanism of ratiometric sensors, which mostly involves the pH-dependent interconversion between the A and B bands of wild-type Aequorea GFP (7), produces little change in quantum yield, and thus, little change in lifetime. By contrast, the intensity change of an optimized pH-independent sensor like Peredox depends instead on a large change in lifetime and quantum yield. Perhaps, in this case, the ligand-dependent conformational change helps to heal a “wound” in the beta barrel surrounding the GFP chromophore. This hypothetical wound, created initially by the circular permutation of the FP that leaves N- and C-termini adjacent to the chromophore, would permit solvent access and collisional quenching that would shorten the lifetime. The conformational change might tighten the association of the scaffold protein and FP and thus increase the lifetime.
Several other nonratiometric single-FP sensors also show a substantial change in fluorescence lifetime, including the cytosolic and endoplasmic reticulum calcium sensors, RCaMP (28) and CatchER (31). Direct mutation of the beta-barrel of the GFP to make it more conformationally sensitive has also successfully produced a single-FP sensor for PKA with a usable lifetime change (6). The single-FP red pH sensor, pHRed, is excitation ratiometric, but also exhibits a large change in lifetime (22).
NAD(P)H autofluorescence should not interfere with the lifetime signals from Peredox, as autofluorescence is typically much dimmer. In addition, the brightest autofluorescence signals are from mitochondria; these are excluded from cell nuclei, and we have not seen differences between the Peredox lifetime in the nucleus and that in the cytosol.
Predictable differences in the dose–responses for lifetime and intensity
In measuring the dose–response curves for Peredox's lifetime response to NAD+/NADH ratio changes, we found a notable shift between the sensor lifetime change and the sensor intensity change: the lactate/pyruvate ratio giving a 50% change in lifetime is approximately 50, compared to a midpoint of approximately 37 for the change in fluorescence intensity (Fig. 4). This change is predictable based on the photon statistics of the lifetime measurement.
Suppose that there are two states of the sensor, bright (NADH-bound) and dim (NAD+-bound)†, the halfway point of the intensity dose–response occurs when these two states are equally occupied. But now consider the halfway point of the lifetime response. The measured mean lifetime is the mean of the lifetime of the two states, but this mean is weighted by the number of photons from each state. When the two states are equally occupied (at the halfway point for the intensity measurements), more photons are coming from the bright state than from the dim state. This means that the measured mean lifetime is closer to the lifetime of the bright state than to the lifetime of the dim state.
To achieve a measured mean lifetime that is halfway between the lifetimes of the two pure states, the distribution between the two states must be shifted toward occupancy of the dim state, until the numbers of photons collected from the two states are equal. This multiplicative shift in the concentration of analyte giving a half-maximal response (Kapparent) is equal to the intensity ratio R of the bright to the dim state (e.g., approximately 2.5-fold in the case of Peredox); but in the case of a Hill coefficient h not equal to one, the shift is R1/h-fold (Supplementary Appendix S1; Supplementary Data are available online at www.liebertpub.com/ars). If analyte binding increases the fluorescence intensity, then Kapparent for the lifetime is lower than Kapparent for the intensity response.
Resting NADH-NAD+ redox in astrocytes is more reduced than neurons
In well-oxygenated, resting hippocampal slices, we found that neurons have an NADH-NAD+ redox that is more oxidized compared with astrocytes; that is, astrocytes have higher NADH levels. The mean NAD+/NADH value in hippocampal neurons was ∼660 and in hippocampal astrocytes was ∼270 (based on Fig. 6; different with p < 0.0001, Mann–Whitney). This finding is generally consistent with the notion that astrocytes are more glycolytic than neurons, with higher rates of conversion of glucose to lactate and lower mitochondrial consumption of pyruvate.
Astrocytes in culture excrete more lactate from glucose than do neurons (net glycolysis), and this lactate is hypothesized to serve as oxidative fuel for neurons (3). In neurons, regulation of glycolytic enzyme levels is thought to depress the early steps of glucose metabolism (11). Both of these observations are consistent with the standing NADH-NAD+ redox difference seen here between astrocytes and neurons. The redox difference also could facilitate a “redox switch” with net transfer of reducing equivalents from astrocytes to neurons (8). Of course, because the oxygenation conditions required in vitro for the maintenance of hippocampal slices are different from those in vivo, the ultimate resolution to questions of neuron and astrocyte redox will require deployment of Peredox in vivo, which should be feasible using 2p-FLIM.
In general, however, the NADH-NAD+ redox reflects not only the level of glucose metabolism but also net lactate export/import, and the level of NADH shuttle activity (4). No single sensor measurement is likely to give a complete story of how metabolism differs between neurons and astrocytes, and future studies will need to combine the use of Peredox with other metabolic sensors, pharmacological agents, and genetic or physiological manipulations [see e.g. (17)]. The ability of Peredox to be quantitatively imaged using 2p-FLIM should facilitate its deployment together with other sensors.
Materials and Methods
Reagents
Unless otherwise specified, all reagents were purchased from Sigma-Aldrich (St. Louis, MO). Plasmids encoding the Peredox sensor are available from addgene.com (www.addgene.org/Gary_Yellen/).
Peredox protein purification
Peredox protein was expressed in cultures of Escherichia coli (BL21, Agilent) grown in LB media for approximately 12 h overnight at 37°C before induction with IPTG for 4 h at room temperature. Cells were lysed with 15% CelLytic B in MOPS buffer containing (in mM) 100 MOPS, 50 KCl, 5 NaCl, and 0.5 MgCl2 at pH 8.0. Lysates were purified using Ni-NTA agarose beads, blocked with 0.1% BSA and 10 mM imidazole in MOPS buffer pH 8.0. Columns were first rinsed thrice with Tris-buffered saline pH 8.0 containing 0.05% Tween-20 and further rinsed thrice with MOPS buffer pH 8.0 containing 10 mM imidazole. Protein was eluted with 500 mM imidazole in MOPS buffer pH 8.0. Protein eluent was dialyzed into MOPS buffer at pH 7.3 and stored at 4°C for a short term (weeks) or −80°C for a long term (months).
Protein concentration was estimated using the alkaline denaturation method and the extinction coefficient for Aequorea chromophores [44,100 M−1 cm−1, (24)].
Protein imaging
Purified Peredox protein was imaged in experimental solutions containing 1 mM total NAD (NADH and NAD+ forms) and 10 mM total lactate and pyruvate. Different ratios of lactate/pyruvate were used to produce different NAD+/NADH ratios by adding purified LDH (5 U/mL; Worthington Biochemical Corp., Lakewood, NJ). The final concentration of protein imaged varied from 2.2 to 6.8 μM. Protein solutions were loaded into borosilicate capillary glass (Warner Instruments, Hamden, CT) pulled to a fine taper and fire-polished until sealed. Pipettes containing protein solutions were imaged on the 2p microscope under circulating perfusion maintained at room temperature (25°C) or elevated temperature (35°C) for comparison with cell and brain slice experiments (also done at 35°C).
Two-photon action cross sections were determined as previously described (23), at room temperature. Fluorescein dissolved in 0.1 M sodium hydroxide was used as the experimental reference standard, and published values for fluorescein quantum yield and the 2p cross section (1) were used to estimate Peredox 2p cross section. Quantum yield for Peredox protein in both the NADH and NAD+-bound states was taken from published estimates (12).
Cell and tissue preparation
HEK293 cells (ATCC, Manassas, VA) were maintained in DMEM (Thermo Fisher, Cambridge, MA) supplemented with 10% FBS (Thermo Fisher), 100 U/mL penicillin, and 100 μg/mL streptomycin (pen-strep; Lonza, Hopkinton, MA). For imaging, cells were plated on glass coverslips precoated in 1 mg mL−1 protamine and transfected (Effectene; Qiagen, Valencia, CA) with a plasmid-encoding Peredox under a universal promotor (CAG). One to four days after transfection, coverslips were transferred to imaging solution containing (in mM) 136 NaCl, 10 HEPES, 2.5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, at pH 7.35, and allowed 15–30 min to equilibrate before experiments began.
All procedures involving animals were approved by the Harvard Medical Area Standing Committee on Animals. For Peredox expression in the hippocampus, male and female wild-type mice (C57BL/6NCrl, Charles River Laboratories, Newton, MA) were injected (stereotaxic intracranial injection) at postnatal day 1 or 2 with a custom adeno-associated vector (AAV; Penn Vector Core, University of Pennsylvania, PA).
For expression of Peredox in both astrocytes and neurons, we found that an AAV2/8 serotype with Peredox expression under a universal promotor (CAG) provided moderate Peredox levels in both cell types.
Between 2 and 3 weeks after injection, mice were anesthetized with isoflurane, decapitated, and brain tissue was harvested. Horizontal slices were cut (7000smz-2, Campden Instruments, Loughborough, England) at a thickness of 275 um, in an ice-cold slicing solution containing (in mM) 87 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 7 MgCl2, 0.5 CaCl2, 75 sucrose, and 10 glucose.
Slices were continuously bubbled with a mix of 95% O2 and 5% CO2. Immediately after slicing, slices were transferred to warm (37°C) artificial cerebral spinal fluid (ACSF) solution containing (in mM) 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, and 10 glucose. For labeling astrocytes, 0.5 μM sulforhodamine 101 (SR101) was included in the ACSF for the first 20 min and then slices were transferred to ACSF without SR101 for the remainder of the incubation at 37°C. Slices were incubated at 37°C for approximately 45 min and thereafter remained at room temperature. Slices from the temporal region of the hippocampus were used for all experiments and were used within 4 h of preparation.
Live-cell microscopy
Protein solutions, cells, and slices were imaged using a custom two-photon microscope system (see 2p-FLIM section) with an Olympus LUMPFLN 60x/W (NA 1.0) objective. Fluorescence emission light was split with an FF562-Di03 dichroic mirror and band-pass filtered for green (FF01-525/50) and red (FF01-641/75) channels (all filter optics from Semrock, Rochester, NY). All Peredox fluorescence photon statistics were measured from emission light collected with the green band-pass filter. Perfusates were maintained at the indicated temperature by an inline heater (SH-27B; Warner Instruments, Hamden, CT). Perfusion rates were approximately 5 mL/min. Unless otherwise indicated (i.e., during lactate/pyruvate calibration), all cells and slices were perfused with 10 mM glucose. During lactate/pyruvate ratio calibrations of HEK293 cells, the total concentration of lactate + pyruvate was kept constant at 10 mM.
For all experiments with HEK cells and hippocampal slices, Peredox was imaged using an excitation wavelength of 800 nm. Images were acquired at 4 frames per second (limited by the scan rate of the galvanometer scanner), and the presented images are averages of 20 frames.
2p-FLIM
Lifetime imaging data were acquired with a modified Thorlabs Bergamo II microscope (Thorlabs Imaging Systems, Sterling, VA), with hybrid photodetectors R11322U-40 (Hamamatsu Photonics, Shizuoka, Japan); the light source was a Chameleon Vision-S tunable Ti-Sapphire mode-locked laser (80 MHz, ∼75 fs; Coherent, Santa Clara, CA). The photodetector signals and laser sync signals were preamplified and then digitized at 1.25 gigasamples per second into a field-programmable gate array board (PC720 with FMC125 and FMC122 modules, 4DSP, Austin, TX).
Laboratory-built firmware and software performed time-correlated single photon counting to determine the arrival time of each photon relative to the laser pulse; the distribution of these arrival times indicates the fluorescence lifetime. Continuous digitization allows for zero dead-time detection of all photons, even at high count rates. For each photodetector, laser cycles with more than one photon detected were excluded from the lifetime analysis; this eliminates the classic “pile-up” distortion of the lifetime (2). Lifetime histograms were fitted using nonlinear least-squares fitting in MATLAB (Mathworks, Natick, MA), with a two-exponential decay convolved with a Gaussian for the IRF (27). Microscope control and image acquisition were performed by a modified version of the ScanImage software written in MATLAB (20) (provided by B. Sabatini and modified by G.Y.).
Analysis
Image analysis was performed with custom MATLAB software. Regions of interest (ROIs) were defined manually for individual cells, and photon statistics (including τEmpirical) was determined for all pixels within the ROI. Data were also output to Excel (Microsoft, Redmond, WA) or Igor (Wavemetrics, Lake Oswego, OR) for further analysis. Dose–response curves for protein and data were fit using the Hill equation of the format f(x) = initial + (final - initial) * xn/(kn + xn), where n is the Hill coefficient and k is the apparent dissociation constant. Calculation of the NAD+/NADH ratio from the lactate/pyruvate ratio was accomplished by establishing the Keq value for LDH at relevant temperature, pH, and ionic strength. First, we determined the value of Keq for ionic strengths of 0.15 (protein assays) and 0.16 (cell assays) by interpolating the published values reported in (26). Then, we used the Q10 for LDH reported by (9) to adjust the Keq values for 35°C and 25°C. Final Keq values used were 8.59 × 10−12 for protein at 35°C, 8.66 × 10−12 for cells at 35°C, and 4.62 × 10−12 for protein at 25°C.
Supplementary Material
Abbreviations Used
- 2p-FLIM
two-photon fluorescence lifetime imaging microscopy
- FP
fluorescent protein
- FRET
Förster resonance energy transfer
- IRF
instrument response function
- LDH
lactate dehydrogenase
- ROI
regions of interest
Acknowledgments
The authors are very grateful to Prof. Bernardo Sabatini for teaching them about 2p microscopy and helping them get started in FLIM. They also thank Prof. Ryohei Yasuda for helping them make their first lifetime measurements and sharing his analysis methodology and code. They are also very grateful to Thorlabs Imaging Systems (especially Jeff Brooker, Adam Larson, Paulo Chaves, and Bill Radtke) for custom modifications and improvements to our microscope. They thank all the members of the Yellen laboratory for helpful discussions, and particular thanks to Dr. Carolina Lahmann and Ms. Veronica Burnham for managing and optimizing the stereotactic surgeries.
They are grateful for support from NIH (DP1 EB016985 and R01 NS055031 to G.Y., F32 NS080455 to R.M.) and the David Mahoney Fellowship (to R.M.).
Author Disclosure Statement
G.Y. has licensed the digitized FLIM technology to Thorlabs, Inc., for whom he is also a consultant. All other authors state that no competing financial interests exist.
This assumption is made for simplicity and may not be true for Peredox. However, the discussion illustrates why the midpoint of the dose–response is shifted, and the argument holds for more complicated cases too.
References
- 1. Albota MA, Xu C, and Webb WW. Two-photon fluorescence excitation cross sections of biomolecular probes from 690 to 960 nm. Appl Opt 37: 7352– 7356, 1998. [DOI] [PubMed] [Google Scholar]
- 2. Becker W. The bh TCSPC Handbook. 6th ed. Berlin, Germany: becker & Hickl GmbH, 2014. [Google Scholar]
- 3. Bélanger M, Allaman I, and Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 14: 724– 738, 2011. [DOI] [PubMed] [Google Scholar]
- 4. Berg JM, Tymoczko JL, Stryer L, Berg JM, Tymoczko JL, and Stryer L. Biochemistry 5th ed. New York: W H Freeman, 2002. [Google Scholar]
- 5. Boens N, Qin W, Basarić N, Hofkens J, Ameloot M, Pouget J, Lefèvre J-P, Valeur B, Gratton E, vandeVen M, Silva ND, Engelborghs Y, Willaert K, Sillen A, Rumbles G, Phillips D, Visser AJWG, van Hoek A, Lakowicz JR, Malak H, Gryczynski I, Szabo AG, Krajcarski DT, Tamai N, and Miura A. Fluorescence lifetime standards for time and frequency domain fluorescence spectroscopy. Anal Chem 79: 2137– 2149, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bonnot A, Guiot E, Hepp R, Cavellini L, Tricoire L, and Lambolez B. Single-fluorophore biosensors based on conformation-sensitive GFP variants. FASEB J 28: 1375– 1385, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brejc K, Sixma TK, Kitts PA, Kain SR, Tsien RY, Ormö M, and Remington SJ. Structural basis for dual excitation and photoisomerization of the Aequorea victoria green fluorescent protein. Proc Natl Acad Sci U S A 94: 2306– 2311, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cerdán S, Rodrigues TB, Sierra A, Benito M, Fonseca LL, Fonseca CP, and García-Martín ML. The redox switch/redox coupling hypothesis. Neurochem Int 48: 523– 530, 2006. [DOI] [PubMed] [Google Scholar]
- 9. Hakala MT, Glaid AJ, and Schwert GW. Lactic dehydrogenase. II. Variation of kinetic and equilibrium constants with temperature. J Biol Chem 221: 191– 209, 1956. [PubMed] [Google Scholar]
- 10. Harvey CD, Ehrhardt AG, Cellurale C, Zhong H, Yasuda R, Davis RJ, and Svoboda K. A genetically encoded fluorescent sensor of ERK activity. Proc Natl Acad Sci U S A 105: 19264– 19269, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, and Bolaños JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol 11: 747– 752, 2009. [DOI] [PubMed] [Google Scholar]
- 12. Hung YP, Albeck JG, Tantama M, and Yellen G. Imaging cytosolic NADH-NAD(+) redox state with a genetically encoded fluorescent biosensor. Cell Metab 14: 545– 554, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jayaraman S, Haggie P, Wachter RM, Remington SJ, and Verkman AS. Mechanism and cellular applications of a green fluorescent protein-based halide sensor. J Biol Chem 275: 6047– 6050, 2000. [DOI] [PubMed] [Google Scholar]
- 14. Kaminski Schierle GS, Bertoncini CW, Chan FTS, van der Goot AT, Schwedler S, Skepper J, Schlachter S, van Ham T, Esposito A, Kumita JR, Nollen EAA, Dobson CM, and Kaminski CF. A FRET sensor for non-invasive imaging of amyloid formation in vivo. Chemphyschem Eur J Chem Phys Phys Chem 12: 673– 680, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Laine R, Stuckey DW, Manning H, Warren SC, Kennedy G, Carling D, Dunsby C, Sardini A, and French PMW. Fluorescence lifetime readouts of Troponin-C-based calcium FRET sensors: a quantitative comparison of CFP and mTFP1 as donor fluorophores. PloS One 7: e49200, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lakowicz JR, editor. Principles of Fluorescence Spectroscopy [Online]. Springer US; Available at: http://link.springer.com/10.1007/978-0-387-46312-4 (accessed on November30, 2015) [Google Scholar]
- 17. Mächler P, Wyss MT, Elsayed M, Stobart J, Gutierrez R, von Faber-Castell A, Kaelin V, Zuend M, San Martín A, Romero-Gómez I, Baeza-Lehnert F, Lengacher S, Schneider BL, Aebischer P, Magistretti PJ, Barros LF, and Weber B. In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab 23: 94– 102, 2016. [DOI] [PubMed] [Google Scholar]
- 18. Magde D, Rojas GE, and Seybold PG. Solvent dependence of the fluorescence lifetimes of xanthene dyes. Photochem Photobiol 70: 737– 744, 1999. [Google Scholar]
- 19. Nimmerjahn A, Kirchhoff F, Kerr JND, and Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods 1: 31– 37, 2004. [DOI] [PubMed] [Google Scholar]
- 20. Pologruto TA, Sabatini BL, and Svoboda K. ScanImage: flexible software for operating laser scanning microscopes. Biomed Eng OnLine 2: 13, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sjöback R, Nygren J, and Kubista M. Absorption and fluorescence properties of fluorescein. Spectrochim Acta A Mol Biomol Spectrosc 51: L7– L21, 1995. [Google Scholar]
- 22. Tantama M, Hung YP, and Yellen G. Imaging intracellular pH in live cells with a genetically encoded red fluorescent protein sensor. J Am Chem Soc 133: 10034– 10037, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tantama M, Martínez-François JR, Mongeon R, and Yellen G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat Commun 4: 2550, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ward WW. Properties of the coelenterate green-fluorescent proteins. In: Bioluminescence and Chemiluminescence: basic Chemistry and Analytical Applications, edited by De Luca M, McElroy DW. New York: Academic, 1981, pp. 235– 242 [Google Scholar]
- 25. Weller J, Kizina KM, Can K, Bao G, and Müller M. Response properties of the genetically encoded optical H2O2 sensor HyPer. Free Radic Biol Med 76: 227– 241, 2014. [DOI] [PubMed] [Google Scholar]
- 26. Williamson DH, Lund P, and Krebs HA. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J 103: 514– 527, 1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yasuda R, Harvey CD, Zhong H, Sobczyk A, van Aelst L, and Svoboda K. Supersensitive Ras activation in dendrites and spines revealed by two-photon fluorescence lifetime imaging. Nat Neurosci 9: 283– 291, 2006. [DOI] [PubMed] [Google Scholar]
- 28. Yellen G. and Mongeon R. Quantitative two-photon imaging of fluorescent biosensors. Curr Opin Chem Biol 27: 24– 30, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhao Y, Hu Q, Cheng F, Su N, Wang A, Zou Y, Hu H, Chen X, Zhou H-M, Huang X, Yang K, Zhu Q, Wang X, Yi J, Zhu L, Qian X, Chen L, Tang Y, Loscalzo J, and Yang Y. SoNar, a highly responsive NAD+/NADH sensor, allows high-throughput metabolic screening of anti-tumor agents. Cell Metab 21: 777– 789, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao Y, Jin J, Hu Q, Zhou H-M, Yi J, Yu Z, Xu L, Wang X, Yang Y, and Loscalzo J. Genetically encoded fluorescent sensors for intracellular NADH detection. Cell Metab 14: 555– 566, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhuo Y, Solntsev KM, Reddish F, Tang S, and Yang JJ. Effect of Ca 2+ on the steady-state and time-resolved emission properties of the genetically encoded fluorescent sensor CatchER. J Phys Chem B 119: 2103– 2111, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Two-photon action cross sections [Online]. [date unknown]. Available at: www.drbio.cornell.edu/cross_sections.html (accessed on December1, 2015)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.