

Abstract
Killer cell immunoglobulin-like receptors (KIRs), expressed on natural killer cells and T cells, have considerable biomedical relevance playing significant roles in immunity, pregnancy and transplantation. The KIR locus is one of the most complex and polymorphic regions of the human genome. Extensive sequence homology and copy number variation makes KIRs technically laborious and expensive to type. To aid the investigation of KIRs in human disease we developed a high-throughput, multiplex real-time polymerase chain reaction method to determine gene copy number for each KIR locus. We used reference DNA samples to validate the accuracy and a cohort of 1698 individuals to evaluate capability for precise copy number discrimination. The method provides improved information and identifies KIR haplotype alterations that were not previously visible using other approaches.
Electronic supplementary material
The online version of this article (doi:10.1186/s13073-016-0358-0) contains supplementary material, which is available to authorized users.
Keywords: Killer cell immunoglobulin-like receptor (KIR), Copy number variation, Haplotype, Real-time quantitative polymerase chain reaction
Background
Complex and multi-allelic copy number variation (mCNV) is abundant in the human genome and is a potential source of genetic diversity in relation to disease [1]. Genes involved in immunity and defence seem to be especially prone to mCNV [2], presumably driven by selection pressure from pathogens. Human leukocyte antigen (HLA) DRB, complement component 4 (C4) loci in the MHC and leukocyte immunoglobulin-like receptor (LILR) loci are recognised examples of multicopy gene families with mCNV linked to disease susceptibility [3–7]. Another genomic region of interest in this regard encompasses the killer cell immunoglobulin-like receptor (KIR) genes [8, 9]. KIR were discovered nearly 20 years ago by serological methods [10, 11]. Subsequently they have been shown to have important physiological and biomedical relevance in wide-ranging conditions including pregnancy, infection, autoimmunity, cancer and transplantation [12]. KIR associations involve epistasis with their variable cognate HLA ligands. Complex interactions of unlinked loci like these may account for some of the heritability void left by genome-wide association studies (GWAS).
KIR genes, which are part of the leukocyte receptor complex (LRC) in human chromosomal region 19q13.4, have evolved rapidly in parallel with their HLA ligands through varying types of selection. As such, KIR genes exhibit high diversity in copy number and haplotypes. Unlike normal homologous recombination, chromosomal crossovers in the KIR cluster may misalign because the genes are closely arranged head-to-tail and they are homologous in sequence to one another. The process, known as non-allelic homologous recombination (NAHR), generates novel expanded and contracted haplotypes with duplication or deletion of whole genes (between ~11 and 18 kb in size), multiple genes and formation of novel fusion genes [3]. Gene dose effects at the mRNA and protein level have been seen for KIR genes, namely KIR2DL2/L3 and KIR3DS1. KIR expression is stochastic and the number of NK cells expressing a given KIR correlates linearly with the total number of copies of the gene carried by the individual [13]. Thus, the overall responsiveness of the NK cell repertoire directly relates to KIR haplotype content. This has implications for NK cell-mediated alloreactivity in hematopoietic stem cell transplantation, where donors with a high proportion of alloreactive NK cells have higher levels of cytolytic activity against leukemic cells [14]. Furthermore, KIR copy number variation (CNV) has been shown to correlate with protection from certain viruses such as HCV and HIV [15, 16].
The current KIR typing techniques that employ specific primers (PCR-SSP) [17–19] or oligonucleotides (PCR-SSO) [20] have drawbacks when applied to large-scale studies of genetically complex diseases; they are time-consuming, expensive and labour-intensive. KIRs are refractory to high throughput methods because of extensive sequence homology, allelic and copy number variation. For this reason, KIR studies have been limited to date by their relatively small scale and they have been ignored in GWAS to date. In addition, recent studies indicate that structural variations in haplotypes have been overlooked. The conventional methods, such as PCR-SSP, PCR-SSO and MALDI-TOF [21] cannot detect such variation as they lack the ability to quantify gene number, instead providing only ‘presence/absence’ status for a gene.
In this paper, we describe a high-throughput method to determine copy number of each KIR locus, using quantitative polymerase chain reaction (PCR) with dual-labelled hydrolysis probes, which we have called qKAT for quantitative KIR semi-automated typing. This method can help simplify disease analysis by identifying unusual haplotypes so that the major haplotypes can be analysed separately. We extend the approach to LILR loci, demonstrating that the underlying strategy of qKAT offers a model for analysing and visualizing other highly variable mCNV regions.
In real-time PCR, the fluorescent threshold value (cycle of quantification, Cq) correlates linearly with logarithmic value of starting DNA copy number [22]. This method can determine the quantity of target DNA sequence specifically and accurately, therefore it has been used extensively for gene quantification, especially in gene expression studies. Compared with complementary DNA quantification in gene expression studies, copy numbers of target gene derived from both chromosomes is slightly different. The relative DNA copy number measured against a reference gene is always an integer ratio. In addition, it is a very small change compared to gene expression (2× and 1.5× for 1–2 and 2–3 copy changes, respectively).
Multiplex quantitative PCR has the advantage of simultaneously amplifying several products in the same tube, using spectrally distinct fluorophores to detect each amplification. The method allows reduction of DNA requirements, reagent costs, human labour and time. Using internal controls increases the reliability of the results. The optimised multiplex assays considerably reduce the cost and setup time by high throughput. Well-to-well variation is minimised in multiplex PCR since target assay and reference assay are run in the same tube at the same time, providing extra confidence in the results.
Methods
Multiplex quantitative PCR assay
For KIR assays, ten multiplex quantitative PCR reactions were carried out in a triplex format that included three probes targeting three different amplicons. The optimisations of primer and probe concentrations are shown in Additional file 1: Figures S1 and S2. The overall performance of each reaction was tested using standard curves (see Additional file 1: Figure S3) and the PCR efficiencies of each reaction are given in Additional file 1: Table S1. Each multiplex reaction detects two KIR genes and one endogenous reference gene (STAT6) that is located on a different chromosome and always has two copies in a diploid genome [23] (Fig. 1) as corroborated using the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/). Altogether, copy numbers of 20 markers were ascertained for 17 KIR genes and their important variants (2DL1–5, 2DS1–3, 2DS4 (separate assays for the gene, full-length variant [FL] and deletion variant [del]), 2DS5, 3DL1–3, 3DS1, 2DP1 and 3DP1). For 3DL1 and 3DL2, two reactions were used to target different parts of the gene to identify known fusion genes [24]. LILR gene copy number was determined using duplex reactions including one LILR target and the reference gene (Additional file 1: Table S2).
Fig. 1.
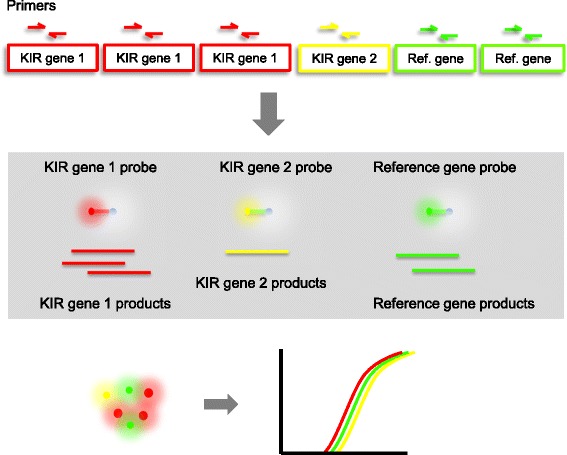
Schematic view of the KIR multiplex qPCR assay qKAT. Sequence specific primers are used for relative quantification of target KIR genes against a reference gene of fixed copy number. Each multiplex qPCR assay detects the simultaneous amplification of two target KIR genes and one reference gene. Two sets of primers target different exons of the two KIR genes and one pair of primers amplify the reference gene. Dual-labelled hydrolysis probes that are specific to each amplicon are used to monitor the PCR amplification in real-time
The reaction mix containing genomic DNA, primers, probes, Taq polymerase and buffer was dispensed into 384-well plates. Five nanograms of each DNA sample were plated into 384-well plates with four replicates. Three control samples of known copy number were included in each run. Multiplex PCR reactions were performed on a Roche LightCycler 480 using absolute quantification settings. Fluorescent signals were collected at the end of each cycle for further analysis.
After PCR amplification, Cq values were calculated using either the Second Derivative Maximum Method or the Fit Points Method. The copy number was determined by relative quantification analysis using the comparative Cq method (also known as delta delta Cq method, ΔΔCq). This method compares the cycle of quantification between the test sample and a calibrator sample with known copy number. In cases where a copy number calibrator sample was not available, the copy number analysis could be performed using the most frequent copy number expected in the samples.
Primer design
Based on PCR-SSP (PCR amplification with sequence-specific primers), our method used primers with 3' ends specifically matching a nucleotide which is unique to a given KIR/LILR gene. The gene-specific primers were designed to detect all the alleles of the given gene (Additional file 1: Table S3). In this assay, 21 primer pairs were used to detect all 16 KIR genes, important KIR gene variants and one reference gene. Primers were designed to produce amplicons in the range of 78–210 bp in size. Compared with the previous PCR-SSP and PCR-SSO methods, the short amplicons allowed the PCR reactions to achieve maximum efficiency. The Tm (melting temperature) of each primer was adjusted to between 48 °C to 57 °C, by adjusting the length of primers.
Initially, 37 primer pairs were designed for all 16 KIR genes and some variants. There was more than one reaction for most genes. Gel electrophoresis was used to check each PCR reaction using a known positive DNA sample and two negative DNA samples (for non-framework genes) and a non-template control. Any reactions with non-specific bands, weak amplification and strong primer-dimer were excluded. Due to the limited choice of unique nucleotides for KIR-specific priming, some primers were designed with GC content of up to 70.6 %.
Probe design
Due to the high sequence similarity between KIR/LILR genes, it is difficult to design specific probes. Dual-labelled probes were designed to exons sequences that are conserved between more than one gene (Additional file 1: Table S4). The same probe could therefore be used in different multiplex assays. Generic probes were feasible because the specificity for each reaction is controlled by the primers and never the same region for different targets was amplified in the same multiplex assay. Using generic probes between different reactions greatly reduced the cost since probes account for a large part of reagent costs. The primer and probe sequences for qKAT are given in Additional file 1 to allow judgment of whether they are appropriate for defining the currently known polymorphisms and variants discovered in the future. Primer and probe combinations used in each reaction are given in Additional file 1: Table S5.
The fluorophores used in the multiplex assays were FAM, Dragonfly Orange and Cy5. The maximum emission wavelengths of the three dyes are: 518 nm, 576 nm and 667 nm, respectively. These three dyes have distant emission spectra, which minimise the fluorescence signal from one dye bleeding into adjacent channels (signal crosstalk). Non-fluorescent black-hole quenchers (BHQ) were used because they have advantages over the other quenchers such as TAMRA, since they absorb the excitation energy from fluorophore and convert it into heat rather than re-emit this energy as light with a different wavelength. This is useful for multiplex PCR reactions, since there is no emitted light from the quencher to interfere with the reporter fluorophores, resulting in less background signals and hence have better signal/background ratios.
Determination DNA copy number using relative quantification analysis
KIR and LILR copy number of genomic DNA sample was determined using comparative Cq (ΔΔCq) relative quantification analysis [25–28]. The relationship between calculated copy number without efficiency correction and true copy number is illustrated in Additional file 1: Figure S4. Calculated copy number without efficiency correction and true copy number diverge significantly as the ΔCq in reference assay increases. This explains that apart from PCR efficiency, the ΔCq in reference assay could affect the copy number calculation. The ΔCq in the reference assay actually reflects the difference of DNA concentrations between sample and calibrator. If the DNA concentration is well quantified and controlled in a reasonable range, then the copy number calculation will not be affected much even without PCR efficiency correction. In addition, the parameters from our assays are slightly better than the assumed values. For example, the PCR efficiencies in the experiments have a mean value of 0.9883 with standard deviation of 0.0470. In this case, the ratio change caused by different PCR efficiencies between different genes are even smaller. Therefore, relative quantification using ΔΔCq method without efficiency correction could be used to determine DNA copy number.
Copy number analysis
Quality control was performed after each PCR run. After checking amplification plots and base lines, failed reactions and outlier values (Cq of reference assay is greater than 32 or data point > 4 standard deviations from the mean ΔCq of the four replicates) were removed. Then the Cq values were exported for comparative Cq calculation using the equations presented above. Zero was assigned to reactions with target assay’s Cq more than 35 and reference assay’s Cq less than 32. The calculation could be performed using either CopyCaller software from Applied Biosystems (Thermo Fisher Scientific) or Excel. CopyCaller provides two additional quality metrics: confidence metric and absolute z-score metric. Confidence metric estimates the confidence that the assigned copy number is the true copy number. Absolute z-score metric estimates how many standard deviations for ΔCq value of one sample varies from the mean ΔCq value assigned with the same copy number. The calculated copy numbers were rounded up to an integer (known as predicted copy number) for further analysis.
Measurement of copy number discrimination
The standard deviation was used to quantify the amount of variation or dispersion in each cluster with the same assigned copy number for each test across different samples (n = 1698). As the sample size increases, the ΔΔCq value tends to form approximately normal distribution. As for normal distribution, approximately 95 % of the values are within 2 standard deviations from the mean and 99.6 % within 3 standard deviations. Assuming all PCR reactions have 100 % efficiency, the ΔΔCq value between one copy and two copies samples will be just 1. To be able to distinguish the difference in more than 99.6 % of the cases, the standard deviations should be less than 0.167 (allowing more than 6 standard deviations within 1). To distinguish the difference in more than 95 % of the cases, the standard deviations should be less than 0.25 (allowing more than 4 standard deviations within 1) (Additional file 1: Table S6). It is progressively more difficult to distinguish the difference between higher copy numbers since the ΔΔCq value narrows.
Assay validation
Currently there is no gold standard method available for KIR typing, especially with copy number information. Data generated from family-based segregation analysis and allele typing could provide information closer to the actual copy number. To validate our method, a reference panel was used including the following DNA samples which had previously been typed with standard methods (PCR-SSP/SSO):
Three extended Centre d’Etude du Polymorphisme Humain (CEPH) families’ DNA (Coriell Cell Repositories, NJ, USA) from Utah were included in this study, CEPH/UTAH Pedigree 1332, 1347 and 1416. There are 15 members in each family. Pedigree data of these families are available from dbLRC [29]. In addition, NA10832, NA10861 and NA11994 from other CEPH/UTAH families were also included because they have unusual copy numbers of KIR genes.
UCLA KIR Exchange panel DNA samples were from UCLA Immunogenetics Center (CA, US, http://www.hla.ucla.edu/cellDna.htm). In this study, UCLA69 to UCLA84 were selected. This cohort has consensus KIR gene presence and absence data generated by PCR-SSP or PCR-SSO methods from several laboratories around the world. Copy number information is not available for these samples.
Two KIR region fully sequenced cell lines, PGF and COX [30], as well as one sequenced CEPH cell line, NA10832 [3], were also included in this study.
A total of 1698 individuals from 339 families from the Human Biological Data Interchange (HBDI) were used to evaluate the precision of a total of 20 KIR quantitative PCR assays. The HBDI panel comprises Caucasian (European-ancestry) families with type 1 diabetes from the United States [31]. At present, qKAT is validated for samples of European-origin only.
KIR haplotype inference in unrelated individuals
To help analyse samples, we developed a new tool, KIR Haplotype Identifier. KIR copy number data from qKAT is used as input and through matrix subtraction the program outputs the possible combinations of haplotypes for each sample. The tool is useful as a quick check for identification of novel or unconventional haplotypes in a cohort. The software processes KIR copy number sample files submitted by the end user. For each sample, a string of values is created through the concatenation of copy numbers provided (referred to as markersig). A second string is created comprising of a series of regular expressions for each marker (referred to as regex). Each regular expression denotes all possible haplotype values. For example, if sample 1 has markers ‘a’, ’b’ and ‘c’ each with copy numbers of 2, 1 and 1, respectively, then the markersig string would be 211 and the regex string would be (2|1|0)(1|0)(1|0), i.e. marker ‘a’ haplotype could be 2 or 1 or 0, marker ‘b’ haplotype could be 1 or 0 and so on. Each regex string is checked against KIR haplotype strings stored in a MySQL database. If a match is found then the associated data (e.g. haplotype, count, frequency, signature, cen motif, te1 motif) is retrieved. The haplotype pair is calculated by subtracting a matrix of the matched string from a matrix of the markersig.
Results
Assay validation
We tested 16 UCLA KIR exchange samples with KIR presence/absence data and three CEPH/UTAH families previously studied for KIR haplotypes. KIR haplotypes were determined by segregation analysis in families (see Additional file 1). For each pedigree, all non-recombinant haplotypes were identified by the Merlin program [32]. The results from KIR copy number assays were compared to the KIR data generated with previous methods [3, 24, 28]. For CEPH family samples, copy numbers were previously determined by segregation analysis of presence and absence data. Some of the duplicated genes were determined by allele typing [28] and fusion genes were determined by inter-gene PCR [3]. Apart from two exceptions described below, our results showed near complete concordance with previous data.
In pedigree 1416, we confirmed the presence of the extended haplotype carrying duplication of KIR3DP1, KIR2DL4 and KIR3DL1/S1 genes (Fig. 2). Pedigree 1416 samples NA10834, NA12241, NA12243, NA12244, NA12245, NA12246 and NA12251 showed one less copy in 3DL1 exon 4 than in exon 9. The previous amplicon region of exon 4 was amplified and sequenced using another pair of primers (Forward 5’-CGCTGTGGTGCCTCGA-3’ and Reverse 5’ACCACGATGTCCAGGGGA-3’). Sequencing results revealed a rare allele 3DL1*056, which contains a SNP in the probe region that disrupts probe binding. The allele has only been observed in this family [33]. As factored in the original design, the second test for KIR3DL1 does not miss this rare allele and, therefore, the allele is identified by discordant results between the two tests for KIR3DL1.
Fig. 2.
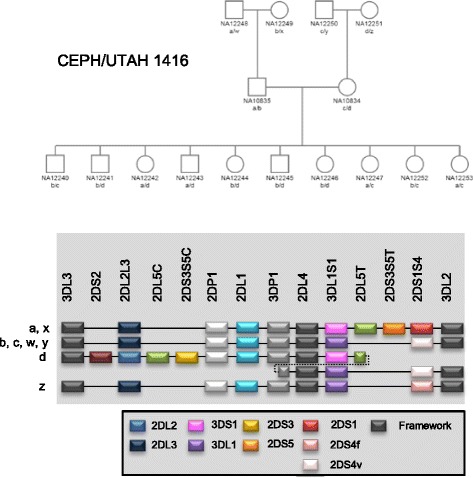
KIR haplotype segregation analysis in CEPH/UTAH pedigree 1416. a, b, c, d, w, x, y and z are the haplotypes deduced by segregation analysis. The gene content of each haplotype is shown. Haplotype c carries the fusion gene KIR2DL5/3DP1 and duplication of KIR3DP1, KIR2DL4 and KIR3DL1/S1 genes [28]
In pedigree 1347, sample NA11882 showed multiple copies for several genes. However only one offspring is available from this person and it seems that the haplotype carrying duplicated genes has not been transmitted to the next generation. Therefore, it was not visible by previous segregation analysis.
For UCLA KIR Exchange panel DNA samples, only presence and absence information was compared since the original data did not include copy number information. There was 100 % concordance between the two methods for KIR presence and absence data. However, additional information was given by copy number data. For example, UCLA76 and UCLA77 have three copies for 3DP1, 2DL4 and 3DL1/S1 loci. Potentially these two samples carry an extended haplotype, described previously [28] (Fig. 3). UCLA80 and UCLA82 have a deletion from 3DL1 to 3DL2, which is similar to a haplotype carrying the fusion gene 3DL1/2v [24] (Fig. 3). For NA10861, NA11994 and the sequenced cell line PGF, COX and NA10832, the copy numbers calculated from our assay were the same as predicted from previous analysis [3, 30].
Fig. 3.

KIR haplotypes for UCLA KIR Exchange panel DNA samples predicted by gene copy number. UCLA76 and UCLA77 have three copies for KIR3DP1, KIR2DL4 and KIR3DL1/S1 loci and potentially carry an extended haplotype (Hap1), described previously [28]. Haplotype 2 of UCLA80 and UCLA82 have a deletion from KIR3DL1 to KIR3DL2, which is similar to a haplotype carrying the fusion gene 3DL1/2v [24]
CNV is seen for LILRA3 and LILRA6 loci but not for other LILR genes [34]. Copy number of LILRA6 had previously been determined for the CEPH family samples using Taqman CNV assays [35]. Our results showed complete concordance with this previous data (Additional file 1: Table S7).
Measurement of copy number clustering
Representative examples of clustering results derived for all the KIR are given in Fig. 4. The standard deviation in each cluster with the same assigned copy number was used to evaluate ability of clear copy number discrimination in the 20 KIR quantitative PCR assays. A total of 1698 DNA samples were used in this analysis (Additional file 1: Table S8). It was possible to confidently distinguish between 3 and 4 copies or even up to 5 copies with the data generated from KIR assays. Moreover, most of the data sets displayed a tighter distribution than a Gaussian distribution (D’Agostino–Pearson normality test). In this situation, the standard deviation usually overestimates the data variability.
Fig. 4.

Calculated copy number plotted against predicted copy number. Each cluster represents samples assigned with the same copy number. The line and error bar represent mean value and standard deviation of each cluster. The KIR gene copy numbers can be determined empirically or, as in this example, by using algorithms incorporated into the CopyCaller software
KIR haplotype inference in unrelated individuals
Family-based segregation analysis can be used to determine gene content on haplotypes. However, in unrelated samples assigning a gene to a haplotype is indefinite when copy number is greater than one for any given locus. For example, an individual carrying two copies of KIR3DS1 and one copy of KIR3DL1 could have two copies of KIR3DS1 on one haplotype and one copy of KIR3DS1 on the other haplotype. Alternatively, the individual could have one copy of KIR3DS1 and KIR3DL1 on the same haplotype with the second copy of KIR3DS1 on the other haplotype. Approaches for inferring KIR haplotype from copy number are detailed in Supplementary Material (Additional file 1: Tables S9–S11). To facilitate analysis of samples for KIR, an online tool was developed using Perl and MySQL called ‘KIR Haplotype Identifier’ (http://www.bioinformatics.cimr.cam.ac.uk/haplotypes/) (Additional file 1: Figure S5). This is a tool for imputing haplotype pairs using observed copy number for each KIR loci to help resolve KIR haplotypes from unphased genotypes of unrelated individuals. The default haplotype library used by this program is based on European-origin KIR gene data but analysis can be carried out using your own KIR haplotype frequency data. This is important when analysing a different population, otherwise the software may not give a proper representation of the samples. The tool outputs the possible combinations of haplotypes for each sample based on the gene content of all haplotypes supplied in the haplotype file. Three output files are generated. The first file (Haplotype Results) lists all possible haplotype pairs for each sample, each haplotypes frequency (from the haplotype file) and the predicted combined frequency of each haplotype pair (Additional file 1: Figure S5). The second file is in the same format as Haplotype Results; however, it lists only haplotypes with the highest combined frequency (Haplotype 1 Frequency × Haplotype 2 Frequency) for each sample. The third file (Log file) contains a list of samples where haplotype pairs could not be assigned. In these cases, possible single haplotypes are listed per sample. These results can be visualised by a ‘KIR Haplotype Resolution Drawing Tool’ developed using R (Additional file 1: Figure S6). The script is available upon request.
Discussion
In this paper, we describe a KIR typing method based on real-time PCR. This method is able to detect the total number of copies of each KIR locus. Clear discrimination between 0, 1, 2, 3 or even 4 copies could be obtained using this method. We extended the approach to LILR loci, demonstrating that the qKAT approach can be used to analyse other mCNV loci.
This method is high-throughput and cost-effective. Using a Roche LightCycler 480 real-time PCR instrument with a 384-well Thermal Block Cycler we could complete our PCR assay in 65 min. A Twister II Plate Handler was used as an automation robotics system (Additional file 1: Figure S7). A MéCour Thermal Plate Stacker was used to keep the stacked plates constantly at 4 °C. With automation, around 22 plates comprising 8448 reactions can be finished within 24 h. Since our full KIR typing assay for each sample requires 40 reactions in total (including quadruplicates), this system can produce full KIR typing for around 210 samples every day.
Copy number information provided by quantitative PCR may be essential for accurate KIR determination. Accurate genotype data are required for population genetic studies and gene dosage effects [36]. Unlike standard genotypes, some KIR genes (e.g. KIR2DL5, KIR2DS3 and KIR2DS5) can be missing and actual genotype cannot be resolved without copy number information (e.g. –A and AA). Furthermore, recently discovered structural variations make typing even more difficult [3, 24, 28]. For truncated haplotypes carrying a multi-locus deletion, conventional methods can only detect them in a homozygote. For extended haplotypes carrying duplicated loci, typing at the allelic level may be helpful when the multiple alleles are different to each other. Specially designed inter-gene PCRs are useful approaches [3, 24, 37] but from our data it seems there may be many more truncated and extended haplotypes [38]. Nevertheless, none of the approaches could provide precise genotype without family data. Recently, pyrosequencing has been used for KIR typing and this can also provide copy number information although there are throughput and cost limitations [24, 39].
Accuracy is extremely important for quantification. We have shown that it is possible for real-time PCR to accurately determine the copy number from genomic DNA. Reference DNA samples were used to validate the accuracy and a large panel of families (1698 samples) to evaluate the precision. Since most CNVs follow Mendelian inheritance, family information can be used to infer copy number in each homologous chromosome after the total copy numbers are obtained from quantitative PCR. This method has been shown to enhance the accuracy of CNV detection [40]. For example, in CEPH family 1347, copy number information assisted in the deduction of gene content for all haplotypes when family data were insufficient to resolve haplotypes for all members with KIR presence/absence data. Our method could be further improved by using probabilistic models to increase confidence of chromosome-specific copy number estimates using family information [41]. This approach can be used for the future development of linkage and association tests that require chromosome-specific copy number information. However, like any other PCR-based method, highly polymorphic sequences always pose challenges for designing primers and probes. As we found with the 3DL1*056 allele in family CEPH1416, there is always the possibility that some rare alleles may be missed due to polymorphism. The primer and probes were designed to avoid known polymorphism in their annealing sites (Additional file 1: Tables S3 and S5) but as more alleles are described, care should be taken to continually review the assays and redesign the primers/probes as required. If an assay is disrupted by a rare SNP (true allele dropout) this will be identified by the loss of linkage with an adjacent gene that is known to be in high linkage disequilibrium; all KIR loci have another KIR locus in tight linkage or have an expected copy number, e.g. framework genes are usually always two copies. One can, therefore, check the data against these predefined ‘standard KIR haplotype rules’ (Additional file 1: Table S13) to identify unexpected results and these samples can be further investigated. Alternatively, inconsistencies can be found using the KIR Haplotype Identifier online tool through the appearance of an unusual haplotype in the results. In rare instances when confirmation is required, a second set of assays for each gene can be used for verification (Additional file 1: Table S14).
There are opportunities for further development of the copy number assay. For example, triplex real-time PCR was used in this assay, but it may be possible to achieve up to heptaplex real-time PCR [42] to improve the throughput. Inclusion of an additional reference gene or multicopy reference could provide superior normalisation for DNA input and avoid potential effects of local genomic changes to the reference gene. Supporting our current choice of reference gene, in our screening with qKAT we have not yet identified a sample exhibiting altered KIR copy number across all KIR loci, including framework genes, indicative of a genomic alteration to the reference gene. Currently, there are other methods to discriminate gene copy number, e.g. DNA microarray, multiplex ligation-dependent probe amplification (MPLA), branched DNA testing, paralogue ratio test (PRT), digital PCR [43] and next generation sequencing (NGS). In addition, KIR haplotyping can be achieved through dye-terminator sequencing of KIR gene amplicons [44]. Comparing current throughput, cost and complexity of assay setup, quantitative PCR has advantages over the others for KIR copy number analysis. Highly repetitive genomic intervals with long stretches of identical sequence, as in the KIR locus, have been less amenable to NGS. The present short read lengths obtained by NGS, or the current inaccuracy of long-read length single molecule sequencing, makes sequence assembly and phasing (haplotype-resolution) problematic for characterisation of mCNV loci, especially when more than two copies of a gene are present. As NGS methods improve and become cheaper, we anticipate that this approach will be useful, particularly at increased scale and for precise typing of KIR alleles at the nucleotide level. The two approaches will complement each other and be useful for cross-validation [45]. qKAT offers a simple solution for, as example, initial assessment KIR disease association at the gene-level or haplotype-level before investing more time in complex analysis at the allele-level. Once an association has been established, allele resolution typing could be informative if a sufficient number of samples are available for statistically powered analysis. qKAT is simple, one-step and flexible, in that a single gene, or combinations of genes, can be typed alone at minimal cost or as required (e.g. KIR A/B haplotype-defining genes). To date, 21 published studies (comprising >20,000 samples in total) including investigations of KIR disease association, function, expression and imputation have utilised the method [13, 31, 34, 38, 43, 45–60]. A KIR typing service using qKAT has also been established at the Addenbrooke’s Hospital Histocompatibility and Immunogenetics (Tissue Typing) laboratory in Cambridge (UK).
Conclusion
This simple, high-throughput and cost-effective direct KIR typing method can be used for disease association studies. In these studies, large numbers of cases and controls are usually needed for KIR-HLA interaction analysis. Therefore, this method allows analysis of large-scale studies that were previously labour-intensive, time-consuming and cost-prohibitive. The underlying strategy of qKAT offers a model for analysing any other highly diverse genomic regions of interest.
Acknowledgements
We thank Janelle Noble for providing DNA samples and Jyothi Jayaraman for technical assistance.
Funding
This work was funded by the Medical Research Council (MRC) and the Wellcome Trust with partial funding from the National Institute of Health (NIH) Cambridge Biomedical Research Centre and NIH Research Blood and Transplant Research Unit (NIHR BTRU) in Organ Donation and Transplantation at the University of Cambridge in collaboration with Newcastle University and in partnership with NHS Blood and Transplant (NHSBT). The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, the Department of Health or NHSBT.
Availability of data and material
The data generated and analysed during this study are available from the corresponding author on request.
Authors’ contributions
WJ, MRLA and JAT designed the assays, performed experiments, analysed the data and wrote the manuscript. CJ contributed to experimental design as well as acquisition and analysis of the data. NS, DD and JAT developed the analysis tools. JT contributed to drafting the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable. The biobank genomic DNA samples from immortalised cell lines were received codified and untraceable to an individual (fully anonymised). The biobanks (HBDI, Coriell Repositories, UCLA International KIR Exchange) hold the original REC approvals for generic research use and the consent documentation.
Abbreviations
- BHQ
Black hole quencher
- CEPH
Centre d’Etude du Polymorphisme Humain
- HBDI
Human Biological Data Interchange
- HLA
Human leukocyte antigen
- HWE
Hardy–Weinberg Equilibrium
- KIR
Killer immunoglobulin-like receptor
- LRC
Leukocyte receptor complex
- MALDI-TOF
Matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry
- mCNV
Multi-allelic copy number variation
- MPLA
Multiplex ligation-dependent probe amplification
- NAHR
Non-allelic homologous recombination
- PCR-SSO
Sequence-specific oligonucleotide PCR
- PCR-SSP
Sequence-specific primed PCR
- qKAT
Quantitative KIR semi-automated typing
Additional file
Supplementary figures S1–S9, supplementary tables S1–S14. (DOCX 2712 kb)
References
- 1.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. 2006;444(7118):444–54. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK. A high-resolution survey of deletion polymorphism in the human genome. Nat Genet. 2006;38(1):75–81. doi: 10.1038/ng1697. [DOI] [PubMed] [Google Scholar]
- 3.Traherne JA, Martin M, Ward R, Ohashi M, Pellett F, Gladman D, et al. Mechanisms of copy number variation and hybrid gene formation in the KIR immune gene complex. Hum Mol Genet. 2010;19(5):737–51. doi: 10.1093/hmg/ddp538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530(7589):177–83. doi: 10.1038/nature16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet. 2007;80(6):1037–54. doi: 10.1086/518257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olsson LM, Holmdahl R. Copy number variation in autoimmunity--importance hidden in complexity? Eur J Immunol. 2012;42(8):1969–76. doi: 10.1002/eji.201242601. [DOI] [PubMed] [Google Scholar]
- 7.Hirayasu K, Arase H. Functional and genetic diversity of leukocyte immunoglobulin-like receptor and implication for disease associations. J Hum Genet. 2015;60(11):703–8. doi: 10.1038/jhg.2015.64. [DOI] [PubMed] [Google Scholar]
- 8.Trowsdale J, Jones DC, Barrow AD, Traherne JA. Surveillance of cell and tissue perturbation by receptors in the LRC. Immunol Rev. 2015;267(1):117–36. doi: 10.1111/imr.12314. [DOI] [PubMed] [Google Scholar]
- 9.Pyo CW, Guethlein LA, Vu Q, Wang R, Abi-Rached L, Norman PJ, et al. Different patterns of evolution in the centromeric and telomeric regions of group A and B haplotypes of the human killer cell Ig-like receptor locus. PLoS One. 2010;5(12):e15115. doi: 10.1371/journal.pone.0015115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Litwin V, Gumperz J, Parham P, Phillips JH, Lanier LL. NKB1: a natural killer cell receptor involved in the recognition of polymorphic HLA-B molecules. J Exp Med. 1994;180(2):537–43. doi: 10.1084/jem.180.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moretta A, Vitale M, Bottino C, Orengo AM, Morelli L, Augugliaro R, et al. P58 molecules as putative receptors for major histocompatibility complex (MHC) class I molecules in human natural killer (NK) cells. Anti-p58 antibodies reconstitute lysis of MHC class I-protected cells in NK clones displaying different specificities. J Exp Med. 1993;178(2):597–604. doi: 10.1084/jem.178.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parham P. MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol. 2005;5(3):201–14. doi: 10.1038/nri1570. [DOI] [PubMed] [Google Scholar]
- 13.Beziat V, Traherne JA, Liu LL, Jayaraman J, Enqvist M, Larsson S, et al. Influence of KIR gene copy number on natural killer cell education. Blood. 2013;121(23):4703–7. doi: 10.1182/blood-2012-10-461442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moretta L, Locatelli F, Pende D, Marcenaro E, Mingari MC, Moretta A. Killer Ig-like receptor-mediated control of natural killer cell alloreactivity in haploidentical hematopoietic stem cell transplantation. Blood. 2011;117(3):764–71. doi: 10.1182/blood-2010-08-264085. [DOI] [PubMed] [Google Scholar]
- 15.Khakoo SI, Thio CL, Martin MP, Brooks CR, Gao X, Astemborski J, et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science. 2004;305(5685):872–4. doi: 10.1126/science.1097670. [DOI] [PubMed] [Google Scholar]
- 16.Pelak K, Need AC, Fellay J, Shianna KV, Feng S, Urban TJ, et al. Copy number variation of KIR genes influences HIV-1 control. PLoS Biol. 2011;9(11):e1001208. doi: 10.1371/journal.pbio.1001208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vilches C, Castano J, Gomez-Lozano N, Estefania E. Facilitation of KIR genotyping by a PCR-SSP method that amplifies short DNA fragments. Tissue Antigens. 2007;70(5):415–22. doi: 10.1111/j.1399-0039.2007.00923.x. [DOI] [PubMed] [Google Scholar]
- 18.Ashouri E, Ghaderi A, Reed EF, Rajalingam R. A novel duplex SSP-PCR typing method for KIR gene profiling. Tissue Antigens. 2009;74(1):62–7. doi: 10.1111/j.1399-0039.2009.01259.x. [DOI] [PubMed] [Google Scholar]
- 19.Martin MP, Carrington M. KIR locus polymorphisms: genotyping and disease association analysis. Methods Mol Biol (Clifton, NJ) 2008;415:49–64. doi: 10.1007/978-1-59745-570-1_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nong T, Saito K, Blair L, Tarsitani C, Lee JH. KIR genotyping by reverse sequence-specific oligonucleotide methodology. Tissue Antigens. 2007;69(Suppl 1):92–5. doi: 10.1111/j.1399-0039.2006.762_3.x. [DOI] [PubMed] [Google Scholar]
- 21.Houtchens KA, Nichols RJ, Ladner MB, Boal HE, Sollars C, Geraghty DE, et al. High-throughput killer cell immunoglobulin-like receptor genotyping by MALDI-TOF mass spectrometry with discovery of novel alleles. Immunogenetics. 2007;59(7):525–37. doi: 10.1007/s00251-007-0222-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6(10):986–94. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- 23.Degenhardt JD, de Candia P, Chabot A, Schwartz S, Henderson L, Ling B, et al. Copy number variation of CCL3-like genes affects rate of progression to simian-AIDS in Rhesus Macaques (Macaca mulatta) PLoS Genet. 2009;5(1):e1000346. doi: 10.1371/journal.pgen.1000346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Norman PJ, Abi-Rached L, Gendzekhadze K, Hammond JA, Moesta AK, Sharma D, et al. Meiotic recombination generates rich diversity in NK cell receptor genes, alleles, and haplotypes. Genome Res. 2009;19(5):757–69. doi: 10.1101/gr.085738.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 28.Martin MP, Bashirova A, Traherne J, Trowsdale J, Carrington M. Cutting edge: expansion of the KIR locus by unequal crossing over. J Immunol. 2003;171(5):2192–5. doi: 10.4049/jimmunol.171.5.2192. [DOI] [PubMed] [Google Scholar]
- 29.Helmberg W, Feolo M, Dunivin R. dbLRC-KIR, a new specialised database for genes of the leucocyte receptor complex. Hum Immunol. 2005;66(8, Supplement 1):17. doi: 10.1016/j.humimm.2005.08.028. [DOI] [Google Scholar]
- 30.Horton R, Coggill P, Miretti MM, Sambrook JG, Traherne JA, Ward R, et al. The LRC haplotype project: a resource for killer immunoglobulin-like receptor-linked association studies. Tissue Antigens. 2006;68(5):450–2. doi: 10.1111/j.1399-0039.2006.00697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Traherne JA, Jiang W, Valdes AM, Hollenbach JA, Jayaraman J, Lane JA, et al. KIR haplotypes are associated with late-onset type 1 diabetes in European-American families. Genes Immun. 2016;17(1):8–12. doi: 10.1038/gene.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30(1):97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 33.Sun JY, Oki A, Senitzer D. Alleles and intron polymorphism of KIR3DL1 shown by combination of allele group-specific primers and sequencing. Tissue Antigens. 2008;72(6):578–80. doi: 10.1111/j.1399-0039.2008.01141.x. [DOI] [PubMed] [Google Scholar]
- 34.Lopez-Alvarez MR, Jones DC, Jiang W, Traherne JA, Trowsdale J. Copy number and nucleotide variation of the LILR family of myelomonocytic cell activating and inhibitory receptors. Immunogenetics. 2014;66(2):73–83. doi: 10.1007/s00251-013-0742-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bashirova AA, Apps R, Vince N, Mochalova Y, Yu XG, Carrington M. Diversity of the human LILRB3/A6 locus encoding a myeloid inhibitory and activating receptor pair. Immunogenetics. 2014;66(1):1–8. doi: 10.1007/s00251-013-0730-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Pascal V, Martin MP, Carrington M, Anderson SK. Genetic control of variegated KIR gene expression: polymorphisms of the bi-directional KIR3DL1 promoter are associated with distinct frequencies of gene expression. PLoS Genet. 2008;4(11):e1000254. doi: 10.1371/journal.pgen.1000254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gomez-Lozano N, Estefania E, Williams F, Halfpenny I, Middleton D, Solis R, et al. The silent KIR3DP1 gene (CD158c) is transcribed and might encode a secreted receptor in a minority of humans, in whom the KIR3DP1, KIR2DL4 and KIR3DL1/KIR3DS1 genes are duplicated. Eur J Immunol. 2005;35(1):16–24. doi: 10.1002/eji.200425493. [DOI] [PubMed] [Google Scholar]
- 38.Jiang W, Johnson C, Jayaraman J, Simecek N, Noble J, Moffatt MF, et al. Copy number variation leads to considerable diversity for B but not A haplotypes of the human KIR genes encoding NK cell receptors. Genome Res. 2012;22(10):1845–54. doi: 10.1101/gr.137976.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moreland AJ, Guethlein LA, Reeves RK, Broman KW, Johnson RP, Parham P, et al. Characterization of killer immunoglobulin-like receptor genetics and comprehensive genotyping by pyrosequencing in rhesus macaques. BMC Genomics. 2011;12:295. doi: 10.1186/1471-2164-12-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kosta K, Sabroe I, Goke J, Nibbs RJ, Tsanakas J, Whyte MK, et al. A Bayesian approach to copy-number-polymorphism analysis in nuclear pedigrees. Am J Hum Genet. 2007;81(4):808–12. doi: 10.1086/520096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang K, Chen Z, Tadesse MG, Glessner J, Grant SF, Hakonarson H, et al. Modeling genetic inheritance of copy number variations. Nucleic Acids Res. 2008;36(21):e138. doi: 10.1093/nar/gkn641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Köppel R, Zimmerli F, Breitenmoser A. Heptaplex real-time PCR for the identification and quantification of DNA from beef, pork, chicken, turkey, horse meat, sheep (mutton) and goat. Eur Food Res Technol. 2009;230(1):125–33. doi: 10.1007/s00217-009-1154-5. [DOI] [Google Scholar]
- 43.Roberts CH, Jiang W, Jayaraman J, Trowsdale J, Holland MJ, Traherne JA. Killer-cell Immunoglobulin-like Receptor gene linkage and copy number variation analysis by droplet digital PCR. Genome Med. 2014;6(3):20. doi: 10.1186/gm537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pyo CW, Wang R, Vu Q, Cereb N, Yang SY, Duh FM, et al. Recombinant structures expand and contract inter and intragenic diversification at the KIR locus. BMC Genomics. 2013;14:89. doi: 10.1186/1471-2164-14-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Norman PJ, Hollenbach JA, Nemat-Gorgani N, Marin WM, Norberg SJ, Ashouri E, et al. Defining KIR and HLA class I genotypes at highest resolution via high-throughput sequencing. Am J Hum Genet. 2016;99(2):375–91. doi: 10.1016/j.ajhg.2016.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beziat V, Liu LL, Malmberg JA, Ivarsson MA, Sohlberg E, Bjorklund AT, et al. NK cell responses to cytomegalovirus infection lead to stable imprints in the human KIR repertoire and involve activating KIRs. Blood. 2013;121(14):2678–88. doi: 10.1182/blood-2012-10-459545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakimuli A, Chazara O, Farrell L, Hiby SE, Tukwasibwe S, Knee O, et al. Killer cell immunoglobulin-like receptor (KIR) genes and their HLA-C ligands in a Ugandan population. Immunogenetics. 2013;65(11):765–75. doi: 10.1007/s00251-013-0724-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pontikos N, Smyth DJ, Schuilenburg H, Howson JM, Walker NM, Burren OS, et al. A hybrid qPCR/SNP array approach allows cost efficient assessment of KIR gene copy numbers in large samples. BMC Genomics. 2014;15:274. doi: 10.1186/1471-2164-15-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beziat V, Traherne J, Malmberg JA, Ivarsson MA, Bjorkstrom NK, Retiere C, et al. Tracing dynamic expansion of human NK-cell subsets by high-resolution analysis of KIR repertoires and cellular differentiation. Eur J Immunol. 2014;44(7):2192–6. doi: 10.1002/eji.201444464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fosby B, Naess S, Hov JR, Traherne J, Boberg KM, Trowsdale J, et al. HLA variants related to primary sclerosing cholangitis influence rejection after liver transplantation. World J Gastroenterol. 2014;20(14):3986–4000. doi: 10.3748/wjg.v20.i14.3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nemat-Gorgani N, Edinur HA, Hollenbach JA, Traherne JA, Dunn PP, Chambers GK, et al. KIR diversity in Maori and Polynesians: populations in which HLA-B is not a significant KIR ligand. Immunogenetics. 2014;66(11):597–611. doi: 10.1007/s00251-014-0794-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones DC, Peacock S, Hughes D, Traherne JA, Allen RL, Barnardo MC, et al. Killer immunoglobulin-like receptor gene repertoire influences viral load of primary human cytomegalovirus infection in renal transplant patients. Genes Immun. 2014;15(8):562–8. doi: 10.1038/gene.2014.53. [DOI] [PubMed] [Google Scholar]
- 53.Cassidy S, Mukherjee S, Myint TM, Mbiribindi B, North H, Traherne J, et al. Peptide selectivity discriminates NK cells from KIR2DL2- and KIR2DL3-positive individuals. Eur J Immunol. 2015;45(2):492–500. doi: 10.1002/eji.201444613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakimuli A, Chazara O, Hiby SE, Farrell L, Tukwasibwe S, Jayaraman J, et al. A KIR B centromeric region present in Africans but not Europeans protects pregnant women from pre-eclampsia. Proc Natl Acad Sci U S A. 2015;112(3):845–50. doi: 10.1073/pnas.1413453112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dunphy SE, Guinan KJ, Chorcora CN, Jayaraman J, Traherne JA, Trowsdale J, et al. 2DL1, 2DL2 and 2DL3 all contribute to KIR phenotype variability on human NK cells. Genes Immun. 2015;16(5):301–10. doi: 10.1038/gene.2015.15. [DOI] [PubMed] [Google Scholar]
- 56.Hydes TJ, Moesker B, Traherne JA, Ashraf S, Alexander GJ, Dimitrov BD, et al. The interaction of genetic determinants in the outcome of HCV infection: evidence for discrete immunological pathways. Tissue Antigens. 2015;86(4):267–75. doi: 10.1111/tan.12650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vukcevic D, Traherne JA, Naess S, Ellinghaus E, Kamatani Y, Dilthey A, et al. Imputation of KIR types from SNP variation data. Am J Hum Genet. 2015;97(4):593–607. doi: 10.1016/j.ajhg.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jamil KM, Hydes TJ, Cheent KS, Cassidy SA, Traherne JA, Jayaraman J, et al. STAT4-associated natural killer cell tolerance following liver transplantation. Gut. 2016 doi: 10.1136/gutjnl-2015-309395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu LL, Landskron J, Ask EH, Enqvist M, Sohlberg E, Traherne JA, et al. Critical role of CD2 co-stimulation in adaptive natural killer cell responses revealed in NKG2C-deficient humans. Cell Rep. 2016;15(5):1088–99. doi: 10.1016/j.celrep.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lopez-Alvarez MR, Jiang W, Jones DC, Jayaraman J, Johnson C, Cookson WO, et al. LILRA6 copy number variation correlates with susceptibility to atopic dermatitis. Immunogenetics. 2016;68(9):743–7. doi: 10.1007/s00251-016-0924-z. [DOI] [PMC free article] [PubMed] [Google Scholar]