

Abstract
RANK/RANKL facilitates migration/invasion via epithelial-mesenchymal transition (EMT) in certain malignant tumors. The relationship and mechanism between RANK/RANKL and EMT in endometrial cancer (EC) cells, however, remain unclear. In this study, we firstly showed that RANK/RANKL activation was correlated with EC staging and EMT markers in human EC tissue specimen. RANK/RANKL promoted migration/invasion and initiated EMT of EC cell lines. Then, protein chip analysis and enzyme-linked immunosorbent assay (ELISA) revealed that the expression and secretion of chemokine ligand 20 (CCL20) was dramatically enhanced in RANKL-treated RANK over-expressed EC cells. Moreover, the higher level of CCL20 in both serum and tumor tissue was detected in orthotopic transplantation mouse models. Finally, we confirmed that CCL20 contributed to invasion and EMT of RANK over-expressed EC cells. In summary, all data supported the hypothesis that RANK/RANKL elevated the expression and secretion of CCL20 in EC cells, which promoted cancer progression through EMT.
Keywords: CCL20, RANK, RANKL, epithelial-mesenchymal transition, endometrial cancer
INTRODUCTION
Endometrial cancer (EC) remains one of the most common gynecological malignancies worldwide. In United States, it ranks fourth place, with about 54,870 incident cases and 10,170 deaths in 2015 [1]. Despite recent medical progresses in surgical treatment for early-stage EC, therapy for advanced EC is still less effective [2, 3]. As we know, metastasis is the primary cause of poor prognosis for EC patients [4]. Therefore, illuminating the mechanisms that facilitate metastasis is of great concern. Based on present studies, cytokines and chemokines existing in the tumor microenvironment were supposed to promote tumor initiation, metastasis and progression [5, 6].
Receptor activator of nuclear factor-κB (RANK) and its ligand RANKL were shown to regulate osteoclast differentiation, bone remodeling and the formation of lymph node. Recent studies reported that RANKL accelerated invasion and metastasis in RANK-expressed cancer cells [7–9]. Also, Palafox et al. [10] found that RANK/RANKL induced EMT in human mammary epithelial cells. Our previous study confirmed that the activation of RANK/RANKL could obviously promote EC metastasis in animal orthotopic transplantation mode [11, 12]. However, it remains elusive whether RANK/RANKL could induce EMT in EC cells.
Some chemokines derived from tumor-associated leukocytes or tumor cells acted as growth factors for cancer cells. They could contribute to tumor metastasis by accelerating angiogenesis, attracting endothelial cells or regulating the motility ability of cancer cells. Among them, CCL20 had proved to be related to invasion and metastasis in some types of cancer [13]. CCL20 was identified in the liver for the first time [14]. It was initially named as liver and activation regulated chemokine (LARC) and alternatively called Exodus-1 or macrophage inflammatory protein-3a (MIP-3a). CCL20 was strongly chemotactic for lymphocytes as well as weakly attracted neutrophils [15]. Accumulating evidence showed that CCL20 was correlated with tumor formation, metastasis or progression in many malignant neoplasms, such as colorectal and breast cancer [16, 17]. In addition, several chemokines including CCL20 had been reported to induce EMT in various tumors [18, 19, 20]. EMT played a key role in tumor progression, which leaded to a more invasive and metastatic phenotype in many human cancers, involving EC [21]. However, the relationship between CCL20 and EMT in EC cells is still unknown.
To clarify the mechanism of RANK/RANKL-induced EMT in EC, we built RANK over-expressed cell lines and animal models. Protein chip analysis and ELISA suggested that the expression and secretion of CCL20 was dramatically enhanced in RANKL-treated RANK over-expressed EC cells. Furthermore, CCL20 could facilitate migration/invasion and promote cancer progression through EMT. This work firstly illustrated mechanisms by which RANK over-expressed EC cells became invasive and metastatic. In addition, it supplied a potential therapeutic target for EC.
RESULTS
RANK/RANKL activation correlates with EC staging and EMT markers in human EC tissue specimens
To clarify the effect of RANK/RANKL on the progression and EMT in EC, we used immunohistochemistry to detect levels of RANK, RANKL and EMT markers in human EC tissue specimens with different stage (I, II, III). The data showed that RANK/RANKL expression was significantly elevated in EC tissue of higher stage (Figure 1A, 1B). Moreover, RANK level was positively connected with N-cadherin (p = 0.0229) and Vimentin (p = 0.0398), but negatively with E-cadherin (p = 0.0118) (Figure 1A, 1C). This indicated that RANK/RANKL activation was related to EMT in EC.
Figure 1. Expression of RANK/RANKL and EMT markers in human EC tissue specimens.
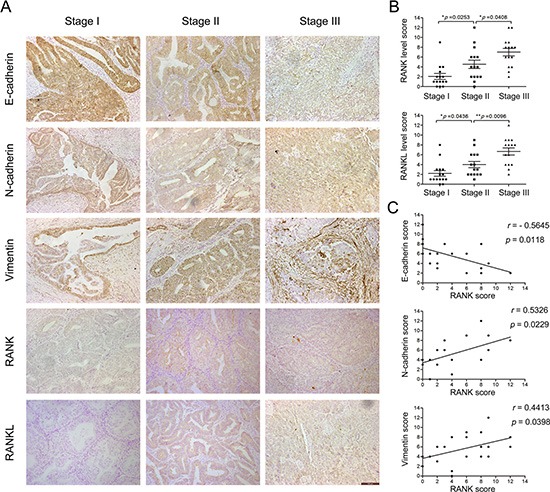
(A) IHC analysis of RANK, RANKL, E-cadherin, N-cadherin and Vimentin in EC (stage I, II, III). Amplification: 200×. Bar = 100 μm. (B) Semi-quantitative analysis of IHC staining determined the levels of RANK and RANKL in each stage of EC. (C) Expression correlations between RANK and E-cadherin, N-cadherin and Vimentin, respectively. Spearman's correlation coefficient test was used to statistical analysis.
Overexpression of RANK contributes to migration and invasion of EC cell lines treated by RANKL
RANK/RANKL was recently shown to promote invasion of cancer cells, but the underlying molecular mechanism remained to be determined. Here, the role of RANK/RANKL in EC cell lines was explored. In order to verify whether RANK/RANKL stimulated EC cells progression, the over-expression plasmid targeting RANK, pIRES2–3FLAG-EGFP-RANK, was inserted in HEC-1A and Ishikawa cells via transient transfection, which exhibited a constitutively active function of RANK receptor. Cells transfected with an empty plasmid, pIRES2–3FLAG-EGFP-CON236, served as the control. These cell lines were named HEC-1ARANK or HEC-1AControl and IshikawaRANK or IshikawaControl. Efficient transfection was detected before performing cellular assays (Figure 2A, 2B).
Figure 2. RANK/RANKL accelerates migration and invasion of EC cells.

(A, B) Overexpression of RANK in HEC-1A and Ishikawa cells revealed by qRT-PCR and western blotting, and the latter was further quantified by densitometry of three times. GAPDH was included as an internal control. **P < 0.01, ***P < 0.001, n = 3. (C, D) Wound-healing migration assays for HEC-1A and Ishikawa cells. The healing of wounds was imaged at 0 h and 48 h, showing that overexpression of RANK enhanced migration. The images were obtained at 100× magnification. Bar = 200 μm. (E, G) Transwell invasion assays. EC cells were grown in upper chamber for 48 h. Invaded cells were fixed, stained and imaged using an inverted microscope at 100 × magnification. Bar = 200 μm. (F, H) Graphs indicated the number of invasive cells for each group. **P < 0.01, n = 3.
In wound-healing migration assay, the wound area was monitored at 48 h after establishing the wound. The wound closure was reduced obviously in RANKL-treated HEC-1ARANK and IshikawaRANK cells when compared with control groups (Figure 2C, 2D).
To further investigate the invasion ability of EC cells, we carried out transwell invasion assay. The average number of cells invaded into the lower chamber was counted under inverted microscope from 5 fields. Comparing to control groups, the invaded number of RANKL-treated RANK over-expressed EC cells was significantly elevated (Figure 2E–2H).
These results collectively demonstrated that the migration and invasion capabilities of EC cells were significantly promoted by RANK/RANKL.
RANK/RANKL initiates EMT in EC cells
EMT was closely correlated with tumor metastasis and progression. To explore the relationship between RANK/RANKL and EMT in EC cells, we microscopically examined the change in morphology of RANK-overexpressed EC cells following stimulation with RANKL. After 48 h of treatment, HEC-1ARANK and IshikawaRANK cells were morphologically transformed toward mesenchymal fibroblastic spindle shape compared with control groups, suggesting a phenotypic transition from epithelial to mesenchymal (data not shown). Then, to test whether this morphological change represented EMT, we detected the expression of several EMT markers from multiple levels. Quantitative real-time PCR analysis indicated that the mRNA level of E-cadherin was decreased, whereas levels of N-cadherin, Vimentin, Snail and Twist were increased in RANKL-treated HEC-1ARANK and IshikawaRANK cells (Figure 3A, 3B). Western blotting and immunofluorescence staining analysis also showed that the overexpression of RANK led to down-regulation of E-cadherin, but up-regulation of N-cadherin, Vimentin, Snail and Twist in EC cells under RANKL treatment (Figure 3C–3E).
Figure 3. RANK/RANKL induces EMT in EC cells.
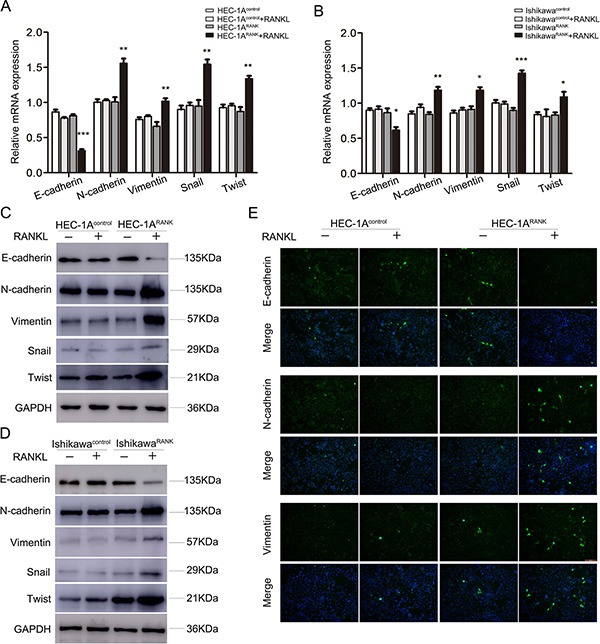
(A, B) HEC-1A and Ishikawa cells were transfected with an empty plasmid or over-expression plasmid targeting RANK. After 48 h stimulation of RANKL, the mRNA levels of E-cadherin, N-cadherin, Vimentin, Snail and Twist were analyzed by qRT-PCR. *P < 0.05, **P < 0.01, ***P < 0.001, n = 3. (C, D) HEC-1A and Ishikawa cells were transfected with an empty plasmid or over-expression plasmid targeting RANK. After 48 h stimulation of RANKL, the protein expression of E-cadherin, N-cadherin, Vimentin, Snail and Twist were detected by western blotting. (E) Immunofluorescence staining showed the expression of E-cadherin, N-cadherin and Vimentin in RANKL-treated HEC-1AControl and HEC-1ARANK cells. The images were obtained under fluorescence microscopy. Bar = 50 μm.
Taken together, our data suggested that RANK/RANKL could induce EMT in EC cells.
RANK/RANKL promotes the expression and secretion of CCL20 in vitro and in vivo
To identify the novel mediators of EMT in EC cells, RANKL-treated HEC-1ARANK and HEC-1AControl cells were analyzed using Human XL Oncology Array Kit from RandD Systems. The expression of CCL20 was elevated in RANKL-treated HEC-1ARANK cell revealed by protein chip analysis (Figure 4A, 4B). Then, ELISA was performed to detect the serum level of CCL20 in different treatment groups. The serum concentration of CCL20 in RANKL-treated HEC-1ARANK and IshikawaRANK cells was prominently higher than that in control groups, and this effect was time-dependent (Figure 4C, 4D). In addition, immunofluorescence staining suggested that CCL20 expression was enhanced in RANKL-treated HEC-1ARANK cell (Figure 4E).
Figure 4. RANK/RANKL increases the expression and secretion level of CCL20 in EC cells.
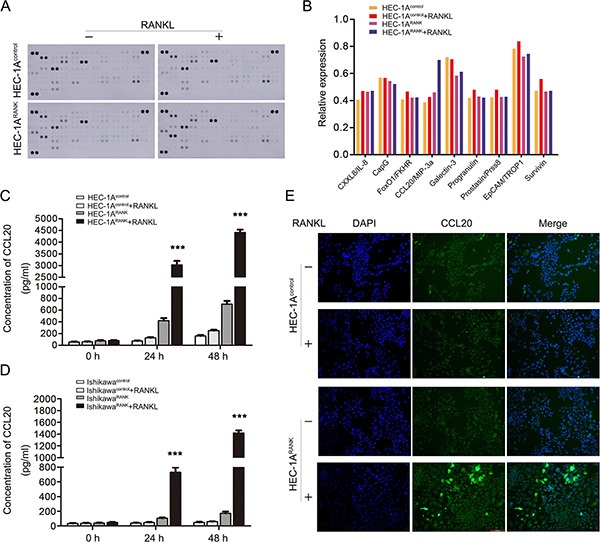
(A) The Human XL Oncology Array detected multiple analytes in cell culture lysates. The lysates were gained from HEC-1AControl and HEC-1ARANK cells under the treatment of RANKL for 48 h. (B) The graph summarized the relative signal intensity of indicated molecules. Among them, CCL20 varied most significantly. (C, D) The cell supernatant was collected from HEC-1A/IshikawaControl cells and HEC-1A/IshikawaRANK cells treated by RANKL for 48 h. The serum concentration of CCL20 was measured with ELISA kit. ***P < 0.001, n = 3. (E) Immunofluorescence staining showed the expression of CCL20 in HEC-1AControl and HEC-1ARANK cells after 48 h stimulation of RANKL. The images were obtained by fluorescence microscopy. Bar = 100 μm.
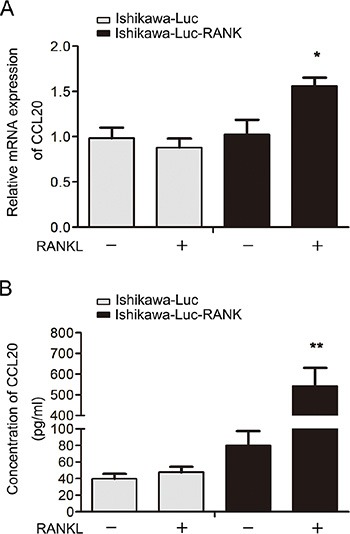
So as to further evaluate the connection between CCL20 and RANK/RANKL in EC, we built animal transplantation models, detecting the expression and secretion of CCL20 in vivo. We gained Ishikawa-Luc/Ishikawa-Luc-Rank light-emitting EC cells to establish orthotopic xenograft nude model. After four weeks’ injection of RANKL, the mice were sacrificed. Tumor tissues were then removed from animals, and grinded to obtain mRNA. qRT-PCR analysis showed that the mRNA level of CCL20 was higher in RANKL-treated group (Figure 5A). Meanwhile, the serum was collected to measure the concentration of CCL20 via ELISA. We discovered that the secretion of CCL20 was also notably increased in RANKL-treated group (Figure 5B).
Figure 5. Expression and secretion level of CCL20 elevates in nude mice orthotopic transplantation modes.
(A) Tumor tissues were removed from tumor-bearing mouse after four weeks’ injection of RANKL. The mRNA expression of CCL20 was detected by qRT-PCR. *P < 0.05, n = 3. (B) Serum from each group was collected when the mice were sacrificed immediately. The serum level of CCL20 was measured using the ELISA kit. **P < 0.01, n = 3.
CCL20 facilitates invasion and EMT of RANK over-expressed EC cells
To better understand the effect of CCL20 on metastasis of EC cells, chamber co-culture assays were carried out. As shown in Figure 6A and 6B, the number of invaded HEC-1ARANK cells was more than HEC-1AControl cells with the co-culture of exogenous CCL20. Besides, when the conditional medium of RANKL-treated HEC-1ARANK cells that included massive endogenous CCL20 was added to lower chambers, HEC-1ARANK cells exhibited stronger invasion ability. Addition of neutralizing antibody against CCL20, however, prominently blocked the capability of CCL20 to promote invasion (Figure 6C, 6D).
Figure 6. CCL20 facilitates invasion and EMT of RANK over-expressed EC cells.
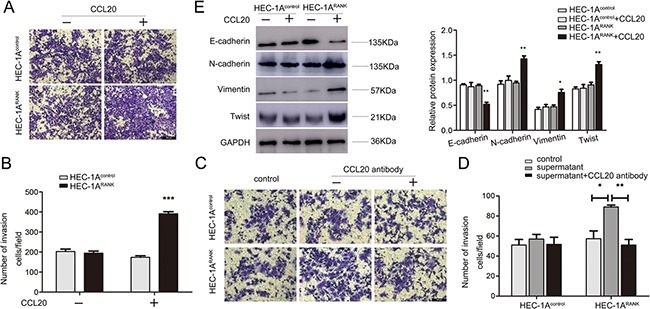
(A) HEC-1AControl and HEC-1ARANK cells were grown in upper chamber without or with the co-culture of exogenous CCL20 (50 ng/ml) for 48 h. Invaded cells were fixed, stained and imaged under an inverted microscope at 100× magnification. Bar = 200 μm. (B) Quantitative analysis of invaded cell number from five random fields. ***P < 0.001, n = 3. (C) HEC-1AControl and HEC-1ARANK cells were grown in upper chamber. Then, added the conditional medium of RANKL-treated HEC-1ARANK cell without or with neutralizing antibody against CCL20 to the lower chambers. After 48 h, representative images were obtained at 200× magnification. Bar = 200 μm. (D) Graphs indicated the number of invaded cells for each group. *P < 0.05, **P < 0.01, n = 3. (E) HEC-1AControl and HEC-1ARANK cells were treated by CCL20 for 48 h. The protein expression of E-cadherin, N-cadherin, Vimentin and Twist were measured. Quantitative analysis of the results was further shown. *P < 0.05, **P < 0.01, n = 3.
In order to clarify the relation between CCL20 and EMT in EC cells, HEC-1AControl and HEC-1ARANK cells were treated with CCL20 for 48 h, and then the levels of EMT markers were measured. Western blotting analysis showed that the expression of E-cadherin was decreased, whereas N-cadherin, Vimentin and Twist were increased in CCL20-stimulated HEC-1ARANK cells (Figure 6E). Collectively, these data indicated that CCL20 could induce EMT in RANK over-expressed EC cells.
DISCUSSION
Tumor metastasis and progression are complex and multistep processes, which make them the leading cause of recurrence and death in cancer patients. Consequently, elucidating molecular mechanisms that promote metastasis is indispensable to seek new therapeutic strategies and improve therapeutic effects of patients with EC [22].
RANK/RANKL is needful for normal physiological processes such as immune responses and bone remodeling [23]. Also, it facilitates invasion and metastasis of human cancers [24]. Our previous study showed that RANK/RANKL expression was higher in EC tissue, and could dramatically promote EC metastasis [11, 12]. Here, we concluded that over-expression of RANK contributed to migration and invasion of EC cell lines under the stimulation of RANKL.
EMT correlates with tumor metastasis and progression. A notable EMT feature is the loss of E-cadherin, which enables a cell to dissolve cell-cell adhesion and break away from its neighbors. Instead, mesenchymal markers like N-cadherin and fibronectin, are increased to facilitate invasion and migration [25, 26]. Recent studies reported that EMT heralded aggressive tumor features and poor prognosis in EC [27, 28]. In this paper, we discovered that there was a morphological transformation in RANKL-treated HEC-1ARANK and IshikawaRANK cells. A down-regulation of E-cadherin and up-regulation of N-cadherin, Vimentin, Snail and Twist were also detected. Thus, we concluded that RANK/RANKL could induce EMT in EC cells.
In reality, cancer metastasis is not due to a single genetic variation, but requires the participation of multiple factors, both within cells and in the tumor microenvironment. Consequently, the important role of chemokines is emerging. Chemokines play an essential part in various biologic events, such as regulating the migration of white blood cells, tissue architecture, Th1/Th2 development and leukocyte homeostasis [29]. Recently, there were several publications supporting that chemokines were critical in tumor metastasis [30–36] and inhibition of them reduced the advancement of metastasis in vivo [37]. Acting in both a paracrine fashion to regulate the activity of neighboring cells and an autocrine manner, chemokines perform various functions. Abnormal secretion of chemokines by tumor-associated macrophages has been involved in tumor cell survival, angiogenesis, migration/invasion and regulation of the immune system interacting with the tumor [38, 39]. Furthermore, chemokines released by the tumor cells themselves provide cancer cells with a ready-made route towards the adjacent tissues or blood stream. Several chemokines including CCL20 have been reported to induce EMT in various tumors. Biswas et al. [18] showed that CXCL13-CXCR5 initiated EMT procession of breast cancer cells. Li et al. [19] indicated that SDF-1/CXCR4 promoted metastasis and EMT in vitro via non-canonical hedgehog pathway in pancreatic cancer. Matsushita et al. [20] found that CXCL16 contributed to liver metastasis of colorectal carcinoma by inducing EMT. Here, we discovered that the expression and secretion of CCL20 was prominently increased in RANKL-treated RANK over-expressed EC cells in vitro and in vivo. In addition, CCL20 accelerated invasion and induced EMT of RANK over-expressed EC cells, and neutralizing antibody against CCL20 could suppress this effect. Therefore, our study presented another model of chemokine-mediated EMT process, in which CCL20 may play a crucial role in EC metastasis and progression (Figure 7). Interestingly, it was true that similar chemokines networks were activated in different tumors. This indicated that underlying damage of tumor immune-surveillance led to subsequent extensive metastasis. Our data supported the hypothesis that CCL20 was elevated in RANKL-treated RANK over-expressed EC cells, and then contributed to immune suppression-mediated cancer cells metastasis via EMT.
Figure 7. A proposed model for the role of CCL20 in RANK/RANKL-induced EMT in EC cells.

RANK/RANKL up-regulates the expression and secretion of CCL20 in EC cells. CCL20 could bind to its solely receptor CCR6. Then, this interplay contributes to the migration/invasion and promotes EC progression through EMT. Furthermore, neutralizing antibody against CCL20 could refrain from this effect.
In conclusion, we summarized that RANK/RANKL up-regulated the expression and secretion of CCL20 in EC cells, which accelerated migration/invasion and promoted cancer progression through EMT. Consequently, CCL20 may represent a promising molecular marker for preventing EC metastasis and progression.
MATERIALS AND METHODS
Clinical samples
45 samples of EC without hormone therapy, radiotherapy, or chemotherapy before surgery were obtained from the International Peace Maternity and Child Health Hospital during 2013 and 2014. The tumor stages and histological grades were established in line with the criteria of Federation International of Gynecology and Obstetrics (FIGO) 2009 staging system [40].
Reagents and antibodies
Recombinant human RANK ligand and recombinant human MIP-3α/CCL20 were purchased from PeproTech (Rocky Hill, NJ, USA). Rabbit monoclonal antibodies (mAb) against E-cadherin and Vimentin were obtained from Cell Signaling Technology (Danvers, MA, USA). Rabbit polyclonal antibody against N-cadherin, Snail and Twist was from Abcam. Mouse mAb against FLAG was purchased from Cell Signaling Technology. Mouse mAb against RANK and goat polyclonal antibody against RANKL were from SANTA CRUZ (Delaware Avenue, CA, USA). Goat polyclonal antibody CCL20, Human XL Oncology Array Kit and Quantikine ELISA Kit were all purchased from R & D Systems (USA).
Immunohistochemistry (IHC) and assessments
The paraffin-embedded EC tissues were sliced into 4 μm section, dewaxed with xylene and rehydrated with graded alcohol. Ethylene diamine tetraacetic acid was used for antigen retrieval, and 3% hydrogen peroxide was used for blocking endogenous peroxidase activity. Then, sections were incubated with primary antibodies against RANK (1:200), RANKL (1:200), E-cadherin (1:400), N-cadherin (1:250) and Vimentin (1:100) at 4°C overnight and with secondary antibodies for 30 minutes at room temperature. Two pathologists blinded to clinical pathological parameters assessed immune-reactivity scores (IRSs) based on Remmele and Stegner [41]. Staining intensity was scored as negative (0), weak (1), moderate (2), or strong (3). The percentage of positive cells was scored as 0 (0–5%), 1 (6–25%), 2 (26–50%), 3 (51–75%), and 4 (76–100%). Then, multiplying these two scores calculates IRS.
Cell culture
Human EC cell lines (HEC-1A and Ishikawa cell) were obtained from the American Type Culture Collection (ATCC, Manassas, vA, USA). HEC-1A and Ishikawa cells were cultured in Dulbecco's modified Eagle medium (DMEM)/F12 (Gibco, Auckland, New Zealand) supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA), 100 μg/ml penicillin and 100 U/ml streptomycin (Gibco). Cells were cultured in a humidified atmosphere of 5% CO2 at 37°C.
Transient transfection
The over-expression plasmid targeting RANK, pIRES2-3FLAG-EGFP-RANK, and the empty plasmid, pIRES2-3FLAG-EGFP-CON236, were purchased from GeneChem Biotech (Shanghai, China). Transient transfection was carried out with 70% confluent EC cells and Lipofectamine 2000 reagents (Invitrogen). Then, both HEC-1A/IshikawaControl cells and HEC-1A/IshikawaRANK cells were treated by 1 ug/ml RANKL (PeproTech, USA) for 48 h.
RNA isolation and quantitative real-time PCR (qRT-PCR) assays
Total RNA was isolated using Trizol reagent (Invitrogen, Shanghai, China) and the quality was assessed using spectrophotometer (Pharmacia Biotech RNA/DNA calculator). The first-strand cDNA was inversely transcribed from total RNA (1 μg) using a reverse transcription kit (TaKaRa, Dalian, China). Then, qRT-PCR was performed to analyze cDNA using SYBR Premix Ex Taq (Takara Biomedical) on an ABI Prism 700 thermal cycler (Applied Biosystems, Foster City, CA, USA). The qRT-PCR conditions for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), E-cadherin, N-cadherin, Vimentin, Snail and Twist were 95°C 2 min, followed by 40 cycles of 95°C 10s, 60°C 30s. Primer sequences were as follows: GAPDH, forward primer 5′-ACAACTTTGGTATCGTGGAAGG-3′ and reverse primer 5′-GCCATCACGCCACAGTTTC-3′; E-cadherin, forward primer 5′-CGAGAGCTACACGTTCACGG-3′ and reverse primer 5′-GGGTGTCGAGGGAAAAATAGG-3′; N-cadherin, forward primer 5′-TGCGGTACAGTGTA ACTGGG-3′ and reverse primer 5′-GAAACCGGGCTA TCTGCTCG-3′; Vimentin, forward primer 5′-TGCCGTT GAAGCTGCTAACTA-3′ and reverse primer 5′-CCAGAG GGAGTGAATCCAGATTA-3′, Snail, forward primer 5′-ACTGCAACAAGGAATACCTCAG-3′ and reverse primer 5′-GCACTGGTACTTCTTGACATCTG-3′; Twist, forward primer 5′-ATTCAAAGAAACAGGGCGTGG-3′ and reverse primer 5′-CCTTTCAGTGGCTGATTGGC-3′. Values of vertical axis represent 2(−ΔCt), and ΔCt is the discrepancy between target genes Ct and GAPDH Ct.
Western blotting analysis
Treated cells were lysed in RIPA buffer containing protease inhibitor phenylmethanesulfonyl fluoride (Beyotime, Nanjing, China). The BCA Protein Assay kit (Beyotime, Nanjing, China) was used to determine protein concentrations. Equal amounts of extracts (50 μg of protein) were fractionated on each lane of a polyacrylamide-sodium dodecyl sulfate (SDS) gels and transferred to polyvinylidenefluoride (PVDF) membrane (Millipore, Billerica, MA). Then membranes were blocked with 5% skimmed milk for 2 h and incubated with antibodies against E-cadherin (1:1000), N-cadherin (1:1000), Vimentin (1:1000), Snail (1:1000), Twist (1:1000), GAPDH (1:1000) and FLAG (1:1000) at 4°C overnight. Subsequently, the membranes were incubated with secondary antibodies for 1 h at room temperature. The target proteins were visualized by adding ECL luminous agent using the image-forming system of Amersham Imager 600. GAPDH antibody was used as the internal standard.
Wound healing assays
The transfected cells were seeded in a 60-mm plate. A 10 μl pipette tip was applied to wound the cell monolayer. After 48 h treatment of RANKL (1 μg/ml), cells were washed with PBS. The images were obtained under an inverted microscope.
Transwell invasion assays
The upper transwell chambers (8-μm pore) were coated with 100 μl of Matrigel at a dilution of 1:6 (BD Biosciences, San Jose CA, USA). A total of 1 × 105-transfected cells were seeded into the top chamber of a 24-well polycarbonate transwell filter (Corning Incorporated, Glendale, AZ, USA). Then, cells were treated with RANKL (1 μg/ml) for 48 h. The numbers of crystal violet-stained cells in five random fields were counted using an inverted microscope.
Protein chip analysis
Lysates from treated human EC cells were extracted with RIPA buffer containing protease inhibitor phenylmethanesulfonyl fluoride (Beyotime, Nanjing, China). Then, 200 μg of cell lysate was run on each array of the Human XL Oncology Array Kit from RandD Systems (Cat. ARY026, USA). The follow-up steps were carried out in accordance with manufacturer's instructions. Array images were collected and analyzed using the Amersham Imager 600 Imaging System.
Enzyme-linked immunosorbent assay (ELISA)
The ELISA kit is an in vitro enzyme-linked immunosorbent assay for quantitative measurement. The supernatants of treated cells were stored at −80°C until assayed for CCL20 with Quantikine ELISA Kit from R & D Systems (Cat. DM3A00, USA). Standards and treated cells samples were both diluted in Calibrator Diluent RD6–21 obtained from the ELISA Kit. The optical density of each well was determined within 30 minutes via a microplate reader setting to 450 nm.
Immunofluorescence assay
HEC-1A cells were grown on coverslips. Transient transfection was performed when cells grew to 70% confluent. Both HEC-1AControl cells and HEC-1ARANK cells were treated by RANKL (1 ug/ml) for 48 h. We fixed the cells with 4% paraformaldehyde for 15 min and blocked them with 0.5% (V/V) Triton X-100 in PBS for 30 min at room temperature. Then cells were incubated with primary antibodies against CCL20 (1:100), E-cadherin (1:200), N-cadherin (1:100) and Vimentin (1:100) at 4°C overnight. Finally, cells were incubated with secondary antibody coupled with fluorophores. Images were obtained by fluorescence microscopy.
In vivo experiments
We gained Ishikawa-Luc/Ishikawa-Luc-Rank light-emitting EC cells to build orthotopic xenograft nude model. The establishment process of animal model was performed in terms of our previously study [12]. After four weeks’ injection of RANKL, the nude mice were sacrificed. Then, tumor tissues were removed from animals, and grinded to obtain mRNA. qRT-PCR was used to analyze the mRNA level of CCL20. Meanwhile, the serum was collected to measure the concentration of CCL20 with ELISA kit.
Statistical analysis
We performed all statistical analyses with SPSS software, version 17.0 (SPSS, Inc., Chicago, IL, USA). The data was analyzed using an unpaired Student's t-test or one-way ANOVA. The correlation detection was performed by Spearman's correlation coefficient test. Each experiment was carried out in triplicate. P-values of < 0.05 were considered statistically significant.
Acknowledgments
We would like to thank the Center Laboratory and Department of Gynecology of International Peace Maternity and Child Health Hospital affiliated to Shanghai Jiao Tong University School of Medicine for excellent technical support.
Footnotes
GRANT SUPPORT
This study was supported by the National Natural Science Foundation of China (No. 81172477, No. 81572547 and No. 81402135), the Project of the Science and Technology Commission of Shanghai Municipality (No. 11ZR1440800 and No. 13JC1401303), and the Project of Outstanding Subject Leaders of the Shanghai Health System (No. XBR2013097).
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Siegel R, Miller K, Jemal A. Cancer statistics. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Di Cristofano A, Ellenson LH. Endometrial carcinoma. Annu Rev Pathol. 2007;2:57–85. doi: 10.1146/annurev.pathol.2.010506.091905. [DOI] [PubMed] [Google Scholar]
- 3.Hill EK, Dizon DS. Medical therapy of endometrial cancer: current status and promising novel treatments. Drugs. 2012;72:705–713. doi: 10.2165/11631840-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Oza AM, Elit L, Tsao MS, Kamel-Reid S, Biagi J, Provencher DM, Gotlieb WH, Hoskins PJ, Ghatage P, Tonkin KS, Mackay HJ, Mazurka J, Sederias J, et al. Phase II study of temsirolimus in women with recurrent or metastatic endometrial cancer: a trial of the NCIC Clinical Trials Group. J Clin Oncol. 2011;29:3278–3285. doi: 10.1200/JCO.2010.34.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leonardi GC, Candido S, Cervello M, Nicolosi D, Raiti F, Travali S, Spandidos DA, Libra M. The tumor microenvironment in hepatocellular carcinoma (review) Int J Oncol. 2012;40:1733–1747. doi: 10.3892/ijo.2012.1408. [DOI] [PubMed] [Google Scholar]
- 6.O'Hayre M, Salanga CL, Handel TM, Allen SJ. Chemokines and cancer: Migration, intracellular signalling and intercellular communication in the microenvironment. Biochem J. 2008;409:635–649. doi: 10.1042/BJ20071493. [DOI] [PubMed] [Google Scholar]
- 7.Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, Karin M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature. 2011;470:548–553. doi: 10.1038/nature09707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santini D, Perrone G, Roato I, Godio L, Pantano F, Grasso D, Russo A, Vincenzi B, Fratto ME, Sabbatini R, Della Pepa C, Porta C, Del Conte A, et al. Expression pattern of receptor activator of NFκB (RANK) in a series of primary solid tumors and related bone metastases. J Cell Physiol. 2011;226:780–784. doi: 10.1002/jcp.22402. [DOI] [PubMed] [Google Scholar]
- 9.Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, Sarosi I, Morony S, Rubin E, Sarao R, Hojilla CV, Komnenovic V, Kong YY, Schreiber M, et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature. 2006;440:692–696. doi: 10.1038/nature04524. [DOI] [PubMed] [Google Scholar]
- 10.Palafox M, Ferrer I, Pellegrini P, Vila S, Hernandez-Ortega S, Urruticoechea A, Climent F, Soler MT, Muñoz P, Viñals F, Tometsko M, Branstetter D, Dougall WC, et al. RANK induces epithelial-mesenchymal transition and stemness in human mammary epithelial cells and promotes tumorigenesis and metastasis. Cancer Res. 2012;72:2879–2888. doi: 10.1158/0008-5472.CAN-12-0044. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, Sun X, Zhang H, Wang Y, Li Y. MPA influences tumor cell proliferation, migration, and invasion induced by RANKL through PRB involving the MAPK pathway in endometrial cancer. Oncol Rep. 2015;33:799–809. doi: 10.3892/or.2014.3651. [DOI] [PubMed] [Google Scholar]
- 12.Wang J, Liu Y, Wang L, Sun X, Wang Y. Clinical prognostic significance and pro-metastatic activity of RANK/RANKL via the AKT pathway in endometrial cancer. Oncotarget. 2015 doi: 10.18632/oncotarget.6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giuliani N, Lisignoli G, Colla S, Lazzaretti M, Storti P, Mancini C, Bonomini S, Manferdini C, Codeluppi K, Facchini A, Rizzoli V, et al. CC-chemokine ligand 20/macrophage inflammatory protein-3a and CC-chemokine receptor 6 are overexpressed in myeloma microenvironment related to osteolytic bone lesions. Cancer Res. 2008;68:6840–6850. doi: 10.1158/0008-5472.CAN-08-0402. [DOI] [PubMed] [Google Scholar]
- 14.Schutyser E, Struyf S, Van Damme J. The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev. 2003;14:409–426. doi: 10.1016/s1359-6101(03)00049-2. [DOI] [PubMed] [Google Scholar]
- 15.Yoshimura T, Matsushima K, Tanaka S, Robinson EA, Appella E, Oppenheim JJ, Leonard EJ. Purification of a human monocyte-derived neutrophil chemotactic factor that has peptide sequence similarity to other host defense cytokines. Proc Natl Acad Sci USA. 1987;84:9233–9237. doi: 10.1073/pnas.84.24.9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marsigliante S, Vetrugno C, Muscella A. CCL20 induces migration and proliferation on breast epithelial cells. J Cell Physiol. 2013;228:1873–1883. doi: 10.1002/jcp.24349. [DOI] [PubMed] [Google Scholar]
- 17.Vicinus B, Rubie C, Stegmaier N, Frick VO, Kölsch K, Kauffels A, Ghadjar P, Wagner M, Glanemann M. miR-21 and its target gene CCL20 are both highly overexpressed in the microenvironment of colorectal tumors: significance of their regulation. Oncol Rep. 2013;30:1285–1292. doi: 10.3892/or.2013.2580. [DOI] [PubMed] [Google Scholar]
- 18.Biswas S, Sengupta S, Roy Chowdhury S, Jana S, Mandal G, Mandal PK, Saha N, Malhotra V, Gupta A, Kuprash DV, Bhattacharyya A. CXCL13-CXCR5 co-expression regulates epithelial to mesenchymal transition of breast cancer cells during lymph node metastasis. Breast Cancer Res Treat. 2014;143:265–276. doi: 10.1007/s10549-013-2811-8. [DOI] [PubMed] [Google Scholar]
- 19.Li X, Ma Q, Xu Q, Liu H, Lei J, Duan W, Bhat K, Wang F, Wu E, Wang Z. SDF-1/CXCR4 signaling induces pancreatic cancer cell invasion and epithelial-mesenchymal transition in vitro through non-canonical activation of Hedgehog pathway. Cancer Lett. 2012;322:169–176. doi: 10.1016/j.canlet.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsushita K, Toiyama Y, Tanaka K, Saigusa S, Hiro J, Uchida K, Inoue Y, Kusunoki M. Soluble CXCL16 in preoperative serum is a novel prognostic marker and predicts recurrence of liver metastases in colorectal cancer patients. Ann Surg Oncol. 2012;19:S518–S527. doi: 10.1245/s10434-011-1993-8. [DOI] [PubMed] [Google Scholar]
- 21.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 22.Liao Y, He X, Qiu H, Che Q, Wang F, Lu W, Chen Z, Qiu M, Wang J, Wang H, Wan X. Suppression of the epithelial-mesenchymal transition by SHARP1 is linked to the NOTCH1 signaling pathway in metastasis of endometrial cancer. BMC Cancer. 2014:487. doi: 10.1186/1471-2407-14-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beleut M, Rajaram RD, Caikovski M, Ayyanan A, Germano D, Choi Y, Schneider P, Brisken C. Two distinct mechanisms underlie progesterone-induced proliferation in the mammary gland. Proc Natl Acad Sci USA. 2010;107:2989–2994. doi: 10.1073/pnas.0915148107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimamura M, Nakagami H, Osako MK, Kurinami H, Koriyama H, Zhengda P, Tomioka H, Tenma A, Wakayama K, Morishita R. OPG/RANKL/RANK axis is a critical inflammatory signaling system in ischemic brain in mice. Proc Natl Acad Sci USA. 2014;111:8191–8196. doi: 10.1073/pnas.1400544111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;342:1234850. doi: 10.1126/science.1234850. [DOI] [PubMed] [Google Scholar]
- 26.Tan EJ, Kahata K, Idås O, Thuault S, Heldin CH, Moustakas A. The high mobility group A2 protein epigenetically silences the Cdh1 gene during epithelial-to-mesenchymal transition. Nucleic Acids Res. 2015;43:162–178. doi: 10.1093/nar/gku1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, Wei J, Lu J, Tong Z, Liao B, Yu B, Zheng F, Huang X, Chen Z, Fang Y, Li B, Chen W, Xie D, et al. Overexpression of Rab25 contributes to metastasis of bladder cancer through induction of epithelial-mesenchymal transition and activation of Akt/GSK-3beta/Snail signaling. Carcinogenesis. 2013;34:2401–2408. doi: 10.1093/carcin/bgt187. [DOI] [PubMed] [Google Scholar]
- 28.Tanaka Y, Terai Y, Kawaguchi H, Fujiwara S, Yoo S, Tsunetoh S, Takai M, Kanemura M, Tanabe A, Ohmichi M. Prognostic impact of EMT (epithelial-mesenchymal-transition)-related protein expression in endometrial cancer. Cancer Biol Ther. 2013;14:13–19. doi: 10.4161/cbt.22625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pirus Ghadjar, Claudia Rubie, Daniel M. Aebersold, Ulrich Keilholz. The chemokine CCL20 and its receptor CCR6 in human malignancy with focus on colorectal cancer. Int J Cancer. 2009;125:741–745. doi: 10.1002/ijc.24468. [DOI] [PubMed] [Google Scholar]
- 30.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verástegui E, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 31.Ding Y, Shimada Y, Maeda M, Kawabe A, Kaganoi J, Komoto I, Hashimoto Y, Miyake M, Hashida H, Imamura M. Association of CC chemokine receptor 7 with lymph node metastasis of esophageal squamous cell carcinoma. Clin Cancer Res. 2003;9:3406–3412. [PubMed] [Google Scholar]
- 32.Wiley HE, Gonzales EB, Maki W, Wu MT, Hwang ST. Expression of CC chemokine receptor-7 and regional lymph node metastasis of B16 murine melanoma. J Natl Cancer Inst. 2001;93:1638–1643. doi: 10.1093/jnci/93.21.1638. [DOI] [PubMed] [Google Scholar]
- 33.Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell-derived Factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62:1832–187. [PubMed] [Google Scholar]
- 34.Murakami T, Maki W, Cardones AR, Fang H, TunKyi A, Nestle FO, Hwang ST. Expression of CXC chemokine receptor-4 enhances the pulmonary metastatic potential of murine B16 melanoma cells. Cancer Res. 2002;62:7328–7334. [PubMed] [Google Scholar]
- 35.Letsch A, Keilholz U, Schadendorf D, Assfalg G, Asemissen AM, Thiel E, Scheibenbogen C. Functional CCR9 expression is associated with small intestinal metastasis. J Invest Dermatol. 2004;122:685–690. doi: 10.1111/j.0022-202X.2004.22315.x. [DOI] [PubMed] [Google Scholar]
- 36.Mashino K, Sadanaga N, Yamaguchi H, Tanaka F, Ohta M, Shibuta K, Inoue H, Mori M. Expression of chemokine receptor CCR7 is associated with lymph node metastasis of gastric carcinoma. Cancer Res. 2002;62:2937–2941. [PubMed] [Google Scholar]
- 37.Zeelenberg IS, Ruuls-Van Stalle L, Roos E. The chemokine receptor CXCR4 is required for outgrowth of colon carcinoma micrometastases. Cancer Res. 2003;63:3833–3839. [PubMed] [Google Scholar]
- 38.Boon T, Van den Eynde B. Tumour immunology. Curr Opin Immunol. 2003;15:129–130. doi: 10.1016/s0952-7915(03)00010-4. [DOI] [PubMed] [Google Scholar]
- 39.Wilson J, Balkwill F. The role of cytokines in the epithelial cancer microenvironment. Semin Cancer Biol. 2002;12:113–120. doi: 10.1006/scbi.2001.0419. [DOI] [PubMed] [Google Scholar]
- 40.Creasman W. Revised FIGO staging for carcinoma of the endometrium. Int J Gynaecol Obstet. 2009;105:109. doi: 10.1016/j.ijgo.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 41.Remmele W, Stegner HE. Recommendation for uniform definition of an immunoreactive score (IRS) for immunohistochemical estrogen receptor detection (ER-ICA) in breast cancer tissue. Pathologe. 1987;8:138–140. [PubMed] [Google Scholar]