

Abstract
Nearly 40% of children with acute myeloid leukemia (AML) suffer relapse due to chemoresistance, often involving upregulation of the oncoprotein STAT3 (signal transducer and activator of transcription 3). In this paper, rhodium(II)-catalyzed, proximity-driven modification identifies the STAT3 coiled-coil domain (CCD) as a novel ligand-binding site, and we describe a new naphthalene sulfonamide inhibitor that targets the CCD, blocks STAT3 function, and halts its disease-promoting effects in vitro, in tumor growth models, and in a leukemia mouse model, validating this new therapeutic target for resistant AML.
Keywords: STAT3, rhodium, leukemia, diazo, protein modifications
AML is an aggressive malignancy. Relapse is common, and relapsed tumors are typically chemoresistant, necessitating fundamentally new therapeutic approaches.[1] STAT3 signaling plays a key role in mediating drug resistance by halting apoptosis and increasing tumorigenicity.[2] As such, STAT3 is a tantalizing target for drug development, either alone or in combination with other chemotherapeutic agents. In this manuscript, we employ a new fingerprinting method—proximity-driven rhodium(II) catalysis—to identify a new inhibitor binding site within the coiled-coil domain (CCD) of STAT3. Furthermore, we describe an optimized compound that targets the coiled-coil domain of STAT3, inhibits STAT3 activity in cells and displays anti-leukemia activity in AML cells in culture and in vivo.
STAT3 is a multidomain protein that is activated by tyrosine phosphorylation (pY705) in response to cytokine–receptor binding (e.g. IL-6 binding to gp130). Reciprocal intermolecular interactions between the C-terminal loop (containing pY705) and the Src homology 2 (SH2) domain facilitate homodimerization (see Fig. 4 for structure). This dimerization drives STAT3 accumulation in the nucleus, DNA binding to the DNA-binding domain, and oncogene transcriptional activation. STAT3 also contains a 4-helix CCD, connecting the DNA-binding and SH2 domains to an N-terminal oligomerization domain. Upregulated STAT3 activation was first reported in cells transformed by the oncogene v-src,[3] and fibroblasts with constitutively-active STAT3 (STAT3-C) developed malignant properties and form tumors in nude mice.[4]
Figure 4.
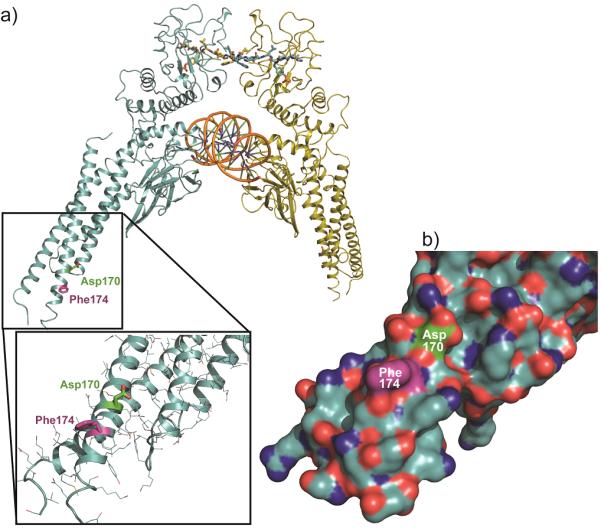
(a) Structure of the pSTAT3 dimer: SH2 domain at the top, bound to a phosphopeptide ligand (wireframe) of the complementary molecule. The central DNA-binding domain bound to dsDNA (orange). The CCD is shown in the inset, with the site of rhodium-catalyzed labeling (Phe174) in purple and the Asp170 residue (green), identified previously as a residue controlling STAT3 function. See text for discussion. (b) STAT3 coiled-coil region, indicating a lack of Lewis-basic (H, M, C) residues (orange).
STAT3 presents the classic problems of “undruggable” protein-protein interfaces. Efforts have typically involved molecules that bind the SH2 domain, mimicking phosphotyrosine interactions,[2, 5–11] though inhibitors of other STAT proteins apparently not involving the SH2 domain have appeared.[12] Our understanding of STAT3 function makes clear that disrupting SH2–phosphopeptide interactions necessary for dimerization will disrupt STAT3 function, and SH2 interactions are essential for STAT3 phosphorylation by upstream kinases. However, the discovery of cell-permeable small-molecule SH2 inhibitors has proven challenging. Beyond the SH2 and DNA-binding domains, other STAT3 domains are relatively unique, and little is known of their function or potential as drug targets.
We identified compelling naphthalene sulfonamides with activity against STAT3, including an initial lead compound (C188, Fig. 1),[13] and compounds from structural optimization, such as C188-9.[2] These compounds inhibit STAT3 binding to an immobilized pY-peptide in surface plasmon resonance (SPR) assays and exhibit other encouraging properties, including inhibition of STAT3 phosphorylation in cells, inhibition of STAT3-dependent gene transcription, and apoptosis induction in AML cell lines. We initially assumed that the activity of these compounds was due to direct binding to the SH2 domain, based on analogy to other STAT3 inhibition efforts and our own computational and medicinal chemistry efforts.
Figure 1.

Structures of selected STAT3 inhibitors.
Further investigations suggested that C188-9 may bind to STAT3 at a site distinct from the SH2 domain. First, a few naphthalene sulfonamides inhibited pSTAT3–DNA binding (Fig. 2), but DNA-binding inhibition is poorly correlated with phosphopeptide-binding inhibition. For example, C188 and MM-206 exhibit dose-dependent inhibition of DNA binding (IC50 = 1–5 μM, Fig. 2c,d), similar to that observed for the SPR-based SH2 binding-inhibition assay. In contrast, C188-9 also inhibits STAT3 binding to its pY-peptide ligand, yet has minimal ability to inhibit STAT3 binding to DNA (IC50 >30 uM), as judged by an SPR-based assay of DNA–STAT3 interactions (c). Furthermore C188-9 and other successful inhibitors have no charge, in contrast to the anionic structure most SH2 small-molecule inhibitors, which mimic a phosphotyrosine dianion. The presumed SH2 domain target—with a prominent cysteine (Cys712) immediately flanking the binding pocket—should be a fruitful target for rhodium(II) conjugates as STAT3 inhibitors. We also failed in attempts to employ rhodium conjugates of naphthalene sulfonamides to improve affinity through rhodium coordination to Lewis-basic side chains found near the SH2 domain.[14–15] While metalloinhibitors failed to deliver on their intended purpose, they facilitated our identification of an allosteric binding site as a new target site for STAT3 inhibitors.
Figure 2.

(a,b) SPR measurement of STAT3 inhibition. STAT3 binding to an immobilized phosphopeptide inhibited by naphthalene sulfonamide inhibitors C188-9 (a) and MM-206 (b). (c) DNA-binding inhibition, measured by SPR. (d) Quantification of DNA-binding inhibition of MM-206.
Rhodium(II) conjugates catalyze reactions of diazo compounds with biomolecules,[16–18] via a metallocarbene intermediate. We previously discovered rhodium–peptide conjugates for protein functionalization,[19] and sought to extend these ideas to STAT3 and to small-molecule ligands, where covalent modification might serve to identify the site of binding of naphthalene sulfonamides. Among several advantages, the method allows biologically-relevant concentrations of a ligand–catalyst, has minimal spurious background labeling, and succeeds for weak, transient interactions. Importantly, the rhodium(II) core does not significantly alter STAT3–small molecule interactions. The rhodium complex, C188-9-Rh2, has in vitro potency similar to that of C188-9 in the SPR assays discussed above, and also inhibits STAT3 phosphorylation in cells and induces apoptosis in AML cell lines (Fig. S3).
To examine rhodium-catalyzed proximity-driven labeling, STAT3 was treated with a rhodium-inhibitor conjugate, C188-9-Rh2 (1 μM) in the presence of excess diazo 1 (1 mM) (Fig. 3). Negative control experiments, such as reactions catalyzed by Rh2(OAc)4, showed minimal modification by diazo reagents, as judged by fluorescent imaging of a blot membrane with a fluorogenic dye (3-azido-7-hydroxycoumarin) and a copper catalyst.[19] In contrast, reactions catalyzed by C188-9-Rh2 gave a strongly fluorescent STAT3 band after azide–alkyne coupling of a fluorogenic azide, indicative of clean modification. Of note, modification of monomeric STAT3 or dimeric pSTAT3 occured with equal facility. Tryptic digestion of the modified STAT3 allowed identification of the site of modification by LC–MS/MS methods (see SI for details). We observed a single new peak with a mass (m+ = 2160.99) consistent with a single modification of an expected digest peptide, Val164–Lys177 (Fig. 3c,d). The identity of this digest peptide could be further confirmed—and the specific site of modification identified—by MS/MS fragmentation of the +1 mod peak. As shown in Fig. 3e, sufficient coverage of b and y ions was observed to conclusively identify phenylalanine (Phe174) as the site of modification. Phenylalanine was previously identified as an efficient reaction site in rhodium-conjugate-catalyzed side-chain modifications.[20]
Figure 3.

(a) Scheme of affinity labeling catalyzed by rhodium(II) conjugates. Recombinant STAT3 (no N-terminal domain, 2 μM) was treated with C188-9-Rh2 (0.5 μM) and diazo 1 (700 μM) in pH 7.4 HEPES. For pSTAT labeling, pSTAT3 (1 μM) and C188-9-Rh2 (1 μM) were used. (b) Blot analysis (3-azido-7-hydroxycoumarin and a copper catalyst) of reactions in the presence of other STAT3 inhibitors. Surface cysteine was blocked with iodoacetimide prior to rhodium reactions. Rxn time for STAT3 = 6 h, for pSTAT3 = 14 h. (c) Extracted ion chromatogram (EIC) of tryptic digest for modified STAT3 for the Val164–Lys177 peptide (m = 1745.80) and +1mod (m = 2160.99). (d) MS spectra at tr = 16.9 min. (e) LC–MS/MS analysis of the Phe174-modified Val164–Lys177 peptide, with expected y and b ions, Daughter ions in bold matched to ±10 ppm.
The modified Phe174 residue sits in the coiled-coil domain (CCD, Fig. 4), quite distal from the expected SH2 domain. We found no evidence for modification of other residues, within the SH2 domain or elsewhere. Modification of Phe174 was inhibited in the presence of MM-206, C188-9, or C188 (Fig. 3b, lanes e–g, j, l), indicating that other naphthalene sulfonamide compounds compete directly for the CCD binding site. In stark contrast, a phosphopeptide SH2 ligand has no effect on the modification (lanes d, k), indicating that SH2 ligands have minimal impact on the distal CCD domain. Similar inhibition was observed with dimeric pSTAT3, which has no accessible SH2 domain. Thus, in addition to identifying the CCD as the site of binding, rhodium-conjugate catalytic modification serves as a direct tool to probe for and establish CCD binding by other inhibitors.
The identification of a unique CCD binding site, while unexpected, is consistent with disparate and puzzling previous observations, including the limited correlation between DNA- and phosphopeptide-binding inhibition. Most compellingly, covalent labeling of the coiled-coil domain with the rhodium-conjugate catalyst is independent of STAT3 dimerization state. Were rhodium-conjugates interacting with the SH2 domain, significantly diminished modification would be expected in the dimeric (phosphorylated) state. Instead, modification efficiency was indistinguishable comparing monomeric STAT3 with phosphorylated dimeric protein that presumably has no available SH2 binding sites (Fig. 3b). Additionally, binding to the coiled-coil domain near Phe174 provides a rationale for the failure of rhodium conjugates to deliver improved STAT3 inhibitory activity due to Lewis-basic coordination (with Met/Cys/His): the coiled coil surface is devoid of Lewis-basic side chains (Fig. 4b).
In 2000, a study[21] described STAT3 mutations within the CCD and presented results that now appear remarkably consistent with our findings. Two examples of STAT3 proteins with a point mutation in the coiled-coil domain had drastically lower SH2-mediated affinity for phosphopeptide recognition sequences in gp130 and EGFR, and these mutants had drastically lower STAT3 phosphorylation stimulated by these receptors.[21] The key residue in the CCD, a point mutant of which exerted the largest effect on STAT3 activity and specifically SH2 affinity, was Asp170. In a coiled coil, this key aspartate (Asp 170) lies one helix turn away from—and thus directly neighbors—the Phe174 that we identified as near the site of naphthalene sulfonamide interactions (Fig. 4a, b). More recent structural data show that the STAT3 tertiary structure—with coiled-coil and SH2 domains on opposite sides of the protein—remains constant regardless of phosphorylation or dimerization state.[22–24] Both the mutants[21] and the results described here point to the coiled-coil surface around Asp170 and Phe174 as crucial for STAT3 function, with point mutations or small-molecule binding having profound effects on the function of the distal SH2 domain. However, it remains for future studies to establish how selective inhibitors of the coiled-coil domain might be. From a drug-development perspective, there may be distinct advantages to targeting domains other than SH2, a large domain family present throughout the genome.[25–27]
Targeting the STAT3 CCD is relevant in models of acute myeloid leukemia (AML). From a brief study synthesizing derivatives of the C188 lead, fluorinated MM-206 (Fig. 1) stood out as a promising candidate. The perfluorophenylsulfonamide group, which has appeared in clinical trials without apparent toxicity concerns,[28–31] was suggested by another STAT3 inhibitor effort.[6] In the SPR-based competitive binding assay (Fig. 2a), MM-206 had IC50 = 1.2 μM, lower than previous-generation inhibitors such as C188 (IC50 = 7.2 μM) and C188-9 (IC50 = 5.0 μM). The inhibitor MM-206 also inhibits inducible STAT3 phosphorylation in AML cell lines (Fig. 5a,b). Following a 30-minute treatment with MM-206 prior to G-CSF stimulation, all three AML cell lines tested exhibited dose-dependent decreases in STAT3 phosphorylation with IC50 0.8–1.9 μM.
Figure 5.

MM-206 inhibits G-CSF-induced STAT3 phosphorylation, induces apoptosis in human AML cells, and slows disease progression in a xenograft model of AML. (a) Histogram of decrease in pSTAT3 in Kasumi-1 cells. (b) MM-206 inhibits G-CSF-induced pY-STAT3 in multiple AML cell lines. Legend values indicate IC50 values. Mean ±SD for n = 3. (c) Apoptosis quantified in Annexin V-FITC-labeled cells treated with MM-206 for 24 h. Spontaneous apoptosis in untreated cells was subtracted to yield the % apoptosis attributable to drug. Data shown for an AML cell line (MV4-11, HL-60) and primary patient-derived AML cells (p198) compared to ALL cell lines (KOPN-8, RS4-11) as a negative control. (d–f) NSG mice were injected iv with 107 MV4-11.ffluc AML cells at day 0. After two weeks, mice received MM-206 (30 mg/kg or vehicl0065), ip daily 5 days per week for 2 weeks (weeks 2–4) or 4 weeks (weeks 2–6). (d) At the time of death, bone marrow was collected and the percentage of human (diseased) AML cells in mouse marrow present was determined as a measure of diease progression. *P<0.05. (e) Kaplan-Meier survival curve shows significantly longer survival for mice treated for 4 weeks. (f) Luminescence images one week after treatment. Colorized signal intensity indicates amount of active disease, from low (blue) to high (red).
To evaluate the therapeutic potential of this new agent, we confirmed its ability to induce apoptosis in AML cell lines and primary tumor cells from pediatric AML patients. A 24-hour treatment with MM-206 increased apoptosis in all AML cell lines and primary samples tested (n = 4, Fig. 5c). Cell viability assays with luciferase-transduced AML cell lines provided similar results. (Fig. S2). In contrast, acute lymphoblastic leukemia (ALL) cell lines—which do not have upregulated STAT3 activity[32]—were much less sensitive to apoptosis induction (Fig. 5c, KOPN-8 and RS4-11), consistent with a STAT3-based mechanism of action.
Finally, we engrafted immunocompromised NOD-SCID-IL2Rγ−/− (NSG) mice with luciferase-expressing MV4-11 AML cells. After 2 weeks, mice were treated for two weeks (10 doses).
Treated cells showed lower luminescence at week 5 relative to control, indicating delayed disease progression. Disease did progress rapidly once treatment stopped, so we tested a 4-week regimen. Again, treament slowed disease progression. Further, there was a significant decrease in the percent of tumor cells in the bone marrow of treated mice (Fig. 5d), and treated mice had significantly prolonged survival (p=0.019 at 10 weeks; Fig. 5e).
Relatively little is known about the functional role of the STAT3 coiled coil. However, on the basis of data from just a few CCD-domain mutants, Cao made a prescient comment that “coiled-coil region of STAT3 may be a useful target for drug design.”[21] This work validates that prediction. It now appears that the N-terminal domain engages in action-at-a-distance, playing a crucial role conveying information between the N-terminal and C-terminal domains, akin to coiled-coil transmembrane domains in extracellular receptors. The inhibitor MM-206 serves as a tool to better understand STAT3 biology, and also validates the CCD as a therapeutic target. Identification of the CCD binding site is made possible by proximity-driven rhodium catalysis—used for the first time here to illuminate binding sites—and inhibition of catalytic modification directly assesses the binding of other STAT3 ligands.
Supplementary Material
Acknowledgements
M.B.M. was supported by a Ruth L. Kirchstein National Service Award (NIH F31CA180696). We acknowledge support from the NIH (5R21CA170625), from the Robert A. Welch Foundation Research Grant C-1680 (Z.T.B.), from the National Science Foundation (CHE-1055569, Z.T.B.), and from the Gillson Longenbaugh Foundation (M.S.R.).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Canner J, Alonzo TA, Franklin J, Freyer DR, Gamis A, Gerbing RB, Lange BJ, Meshinchi S, Woods WG, Perentesis J, Horan J. Cancer. 2013;119:4162. doi: 10.1002/cncr.28342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Redell MS, Ruiz MJ, Alonzo TA, Gerbing RB, Tweardy DJ. Blood. 2011;117:5701. doi: 10.1182/blood-2010-04-280123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yu CL, Meyer DJ, Campbell GS, Larner AC, Carter-Su C, Schwartz J, Jove R. Science. 1995;269:81. doi: 10.1126/science.7541555. [DOI] [PubMed] [Google Scholar]
- [4].Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Cell. 1999;98:295. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- [5].Liu LJ, Leung KH, Chan DSH, Wang YT, Ma DL, Leung CH. Cell Death Dis. 2014;5:e1293. doi: 10.1038/cddis.2014.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhang X, Yue P, Page BDG, Li T, Zhao W, Namanja AT, Paladino D, Zhao J, Chen Y, Gunning PT, Turkson J. Proc. Nat. Acad. Sci. U.S.A. 2012;109:9623. doi: 10.1073/pnas.1121606109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shahani VM, Yue P, Haftchenary S, Zhao W, Lukkarila JL, Zhang X, Ball D, Nona C, Gunning PT, Turkson J. ACS Med. Chem. Lett. 2011;2:79. doi: 10.1021/ml100224d. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [8].Shahani VM, Yue P, Fletcher S, Sharmeen S, Sukhai MA, Luu DP, Zhang X, Sun H, Zhao W, Schimmer AD, Turkson J, Gunning PT. Bioorg. Med. Chem. 2011;19:1823. doi: 10.1016/j.bmc.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Page BDG, Fletcher S, Yue P, Li Z, Zhang X, Sharmeen S, Datti A, Wrana JL, Trudel S, Schimmer AD, Turkson J, Gunning PT. Bioorg. Med. Chem. Lett. 2011;21:5605. doi: 10.1016/j.bmcl.2011.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kraskouskaya D, Duodu E, Arpin CC, Gunning PT. Chem. Soc. Rev. 2013;42:3337. doi: 10.1039/c3cs35449k. [DOI] [PubMed] [Google Scholar]
- [11].Morlacchi P, Robertson FM, Klostergaard J, McMurray JS. Future Med. Chem. 2014;6:1909. doi: 10.4155/fmc.14.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhou L, Kawate T, Liu X, Kim YB, Zhao Y, Feng G, Banerji J, Nash H, Whitehurst C, Jindal S, Siddiqui A, Seed B, Wolfe JL. Biorg. Med. Chem. 2012;20:750. doi: 10.1016/j.bmc.2011.12.006. [DOI] [PubMed] [Google Scholar]
- [13].Xu X, Kasembeli MM, Jiang X, Tweardy BJ, Tweardy DJ. PLoS ONE. 2009;4:e4783. doi: 10.1371/journal.pone.0004783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vohidov F, Knudsen SE, Leonard PG, Ohata J, Wheadon MJ, Popp BV, Ladbury JE, Ball ZT. Chem. Sci. 2015;6:4778. doi: 10.1039/c5sc01602a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kundu R, Cushing PR, Popp BV, Zhao Y, Madden DR, Ball ZT. Angew. Chem. Int. Ed. 2012;51:7217. doi: 10.1002/anie.201202291. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kundu R, Cushing PR, Popp BV, Zhao Y, Madden DR, Ball ZT. Angew. Chem. 2012;124:7329. doi: 10.1002/anie.201202291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tishinov K, Schmidt K, Häussinger D, Gillingham DG. Angew. Chem. Int. Ed. 2012;51:12000. doi: 10.1002/anie.201205201. [DOI] [PubMed] [Google Scholar]; Tishinov K, Schmidt K, Häussinger D, Gillingham DG. Angew. Chem. 2012;124:12166. doi: 10.1002/anie.201205201. [DOI] [PubMed] [Google Scholar]
- [17].Antos JM, McFarland JM, Iavarone AT, Francis MB. J. Am. Chem. Soc. 2009;131:6301. doi: 10.1021/ja900094h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ball ZT. Acc. Chem. Res. 2013;46:560. doi: 10.1021/ar300261h. [DOI] [PubMed] [Google Scholar]
- [19].Vohidov F, Coughlin JM, Ball ZT. Angew. Chem. Int. Ed. 2015;54:4587. doi: 10.1002/anie.201411745. [DOI] [PubMed] [Google Scholar]; Kundu R, Cushing PR, Popp BV, Zhao Y, Madden DR, Ball ZT. Angew. Chem. 2012;124:7329. doi: 10.1002/anie.201202291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Popp BV, Ball ZT. Chem. Sci. 2011;2:690. [Google Scholar]
- [21].Zhang T, Kee WH, Seow KT, Fung W, Cao X. Mol. Cell. Biol. 2000;20:7132. doi: 10.1128/mcb.20.19.7132-7139.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Neculai D, Neculai AM, Verrier S, Straub K, Klumpp K, Pfitzner E, Becker S. J. Biol. Chem. 2005;280:40782. doi: 10.1074/jbc.M507682200. [DOI] [PubMed] [Google Scholar]
- [23].Ren Z, Mao X, Mertens C, Krishnaraj R, Qin J, Mandal PK, Romanowski MJ, McMurray JS, Chen X. Biochem. Biophys. Res. Commun. 2008;374:1. doi: 10.1016/j.bbrc.2008.04.049. [DOI] [PubMed] [Google Scholar]
- [24].Nkansah E, Shah R, Collie GW, Parkinson GN, Palmer J, Rahman KM, Bui TT, Drake AF, Husby J, Neidle S, Zinzalla G, Thurston DE, Wilderspin AF. FEBS Lett. 2013;587:833. doi: 10.1016/j.febslet.2013.01.065. [DOI] [PubMed] [Google Scholar]
- [25].Jones RB, Gordus A, Krall JA, MacBeath G. Nature. 2006;439:168. doi: 10.1038/nature04177. [DOI] [PubMed] [Google Scholar]
- [26].Mace PD, Wallez Y, Dobaczewska MK, Lee JJ, Robinson H, Pasquale EB, Riedl SJ. Nat. Struct. Mol. Biol. 2011;18:1381. doi: 10.1038/nsmb.2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Taylor JD, Ababou A, Fawaz RR, Hobbs CJ, Williams MA, Ladbury JE. Proteins: Struct., Funct., Bioinf. 2008;73:929. doi: 10.1002/prot.22119. [DOI] [PubMed] [Google Scholar]
- [28].Sargent JM, Elgie AW, Williamson CJ, Hill BT. Anti-Cancer Drugs. 2003;14:467. doi: 10.1097/00001813-200307000-00013. [DOI] [PubMed] [Google Scholar]
- [29].Kluza J, Mazinghien R, Irwin H, Hartley JA, Bailly C. Anti-Cancer Drugs. 2006;17:155. doi: 10.1097/00001813-200602000-00006. [DOI] [PubMed] [Google Scholar]
- [30].Kirby S, Gertler SZ, Mason W, Watling C, Forsyth P, Aniagolu J, Stagg R, Wright M, Powers J, Eisenhauer EA. Neuro-Oncology. 2005;7:183. doi: 10.1215/S1152851704000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rubenstein SM, Baichwal V, Beckmann H, Clark DL, Frankmoelle W, Roche D, Santha E, Schwender S, Thoolen M, Ye Q, Jaen JC. J. Med. Chem. 2001;44:3599J. doi: 10.1021/jm000478d. Med. Chem. [DOI] [PubMed] [Google Scholar]
- [32].Furuichi Y, Goi K, Inukai T, Sato H, Nemoto A, Takahashi K, Akahane K, Hirose K, Honna H, Kuroda I, Zhang X, Kagami K, Hayashi Y, Harigaya K, Nakazawa S, Sugita K. Cancer Res. 2007;67:9852. doi: 10.1158/0008-5472.CAN-07-0105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.