

Abstract
We examined an extended, consanguineous family with seven individuals with severe intellectual disability and microcephaly. Further symptoms were hearing loss, vision impairment, gastrointestinal disturbances, and slow and asymmetric waves in the EEG. Linkage analysis followed by exome sequencing revealed a homozygous variant in SPATA5 (c.1822_1824del; p.Asp608del), which segregates with the phenotype in the family. Molecular modelling suggested a deleterious effect of the identified alterations on the protein function. In an unrelated family, we identified compound heterozygous variants in SPATA5 (c.[2081G > A];[989_991delCAA]; p.[Gly694Glu];[.Thr330del]) in a further individual with global developmental delay, infantile spasms, profound dystonia, and sensorineural hearing loss. Molecular modelling suggested an impairment of protein function in the presence of both variants.
SPATA5 is a member of the ATPase associated with diverse activities (AAA) protein family and was very recently reported in one publication to be mutated in individuals with intellectual disability, epilepsy and hearing loss. Our results describe new, probably pathogenic variants in SPATA5 that were identified in individuals with a comparable phenotype. We thus independently confirm that bi-allelic pathogenic variants in SPATA5 cause a syndromic form of intellectual disability, and we delineate its clinical presentation.
Electronic supplementary material
The online version of this article (doi:10.1186/s13023-016-0509-9) contains supplementary material, which is available to authorized users.
Keywords: ARID, Microcephaly, Hearing loss, Hypotonia, NGS
Main text
Intellectual disability is characterized by significant limitations in both intellectual functioning and in adaptive behavior and has a prevalence of about 2 % of the population [1]. The cause for intellectual disability is often genetic and therefore, it is one of the most common reasons for genetic counselling and poses a major socio-economic burden worldwide. Recent studies have shown that in many cases point mutations, which occur de novo or are inherited through autosomal recessive or X-linked traits, are responsible for the phenotype [2, 3]. Very recently, several affected individuals with various variants in SPATA5 (MIM 616577) have been reported [4, 5]. In our cohort of about 200 families with intellectual disability, we identified a homozygous deletion of one amino acid in SPATA5 in a consanguineous family with seven affected members. In an unrelated affected individual with sporadic global developmental delay and epileptic encephalopathy we identified compound heterozygous variants in SPATA5 in the context of clinical exome sequencing.
This study was approved by the Ethics Committees of the University Bonn and the University Erlangen-Nuremberg in Germany and informed consent of all examined persons or of their guardians recruited in the course of this study was obtained. Inclusion of a single individual followed at the Children’s Hospital of Philadelphia, USA was IRB-exempt.
Family MR003 is a large Syrian family with multiple consanguineous marriages and seven affected individuals (Fig. 1). Affected individuals IV-12 and IV-13 are uncle and aunt of the affected individuals V-1 to V-5. Pregnancy, delivery, and perinatal period were reported to be uneventful for all individuals of family MR003 except for individuals IV-13, V-3, and V-4. Individual IV-13 was born with cyanosis and neonatal hypotonia; individuals V-3 and V-4 had neonatal hypotonia after an unremarkable delivery. All affected individuals reached developmental mile stones including gross motor milestones such as unsupported sitting and walking at a late normal range. Affected individuals were examined at the age of 41 years (IV-12), 39 years (IV-13), 22 years (V-1), 20 years (V-2), 12 years (V-3), 5 years (V-2) and 2 years (V-5). Common symptoms of all affected members are moderate to severe intellectual disability, severely limited receptive and expressive speech, and microcephaly (Additional file 1: Table S1). Seizures were not reported in either of the family members. EEG in V-1 showed slow and asymmetric fronto-central waves. Individuals IV-12, IV-13, and V-2 presented with hearing impairment as determined by Auditory Evoked Potential, other affected individuals were not tested, thus, we cannot exclude mild hearing impairment in these individuals. Individuals IV-12 and IV-13 presented with vision impairment especially at night. Gastrointestinal disturbances, such as constipation, are observed in individuals IV-12 and IV-13. All affected individuals demonstrate autistic features including reduced eye contact. Additionally, V-2 shows stereotypic movements and V-1 has some degree of auto-aggression and is easily frightened. Individual V-3 presented with central muscular hypotonia and individuals IV-12 and IV-13 have a decreased ability to ambulate, like through a combination of central muscular hypotonia and decreased coordination. All affected individuals have a normal sleep pattern. Facial features include a long nose with underdeveloped alae nasi and arched eyebrows. Apart from presenting with microcephaly, growth parameters were in the normal range for all individuals.
Fig. 1.
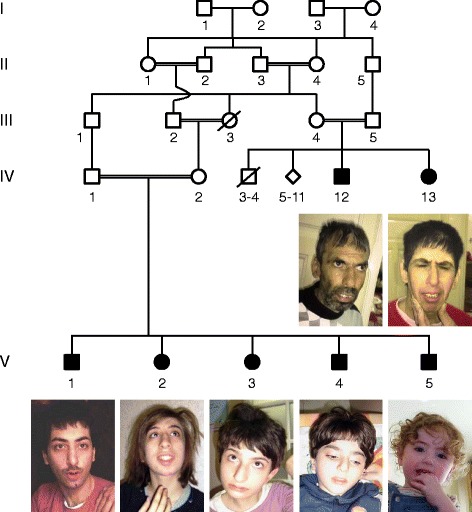
Pedigree and pictures of family MR003
Metabolic work-up of individual V-2 showed normal amino acids and urinary organic acids. Karyotype and ocular fundus was unremarkable. Brain MRI for IV-13 at an adult age indicated brain atrophy. Brain MRIs for individuals V-1 and V-2 at ages of 2 and 4 years, respectively, were unremarkable.
In family B, an outbred family of European ancestry, only one child, B1, is affected (Fig. 2a). The proband was born after a pregnancy complicated by maternal diabetes mellitus. Delivery was uneventful at 41 weeks of gestation. The affected individual was hypotonic and profoundly delayed with failure to achieve any major developmental milestones. He was referred to early intervention for developmental delay at a young age. He failed initial brainstem auditory evoked responses (BAERS) as a neonate. A hearing test was repeated later on and he was diagnosed with sensorineural hearing loss. At 8 months he had a percutaneous gastrostomy tube placed for failure to thrive. The proband started having infantile spasms at the age of 8 months. A head CT and brain MRI were unremarkable. A video EEG showed hypsarrythmia. He was treated with Lacosamide and Levetiracetam. At 17 months he was admitted to the Children’s Hospital of Philadelphia for break-through seizures in the setting of pneumonia, but has remained seizure-free with an unremarkable follow-up EEG. Ophthalmologic examination revealed poor vision likely secondary to cortical blindness. Cardiology evaluation noted a fenestrated atrial septal defect. The proband had a movement disorder, which developed around 10 months of age, characterized by profound dystonic posturing of upper limbs greater than lower limbs with neck involvement in the absence of spasticity. At 17 months, the proband was unable to sit independently or roll over. He was able to hold objects intermittently but unable to transfer them. He did not smile or coo. His growth parameters were in the low normal range (weight <5th percentile, length 9th percentile and head circumference 5th percentile). He had divergent strabismus, low nasal bridge and broad eyebrows, roving eye movements, scoliosis, and a sacral tuft of hair.
Fig. 2.
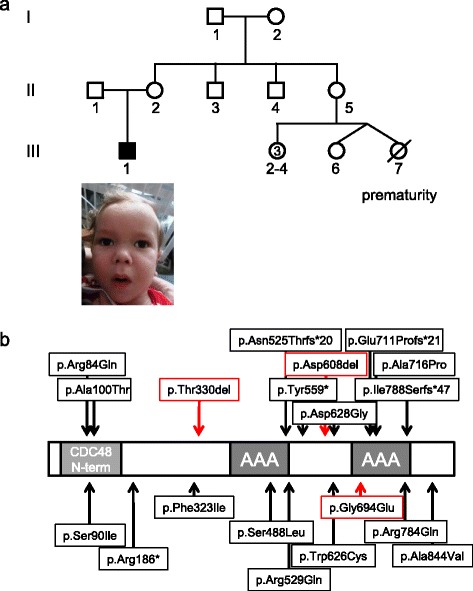
a Pedigree and picture of family B. b Schematic structure of SPATA5 and identified alterations. Previously reported variants are indicated in black, the variants identified in this study c.1822_1824del; p.Asp608del, c.2081G > A; p.Gly694Glu and c.989_991delCAA; p.Thr330del are indicated in red
Linkage analysis for family MR003 was performed using the genotype data of Human610-Quad DNA Analysis BeadChips (Illumina, San Diego, CA, USA) and resulted in the identification of single candidate region on chromosome 4:122,982,313-133,038,675 bp, with a size of 10 Mb as previously published [6]. Whole-exome sequencing (WES) was performed on an Illumina Genome Analyzer IIx (Illumina, San Diego, CA, USA) after enrichment with the SureSelect Human All Exon Kit (Agilent, Santa Clara, Ca, USA) as described previously [7]. Data was filtered for rare variants that were annotated with a minor allele frequency of ≤0.01 in the databases of 1000 Genomes [8] and Exome Variant Server (EVS), and that are within the candidate region. Then variants were prioritized according to predicted effect on protein sequence, and according to function of the encoded protein. Variants were validated and tested for segregation using Sanger sequencing. We identified a single homozygous candidate variant in SPATA5 (NM_145207): c.1822_1824del; p.Asp608del that was predicted as damaging by in silico prediction tools.
Genomic DNA of affected individual B1 was extracted from blood with the AutoGen DNA extraction kit following manufacturer’s guidelines (AutoGen, Holliston, MA, USA). The WES library was prepared using the SureSelectXT Human All Exon V5 kit following standard manufacturer protocol (Agilent Technologies, Santa Clara, CA, USA) and sequenced on the Illumina HiSeq 2500 using the 2 × 100 bp kit, with the targeted average coverage of 150x in the proband and 100x in the mother (Illumina, San Diego, CA, USA). Alignment and variant calling were performed with an in-house bioinformatics pipeline. Variants with a minor allele frequency of <0.005 in the Exome Aggregation Consortium database and expected to affect coding/splicing of the protein, or were present in the Human Gene Mutation Database (HGMD) [9] were included in the analysis using the Bench Lab NGS software (Cartagenia, Cambridge, MA, USA). A pair of variants in SPATA5: c.989_991delCAA; p.Thr330del and c.2081G > A; p.Gly694Glu was identified based on their compound heterozygous inheritance. The proband’s mother was heterozygous for the p.Thr330 deletion. The p.Gly694Glu variant was not present in the mother; it is presumably inherited from the father who was not available. The c.989_991delCAA; p.Thr330del variant was previously described in a proband with overlapping phenotype [4].
To determine the effect of the identified alterations on the phenotype the amino acid sequence of SPATA5 was analysed using PFAM [10] and two AAA ATPase domains spanning residues 390–523 and 664–796, respectively, were identified. Then, modeling was performed with Modeller 9.9 [11] using the crystal structure of the hexamieric ATPase p97 (PDB code: 3CF2) [12] as a template. The resulting model was verified using PROCHECK [13] and WHATCHECK [14] and revealed a good stereochemistry and no steric clashes. The deletions p.Asp608del and p.Thr330del were created with ModLoop [15, 16]. RasMol [17] was used for structure analysis and visualization.
The SPATA5 model (Fig. 3a) reveals that p.Asp608 is located at the subunit interface and forms stabilizing interactions with p.Lys517 of the adjacent subunit (Fig. 3b). Deletion of p.Asp608 causes a loss of the salt-bridge with p.Lys517 (Fig. 3c). In addition, the shorter loop in the mutant causes an unfolding of the α-helix, which is present N-terminally adjacent to the site of variant in the wildtype (Fig. 3b, c). These effects are expected to reduce the stability of both the hexamer and also of the individual subunits consequently leading to reduced enzymatic activity.
Fig. 3.
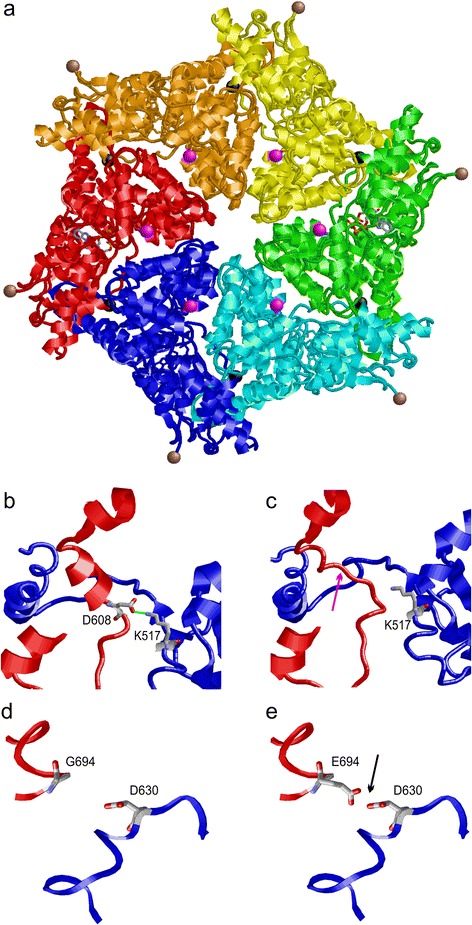
Structure of the AAA ATPase domains of SPATA5. a Model of the hexameric quaternary structure of SPATA5 showing the individual subunits in different colours. ADP-molecules bound to the ATPase domains are shown in stick presentation and coloured according to their atom type. p.Asp608 and p.Gly608Glu are located at the subunit interface and are depicted as black and magenta balls, respectively. pThr330 is located N-terminally adjacent to the globular domain and is shown as brown ball. b Enlargement showing the stabilizing interactions of p.Asp608 (D608) with p.Lys517 (K517) of the adjacent subunit (both residues are shown in stick presentation). The salt-bridge between both residues is shown in green and the subunits are coloured in red and blue respectively. c Deletion of p.Asp608 (D608) results in a loss of the helical secondary structure (pink arrow) and of the intersubunit salt-bridge. d Enlargement showing the location of pGly694 (G694) at the subunit interface. e Replacement of p.Gly544 by glutamate (E694) results in electrostatic repulsion (black arrow) with p.Asp630 (D630) of the adjacent subunit
Similar to p.Asp608, p.Gly694 is also located at the subunit of the interfaces in the hexameric enzyme (Fig. 3d). A replacement of p.Gly694 by glutamate causes electrostatic repulsion with p.Asp630 of the adjacent protein subunit (Fig. 3e). Therefore, this alteration is expected to decrease hexamer stability in a similar fashion as p.Asp608del. However, the effect on the fold of the individual subunits appears less severe than for p.Asp608del.
Due to lack of sequence similarity to known proteins, the sequence stretch around p.Thr330 cannot be modelled in atomic detail. The location p.Thr330 in the N-terminal proximity of the globular AAA ATPase domain suggests that a deletion of this residue might still affect protein function and stability, although to a lesser extent compared to the p.Asp608del or p.Gly694Glu variants. The p.Thr330del variant was previously described as pathogenic [4].
SPATA5 encodes a protein of 892 amino acids, which is part of the ATPase associated with diverse activities (AAA) protein family. Proteins of this family are characterized by their ATPase domain, which has a length of 200–250 amino acids and is highly conserved. AAA proteins usually form hexamer or heptamer rings with a central pore [18]. By hydrolysing ATP they can generate mechanical force, which is used for conformational remodelling of proteins or polynucleotides. Thus, members of the AAA protein family are thought to play an important role in protein degradation, DNA replication, membrane fusion events and in the movement of microtubules in various cellular compartments [18].
Variants in LONP1, a member of the closely related AAA+ family, have been reported to be causative for CODAS syndrome (MIM 600373). CODAS syndrome is characterized by intellectual disability, muscular hypotonia, epilepsy, sensorineural hearing loss, short stature, and skeletal abnormalities [19]. In addition, mutations in genes encoding for members of the AAA protein family have been reported in additional disorders including Zellweger syndrome (MIM 214100, PEX1), hereditary spastic paraplegia type 7 (MIM 607259, SPAST, SPG7), early-onset torsion dystonia (MIM 605204, TOR1A), and Paget disease with frontotemporal dementia (MIM 601023, VCP) [20–25]. Although these disorders differ significantly in their clinical presentation, neurological features are the major phenotype in all these disorders.
Very recently, several individuals with variants in SPATA5 presenting with symptoms of microcephaly, intellectual disability, seizures and hearing loss were reported [4, 5]. All variants in SPATA5 reported to date are located in important regions with an emphasis on the CDC48 N-terminal domain, the AAA domains or the subunit interface (Fig. 2b). Two of the variants reported here (p.Asp608del and p.Gly694Glu) are located at the subunit interface along with three previously reported variants and our modelling showed an impact on subunit oligomerization. The other variant reported here (p.Thr330del) was previously reported in individuals with a similar phenotype [4].
We conclude that the variants we identified in SPATA5 are causative for the phenotype in the affected individuals. Our findings suggest that bi-allelic pathogenic variants in SPATA5 may cause a phenotype with some distinct features. All individuals present with intellectual disability and gross motor delay out of proportion to the degree of intellectual impairment. Other neurological features can be variable. While the affected individual B1 presented with a profound movement disorder and epileptic encephalopathy characterized by infantile spasms with hypsarrhythmia, none of the seven affected individuals in family MR003 had seizures or dystonia. For many previously reported individuals movement disorders such as hypotonia, dystonia or spasticity have been described, as well. Most individuals have microcephaly, sensorineural hearing loss and cortical vision impairment. Autistic features were common in examined individuals. Neuroimaging shows non-specific abnormalities such as diffuse or general atrophy or a thin corpus callosum in some individuals while other individuals have a normal MRI. Many individuals also present with gastrointestinal issues. We suggest that pathogenic SPATA5 variants can be identified in individuals with complex neurodevelopmental phenotypes with obligatory intellectual disability, particularly in individuals with concomitant hearing and vision impairment. Our report expands the known features of SPATA5 encephalopathy and suggests that some of the clinical features, such as the presence or absence of epilepsy, may be mutation specific. We expect that further cases will add to the delineation of the SPATA5 phenotype.
Acknowledgements
We are grateful to the patients and their families for their participation in the study. This study was supported by the German Intellectual disability Network (MRNET) through a grant from the German Ministry of Research and Education to A. Reis (01GS08160), and by the Deutsche Forschungsgemeinschaft (DFG) (AB393/1-2 and AB393/2-2) through a grant from to R. Abou Jamra.
Funding
Supported by the German Intellectual disability Network (MRNET) through a grant from the German Ministry of Research and Education to A. Reis (01GS08160), and by the Deutsche Forschungsgemeinschaft (DFG) (AB393/1-2 and AB393/2-2).
Availability of data and materials
WES raw data supporting the conclusions of this article are not available due to patient confidentiality.
Authors’ contributions
RB analyzed and interpreted NGS data and prepared manuscript; AIN analyzed and interpreted NGS data and prepared manuscript; HT analyzed NGS data; IDK recruited and phenotyped family B and revised manuscript; LM recruited and phenotyped family B and revised manuscript; IH recruited and phenotyped family B and revised manuscript; DRM recruited and phenotyped family B and revised manuscript; AR achieved funding and supervised project; AS analyzed and interpreted NGS data and prepared manuscript; HS performed molecular modelling; RAJ recruited family MR003, coordinated project and prepared manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Consent to publish pictures and clinical data was obtained.
Ethics approval and consent to participate
This study was approved by the Ethics Committees of the University Bonn and the University Erlangen-Nuremberg in Germany and informed consent of all examined persons or of their guardians recruited in the course of this study was obtained. Inclusion of a single individual followed at the Children’s Hospital of Philadelphia, USA was IRB-exempt.
Web resources
1000 genomes project, http://www.1000genomes.org/
dbSNP, http://www.ncbi.nlm.nih.gov/SNP/
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, https://esp.gs.washington.edu/drupal/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://www.genome.ucsc.edu
Abbreviations
- AAA
ATPase associated with diverse activities
- ARID
Autosomal recessive intellectual disability
- BAERS
Brainstem auditory evoked responses
- EEG
Electroencephalography
- HGMD
Human gene mutation database
- WES
Whole exome sequencing
Additional file
Comparison of phenotypes of individuals with mutations in SPATA5. (DOCX 15 kb)
References
- 1.Leonard H, Wen X. The epidemiology of mental retardation: challenges and opportunities in the new millennium. Ment Retard Dev Disabil Res Rev. 2002;8:117–34. doi: 10.1002/mrdd.10031. [DOI] [PubMed] [Google Scholar]
- 2.Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, Hosseini M, Behjati F, Haas S, Jamali P, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- 3.Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di Donato N, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–82. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 4.Tanaka AJ, Cho MT, Millan F, Juusola J, Retterer K, Joshi C, Niyazov D, Garnica A, Gratz E, Deardorff M, et al. Mutations in SPATA5 Are Associated with Microcephaly, Intellectual Disability, Seizures, and Hearing Loss. Am J Hum Genet. 2015;97:457–64. doi: 10.1016/j.ajhg.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurata H, Terashima H, Nakashima M, Okazaki T, Matsumura W, Ohno K, Saito Y, Maegaki Y, Kubota M, Nanba E, et al. Characterization of SPATA5-related encephalopathy in early childhood. Clin Genet 2016. doi: 10.1111/cge.12813 [DOI] [PubMed]
- 6.Abou Jamra R, Wohlfart S, Zweier M, Uebe S, Priebe L, Ekici A, Giesebrecht S, Abboud A, Al Khateeb MA, Fakher M, et al. Homozygosity mapping in 64 Syrian consanguineous families with non-specific intellectual disability reveals 11 novel loci and high heterogeneity. Eur J Hum Genet. 2011;19:1161–6. doi: 10.1038/ejhg.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abou Jamra R, Philippe O, Raas-Rothschild A, Eck SH, Graf E, Buchert R, Borck G, Ekici A, Brockschmidt FF, Nothen MM, et al. Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am J Hum Genet. 2011;88:788–95. doi: 10.1016/j.ajhg.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Genomes Project C. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133:1–9. doi: 10.1007/s00439-013-1358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–30. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Webb B, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2014;1137:1–15. doi: 10.1007/978-1-4939-0366-5_1. [DOI] [PubMed] [Google Scholar]
- 12.Davies JM, Brunger AT, Weis WI. Improved structures of full-length p97, an AAA ATPase: implications for mechanisms of nucleotide-dependent conformational change. Structure. 2008;16:715–26. doi: 10.1016/j.str.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 13.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–86. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 14.Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 15.Fiser A, Do RK, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–73. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fiser A, Sali A. ModLoop: automated modeling of loops in protein structures. Bioinformatics. 2003;19:2500–1. doi: 10.1093/bioinformatics/btg362. [DOI] [PubMed] [Google Scholar]
- 17.Sayle RA, Milner-White EJ. RASMOL: biomolecular graphics for all. Trends Biochem Sci. 1995;20:374. doi: 10.1016/S0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]
- 18.Bar-Nun S, Glickman MH. Proteasomal AAA-ATPases: structure and function. Biochim Biophys Acta. 2012;1823:67–82. doi: 10.1016/j.bbamcr.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 19.Strauss KA, Jinks RN, Puffenberger EG, Venkatesh S, Singh K, Cheng I, Mikita N, Thilagavathi J, Lee J, Sarafianos S, et al. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am J Hum Genet. 2015;96:121–35. doi: 10.1016/j.ajhg.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–81. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- 21.Portsteffen H, Beyer A, Becker E, Epplen C, Pawlak A, Kunau WH, Dodt G. Human PEX1 is mutated in complementation group 1 of the peroxisome biogenesis disorders. Nat Genet. 1997;17:449–52. doi: 10.1038/ng1297-449. [DOI] [PubMed] [Google Scholar]
- 22.Reuber BE, Germain-Lee E, Collins CS, Morrell JC, Ameritunga R, Moser HW, Valle D, Gould SJ. Mutations in PEX1 are the most common cause of peroxisome biogenesis disorders. Nat Genet. 1997;17:445–8. doi: 10.1038/ng1297-445. [DOI] [PubMed] [Google Scholar]
- 23.Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997;17:40–8. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- 24.Hazan J, Fonknechten N, Mavel D, Paternotte C, Samson D, Artiguenave F, Davoine CS, Cruaud C, Durr A, Wincker P, et al. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat Genet. 1999;23:296–303. doi: 10.1038/15472. [DOI] [PubMed] [Google Scholar]
- 25.Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, De Michele G, Filla A, Cocozza S, Marconi R, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–83. doi: 10.1016/S0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
WES raw data supporting the conclusions of this article are not available due to patient confidentiality.