

Abstract
Circular RNAs (circRNAs) arise during post‐transcriptional processes, in which a single‐stranded RNA molecule forms a circle through covalent binding. Previously, circRNA products were often regarded to be splicing intermediates, by‐products, or products of aberrant splicing. But recently, rapid advances in high‐throughput RNA sequencing (RNA‐seq) for global investigation of nonco‐linear (NCL) RNAs, which comprised sequence segments that are topologically inconsistent with the reference genome, leads to renewed interest in this type of NCL RNA (i.e., circRNA), especially exonic circRNAs (ecircRNAs). Although the biogenesis and function of ecircRNAs are mostly unknown, some ecircRNAs are abundant, highly expressed, or evolutionarily conserved. Some ecircRNAs have been shown to affect microRNA regulation, and probably play roles in regulating parental gene transcription, cell proliferation, and RNA‐binding proteins, indicating their functional potential for development as diagnostic tools. To date, thousands of ecircRNAs have been identified in multiple tissues/cell types from diverse species, through analyses of RNA‐seq data. However, the detection of ecircRNA candidates involves several major challenges, including discrimination between ecircRNAs and other types of NCL RNAs (e.g., trans‐spliced RNAs and genetic rearrangements); removal of sequencing errors, alignment errors, and in vitro artifacts; and the reconciliation of heterogeneous results arising from the use of different bioinformatics methods or sequencing data generated under different treatments. Such challenges may severely hamper the understanding of ecircRNAs. Herein, we review the biogenesis, identification, properties, and function of ecircRNAs, and discuss some unanswered questions regarding ecircRNAs. We also evaluate the accuracy (in terms of sensitivity and precision) of some well‐known circRNA‐detecting methods. WIREs RNA 2015, 6:563–579. doi: 10.1002/wrna.1294
For further resources related to this article, please visit the WIREs website.
INTRODUCTION
Following the discovery of the first circular RNA (circRNA) molecules,1 several types of RNA circles have been detected in various organisms. Unlike plant viroids and the hepatitis delta virus,1, 2 which have circular single‐stranded RNA (ssRNA) genomes, several transcribed RNA molecules, including tRNAs, rRNAs, and mRNAs, can also be circularized via ribozymal activity (Group I3 and Group II4 introns), archaeal splicing,5, 6, or spliceosomal machinery.7, 8, 9 In the past, observed circRNAs (especially in animals) were usually thought to be by‐products of pre‐mRNA processing, and were therefore interpreted to be results of missplicing.7, 8, 9 Recently, advances in high‐throughput RNA sequencing (RNA‐seq) have created unprecedented opportunities to globally investigate transcriptomes, revealing the existence of a large amount of previously unidentified circRNAs, especially of exonic circRNAs (ecircRNAs).10 Genome‐wide analysis of RNA‐seq data revealed that ecircRNAs are abundant in mammalian transcriptomes, and some of them are evolutionarily conserved in terms of sequence and expression,10, 11, 12 suggesting that they possess cellular function. The most prominent examples of functional ecircRNAs are human CDR1as/ciRS‐7 and circRNA of mouse Sry, which were experimentally validated to function as miRNA sponges, and are thereby involved in gene expression regulation.13, 14 Moreover, some ecircRNAs were originated from genes related to splicing factors,15 DNA methyltransferases,15 and important diseases such as dystrophy16 and cancers.7, 9 Although the relationship between ecircRNAs and their linear counterparts is mostly unknown, an understanding of such an association with disease‐forming processes will help considerably in developing efficient diagnostic methods or even therapies. Of particular note, ecircRNAs were shown to be more stable than their linear counterparts in plasma17 and saliva,18 suggesting their potential as diagnostic biomarkers.
Generally, an ecircRNA event can be detected by aligning expressed sequences (e.g., RNA‐seq reads) against the reference genome. After that, nonco‐linear (NCL) junctions are determined on the basis of the presence of two connected exons in which the exon order is topologically inconsistent with the reference genome. Although varied bioinformatics methods based on RNA‐seq data have been developed and used to identify thousands of ecircRNA candidates in diverse species (Table 1), there remain several challenges. For example, an observed NCL junction site may also be formed by other types of NCL event (e.g., trans‐splicing events and genetic rearrangements) or various types of false positives (e.g., sequencing errors, alignment errors, and in vitro artifacts). In addition, the identification results may drastically differ between different circRNA‐detecting methods and data derived from different RNA treatments, resulting in biased interpretations for ecircRNA analysis. To date, there is no competent method that can simultaneously account for the abovementioned issues during the identification of NCL events (including circRNAs). Moreover, there remains a need to evaluate the sensitivity and precision of currently available methods for circRNA identification.
Table 1.
Recently Published Studies for Detecting circRNAs on the Basis of RNA Sequencing Data
| Study | Exonic/intronic circRNA | Treatment of RNA Library | Number of Detected circRNA Events | Method for circRNA Identification |
|---|---|---|---|---|
| Pseudo reference based | ||||
| Salzman et al. (2012)10 | Exonic | rRNA− | >880 human genes and >1000 mouse genes contain circRNAs | In‐house pipeline |
| Salzman et al. (2013)19 | Exonic | rRNA−&polyA− | 46,866 events in 8466 human genes | In‐house pipeline |
| Zhang et al. (2013)20 | Intronic | rRNA−&polyA−; rRNA−&RNase R+ | 103 events (human) | The in‐house pipeline |
| Zhang et al. (2014)15 | Exonic | rRNA−&polyA−; rRNA−&RNase R+ | 1662 events (human) | CIRCexplorer |
| Fragment based | ||||
| Jeck et al. (2013)11 | Exonic | rRNA−&RNase R+ | 7771 events (human); 646 events (mouse) | MapSplice |
| Memczak et al. (2013)13 | Exonic | rRNA− | 1903 events (human); 1111 events (nematode) | find_circ |
| Ashwal et al. (2014)21 | Exonic | rRNA−&RNase R+ | 3117 events (fruit fly) | In‐house pipeline |
| Hoffmann et al. (2014)22 | Exonic | rRNA‐ | 1712 events (human) | segemehl |
| Guo et al. (2014)23 | Exonic | rRNA− | 7112 events (human); 635 events (mouse) | In‐house pipeline |
| Westholm et al. (2014)24 | Exonic | rRNA− | 2513 (fruit fly) | In‐house pipeline |
| Bachmayr‐Heyda et al. (2015)25 | Exonic | rRNA− | 1812 (human) | find_circ |
| Gao et al. (2015)26 | Both | rRNA− | 3000–10,000 (human) | CIRI |
In this review, we focus on the discussion of the biogenesis, identification, properties, and function of ecircRNAs. We also describe our generation of artificial paired‐end RNA‐seq reads from a mix of simulated intragenic NCL transcripts and well‐annotated co‐linear transcripts to evaluate the sensitivity and precision of five well‐known circRNA‐detecting tools: TopHat‐Fusion,27 MapSplice,28 segemehl,22 find_circ,13 and CIRI.26 Some unanswered questions regarding ecircRNA biogenesis, identification, and function are also discussed.
BIOGENESIS OF EXONIC CIRCRNAS
Models of ecircRNA Formation
Eukaryotic exonic regions of pre‐mRNAs are typically disrupted by intron(s), and spliceosomes are responsible for the removal of introns from pre‐mRNAs. A generally accepted model depicts a two‐step spliceosome action29: (1) the branch point (BP, usually an adenosine) attacks the 5′ splice site (5′SS) via its 2′‐hydroxyl group, forming a 2′‐5′ phosphodiester bond and a free 3′‐hydroxyl end on the 5′ exon; and (2) the newly generated 3′‐hydroxyl attacks the 3′ splice site (3′SS), and a 3′‐5′ phosphodiester bond is formed to ligate the two exons; meanwhile, the intron lariat with a 2′‐5′ linkage is excised. Strict control of the choice of splice site pairs is important, as it ensures the accuracy of mRNA products and consequent processes. However, splicing machinery also shows certain degrees of flexibility (sometimes described as aberrant) in splice site choice. Variance in splice site pairing (or alternative splicing) often creates varied transcript isoforms. As many ecircRNAs contain exons from coding genes and are connected by canonical splice sites, it is generally believed that ecircRNAs are produced through spliceosomal splicing mechanisms. Some evidence has been proposed to support this scenario. One line of direct evidence supporting the essential role of the spliceosomal machinery in ecircRNA biogenesis comes from the use of splice inhibitor isoginkgetin.30 Following isoginkgetin treatment, both linear and circular isoforms were significantly reduced in nascent RNA pools. Mutagenesis analyses show that both 5′ and 3′ splice signals from the circular junction are essential for exon circularization.21, 30 These results supported the hypothesis that spliceosome‐mediated pre‐mRNA splicing may involve backsplicing (or reverse splicing), which connects a downstream splice donor site (5′ splice site) to an upstream acceptor splice site (3′ splice site) and forms an ecircRNA. Comparison of backsplicing and canonical (or linear) splicing further indicated that although canonical splicing factors can control both processes, the splicing regulatory rules for circRNA biogenesis are different from those for linear splicing.31 In addition, it was proposed that linear splicing and circularization may compete for limited splicing factors—introducing flanking exons with strong 5′ and 3′ splice sites dramatically decreases circularization efficiency.21 In addition, the minigene system, which is frequently used to investigate the splicing mechanism, is also used to investigate ecircRNA biogenesis. A recent study showed that, in addition to canonical splice signals, important signal sequences in the spliceosomal machinery (such as poly‐pyrimidine tracts) also influence circularization; however, the involvement of the branch point is less conclusive.30 Other studies demonstrated that changes in encircled exonic sequences can abolish circRNA formation,32 but in some cases it does not affect circularization.30 Several models that were proposed to explain the possible formation of ecircRNAs are discussed below (see also Figure 1).
Figure 1.
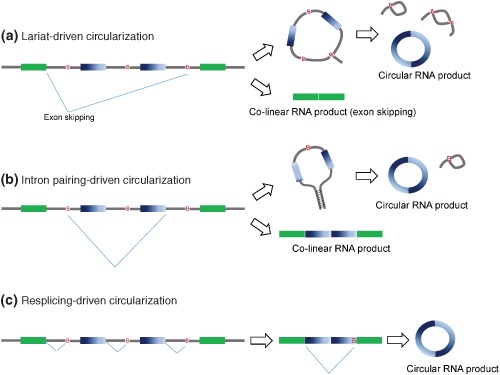
Possible models of ecircRNA biogenesis. (a) Lariat‐driven circularization, (b) Intron‐pairing‐driven circularization, and (c) Resplicing‐driven circularization. B, branch point.
Lariat‐driven Circularization
In an exon skipping (cassette‐on) event, the spliced intron lariat also contains the skipped exon(s) (Figure 1(a)). If further splicing occurs within the lariat before the unraveling of the lariat by debranching enzymes, a stable RNA circle enclosing the skipped exons can be generated.11 Meanwhile, a linear transcript excluding the skipped exon(s) is also produced. Exon skipping was suggested to be the cause of circRNA formation in some early cases since the linear counterparts of such skipping events were detected.33, 34 Genome‐wide analysis of RNA‐seq data from a human fibroblast cell line revealed that, for 45% of 7771 predicted circRNAs, the corresponding linear isoforms also exhibited exon skipping events,11 suggesting that RNA circularization was correlated with exon skipping. However, such a trend was not observed in a separate study on different biosamples.10
Intron Pairing‐driven Circularization
In this model, the formation of ecircRNAs is independent of exon skipping. It differs from the lariat‐driven circularization model by the choice of splice site pairs and the lack of knowledge about the corresponding linear product(s). It was suggested that intronic motifs might border the circularized exons(s) and thereby join the circularized exons(s)11 (Figure 1(b)). Distinguishing between lariat‐ and intron pairing‐driven ecircRNAs is difficult, because the corresponding products/intermediates are likely to be short‐lived, as a result of degradation through nonsense‐mediated decay or by debranching enzymes.
Resplicing‐driven Circularization
In the presence of proper cis‐ and trans‐splicing elements, resplicing may take place on spliced mRNAs (Figure 1(c)). Exonic circRNAs may be generated by a two‐step splicing pathway, in which the initial splicing removes canonical splice sites and thereby resplicing makes use of cryptic splice sites on the spliced mRNAs for circularization in an exon skipping fashion.35 Resplicing is likely to be merely an aberrant splicing event of pre‐mRNAs, which is often detected in cancers. For example, two of the most notable resplicing events were detected on human TSG101 and FHIT mRNAs in cancer cells, which were suggested to arise from cancer‐specific aberrant splicing.35 The occurrence of resplicing (whether it generates circRNAs or not) is not well documented. It is also unclear how frequently resplicing occurs at canonical splice sites.
Criteria for Exon Circularization
The Involvement of Reverse Complement Sequences
It has been shown that base pairing between the reverse complementary sequences (RCSs) in flanking introns can bring the downstream 5′SS into the proximity of the upstream 3′SS (Figure 2(a)), leading to circularization of the mouse Sry gene.8, 36 Several transcriptome‐wide analyses also indicated a significant correlation between the presence of flanking intronic RCSs (especially inverted Alu elements in primates; Figure 2(a)) and exon circularization.11, 15, 37 Extensive mutagenesis of expression plasmids revealed that short (30–40 nt) inverted repeats (e.g., Alu elements) are sufficient for ecircRNA generation.32 However, it was observed that not all intronic repeats could support exon circularization; on the contrary, enhancing the stability of base‐pairing sometimes might impede circRNA formation.32 In addition, if multiple copies of RCSs are present in a single gene, the competition for base pairing among RCSs may affect circularization efficiency, and even result in alternative circularization, bringing more diversity of circular transcripts from a single gene.15 It was demonstrated that circRNAs can be predicted by scoring the presence of RCSs in the bracketing introns.37 Although exon circularization is highly correlated with intronic RCSs, several studies using the minigene system showed that human ecircRNAs are not always bracketed by Alu or RCSs‐containing introns, further indicating that RCSs can enhance,15, 30, 31, 32 but are not essential for,30, 31 ecircRNA production. The bracketing introns of ecircRNAs are also highly enriched for RCSs in animals that are not rich in repeats, such as Caenorhabditis elegans 37 and Drosophila 24, 31; however, it is formally possible that ecircRNA biogenesis may be regulated by different cis elements in different species.
Figure 2.
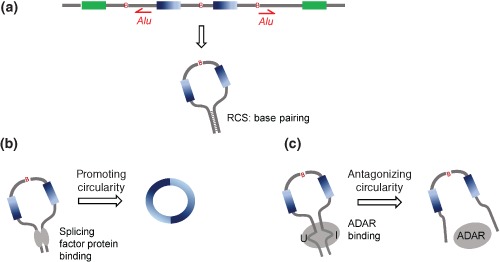
Regulation of exon circularization. (a) The presence of flanking intronic RCSs (e.g., Alu elements) can lead to exon circularization. (b) Some splicing factors (e.g., QKI and MBL) can promote ecircRNA generation. (c) ADAR proteins can antagonize circRNA production. RCS, reverse complementary sequence.
Regulatory Factors for Circularization
Although ecircRNA biogenesis can be viewed as a mode of alternative splicing, it remains unknown whether factors involved in alternative splicing regulation are also involved in ecircRNA biogenesis. A recent study appeared to partially answer this question by demonstrating that a considerable number of ecircRNAs are dynamically regulated by Quaking (QKI), an alternative splicing factor, during the human epithelial‐mesenchymal transition.38 The addition of QKI binding motifs to flanking introns can significantly induce circRNA formation,38 suggesting that QKI is an important regulator of circularization. Another regulator of circRNA biogenesis is muscleblind (MBL/MBNL1), a splicing factor that was found to be circularized in flies and humans.21 This circRNA contains multiple MBL‐binding motifs in its flanking introns, which are specifically bound by MBL. Downregulation of MBL can result in a remarkable decrease in circularization.21 Both QKI and MBL promote ecircRNA generation by bringing the 5′ SS closer to the upstream 3′ SS (Figure 2(b)). On the other hand, the double‐strand RNA‐editing enzyme – adenosine deaminase acting on RNA (ADAR) proteins, which tend to bind and mediate A‐to‐I editing on inverted Alu repeats, were also demonstrated to regulate circRNA biogenesis.37 Disruption of ADAR expression could result in a significant increase in circRNA expression in C. elegans and human,12, 37 suggesting that ADAR proteins might play an antagonistic role in circRNA production (Figure 2(c)).
IDENTIFICATION OF EXONIC CIRCRNAS
Strategies to Identify ecircRNAs
Many RNA‐seq‐based bioinformatics tools have been developed to identify ecircRNA candidates. Table 1 summarizes some recently published studies on the detection of ecircRNAs. Basically, ecircRNAs are detected by comparing the reference genomes with RNA‐seq reads, and then extracting matches comprised of sequence segments topologically inconsistent with the corresponding DNA sequences in the reference genome. According to the dependency on genome annotation (i.e., annotated exon–intron boundaries), these tools can be classified into two categories: pseudo‐reference‐ and fragment‐based strategies (Figure 3 and Table 1). For the pseudo‐reference‐based strategy, genome annotation is required. All possible combinations of pseudo references are constructed; each of them is comprised of two well‐annotated exons in which the exon order is topologically inconsistent with the reference genome (Figure 3(a)). A pseudo reference is regarded as a circRNA candidate if it has at least one read that maps to its NCL junction site (Figure 3(a)). On the other hand, the fragment‐based strategy detects circRNAs without the help of genome annotation. RNA‐seq reads (each paired‐end read is viewed as two ‘single’ reads) are split into two or more segments, and each segment is mapped to the reference genome; segmented reads mapped in an NCL manner are retained (Figure 3(b)). There are two major limitations for pseudo‐reference‐based methods: first, they cannot identify circRNAs with unannotated exon junctions; and second, they are not suitable for detection of circRNAs in the genomes that are incomplete or poorly annotated. On the other hand, the fragment‐based methods can be used to identify NCL junctions at a single nucleotide resolution in the absence of any existing genome annotation. However, as segmented reads are smaller than full‐length reads, such an approach is more likely to yield alignment errors (or ambiguity) than the pseudo‐reference‐based strategy while performing read‐to‐genome alignment. In addition, NCL junctions that do not match annotated exon boundaries tend to be unreliable and are more likely to originate from missplicing.39, 40, 41, 42
Figure 3.
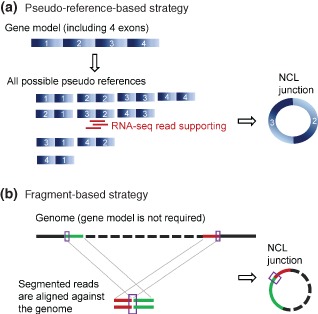
Two RNA‐seq‐based strategies for detecting NCL junctions of ecircRNA candidates: (A) pseudo‐reference‐based and (B) fragment‐based strategies. The former identifies NCL junction sites at annotated exon junctions; whereas the latter does not. NCL, nonco‐linear.
Certain additional criteria are often applied to improve the accuracy of ecircRNA identification. For example, if paired‐end reads are used to identify ecircRNAs, both ends of each matched read should (i) be mapped to the circle predicted by the circular junction and (ii) be in the correct orientation (Figure 4(a)). From the mapping patterns of paired‐end reads, various scenarios for circles, such as circles containing a single exon, partial fragment(s) of an annotated exon, or an intron‐containing fragment, can be depicted (Figure 4(b)). In addition, fragment‐based strategies generally consider only NCL junctions that are flanked by the GT‐AG canonical splice sites for improving accuracy.
Figure 4.
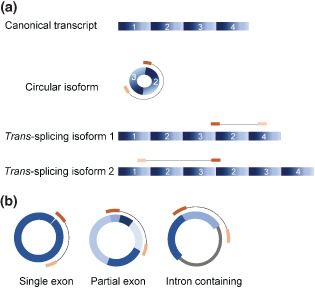
Usage of paired‐end RNA sequencing reads for identifying circRNAs. (a) Removal of noncircular RNA events. Noncircular RNA events (e.g., trans‐splicing events in the figure) can be distinguished if the paired‐end of a read spanning a NCL junction maps outside the predicted circle. (b) Possible scenarios for circles based on the mapping of paired‐end reads (from left to right): circles containing a single exon, partial fragment(s) of an annotated exon, or an intron‐containing fragment.
Moreover, the observations that circRNAs are non‐polyadenylated10, 19, 23 or RNase R‐resistant11, 13, 43 have been exploited by many studies to increase the accuracy of ecircRNA identification (Table 1). Circular‐junction candidates are often detected from an rRNA‐depleted total RNA library, and filtered by comparison to candidates detected in other library sets treated with either RNase R6, 11, 20 or poly(dT).20, 24 However, although such approaches detect abundant circles, the following matters need to be borne in mind. First, not all backsplicing events show enrichment in RNase R‐treated RNA libraries. For example, CDR1as/ciRS‐7 was not enriched after RNase R treatment.11 Second, circRNAs and trans‐splicing events sometimes share the same NCL junctions,41 and such junctions are therefore present in both poly(A)‐ and non‐poly(A)‐selected RNA‐seq data. Third, not all mRNAs lacking poly(A) are circular; for example, certain replication‐dependent histone genes are co‐linear transcripts without poly(A) tails.44 Fourth, the amount of circRNA events are often amplified in the treated RNA‐seq data as compared to the data from untreated samples, raising the concern that a considerable number of detected events may represent rarely but pervasively occurring ‘background’ NCL junctions derived from splicing errors.11 Finally, such treated data are sensitive to endonuclease contamination, and may also be less effective for identifying longer exons.11, 45, 46
Difficulties of ecircRNA Identification
Identification of ecircRNAs often suffers as a result of three major challenges: (1) discrimination between circRNAs and other types of NCL events, such as trans‐splicing and genetic rearrangements; (2) removal of false positives arising from sequencing errors, alignment errors, and in vitro artifacts; and (3) biased identification of ecircRNAs from different bioinformatics methods or the use of sequencing data from different treatments. In fact, these difficulties are common in the detection of all types of NCL RNAs.41
Discrimination between circRNAs and Other Types of NCL Events
Read‐supported NCL junctions provide the major evidence for the identification of circRNAs. However, NCL junctions can also be formed by trans‐splicing or genetic rearrangements. Of these three types of NCL events, both circRNA and trans‐splicing events are generated during post‐transcriptional processes (which may be designated as ‘PtNCL’ events). As somatic recombination events are less likely to (1) occur in multiple biological samples or (2) be conserved across multiple species, PtNCL events can be distinguished from genetic rearrangements by this simple rule.40, 41 More effective approaches utilize integration analysis of genome sequencing data and RNA‐seq data to detect potential rearrangement events.47, 48, 49, 50 Nevertheless, most of these methods were specifically designed to identify NCL events that consist of sequence fragments from two or more different genes. There is no currently available tool that can be directly utilized to distinguish between the PtNCL events and genetic rearrangements. For discriminating between circRNAs and trans‐splicing events, trans‐splicing events can be detected if the paired‐end of a read spanning a NCL junction maps outside the predicted circle (Figure 4(a)).10, 23 On the other hand, it is believed that most circRNAs are non‐polyadenylated10, 19, 23 or RNase R‐resistant,11, 13, 43 while trans‐spliced RNA products are not. Some studies thus used such biochemical properties to filter out potential trans‐splicing events.10, 15, 23, 41 However, some NCL events can be observed in both poly(A)‐depleted (or RNase R‐treated) and poly(A)‐selected libraries. There are two scenarios for this observation. First, RNase R or poly(A)‐depleted treatments may not completely deplete linear RNAs. Second, circRNA and trans‐splicing events may share the same NCL junctions.41 Currently, there is no systematic approach to effectively distinguish between these three types of NCL events (circRNA, trans‐splicing, and genetic rearrangement events).
Removal of False Positives
As stated above, circRNAs are one class of NCL RNAs. Detecting all types of NCL event often suffers from false positives arising from sequencing errors, alignment errors, and in vitro artifacts. These false positives can severely affect the accuracy of detecting NCL RNAs. In general, false positives caused by sequencing errors can be reduced by increasing the number of RNA‐seq reads that support the NCL junctions, or by eliminating skew mapping between reads and the corresponding NCL junctions. However, as most circRNAs are expressed at a relatively lower level compared with co‐linear mRNAs,19, 23, 46 such approaches may sacrifice a considerable number of true positives unless the sequencing depth is very deep. Furthermore, as paralogous genes or repetitive sequences are prevalent in genomes, ambiguous alignments during short‐read mapping are often misinterpreted as NCL events. In particular, sequencing errors within repetitive sequences can increase the chances of mapping errors, and result in misidentified backsplicing junctions.24 A recent study suggested that comparison of different alignment results can effectively eliminate ambiguous alignments.41 Nevertheless, it is still very difficult to determine whether an observed NCL junction arises from ambiguous alignments in incomplete or draft genomes. For circRNA detection, a previous study eliminated potential alignment errors by controlling for the alignment quality of both ends of RNA‐seq reads that were mapped inside a circle candidate.19 However, it remains a major challenge to effectively remove alignment errors without losing sensitivity.
Finally, spurious NCL events may also be generated from artificial RNA‐seq reads that are produced during cDNA library construction.40, 41, 51, 52 As RNA‐seq data are generally derived from reverse transcriptase (RT)‐based sequencing approaches, RT artifacts, such as template switching, often impede the accurate identification of NCL events. Reverse transcriptase may switch templates in the process of reverse transcription, either to a different RNA molecule or to a different location on the same template.51, 53 Switching may occur on DNA or RNA templates, and such experimental artifacts (or so‐called ‘template switching events’) frequently emerge in cDNA products.51, 53 Several studies have demonstrated that the majority of NCL events extracted from mRNAs were generated from experimental artifacts.41, 52 Unfortunately, it is difficult to distinguish such artifacts from genuine NCL events by simple experimental validations, not mention to the NCL RNA candidates merely identified by bioinformatics strategies without any experimental validation. Recently, some NCL events that previously passed RT‐PCR validations were subsequently confirmed by more careful validations to be originated from in vitro artifacts.41 Previous studies have indicated that increasing the primer annealing temperature during reverse transcription may reduce the emergence of template switching events.53, 54 However, such experiments were shown to be insufficient to eliminate template switching‐derived NCL events.40, 51 It was also demonstrated that such RT artifacts cannot be easily removed by controlling for canonical splice signals encompassing the NCL junctions or the depth of RNA‐seq reads supporting the NCL junctions.40, 41, 52 Several studies demonstrated that RTase‐dependent RNA products were likely to be RT artifacts, suggesting that comparisons of different RTases products could effectively detect such artifacts.40, 41, 51 Alternatively, some non‐RTase‐based experiments, such as Northern blot and RNase protection assay,55 can be applied to the detection of RT artifacts, although these validations are more expensive and time consuming than RTase‐based ones. To date, there is only one systematic approach that can detect NCL RNAs while controlling for experimental artifacts.52 Unfortunately, this approach is based on Drosophila hybrid mRNAs (Drosophilia melanogaster females vs. Drosophilia sechellia males) and a mixed mRNA‐negative control sample,52 and thus cannot be applied to human studies.
Biased Identification of circRNAs
There are many discrepancies among circRNA candidates identified by different methods,23 with the major contributing factor being that different methods used different detection rules to identify circRNAs.56 Such discrepancies between results also imply that a considerable proportion of detected circRNA candidates are merely false positives. In addition, using RNA‐seq data derived using different RNA‐library treatments to detect circRNA candidates may also yield different results. To examine this issue, we used five well‐known circRNA‐detecting methods, TopHat‐Fusion,27 MapSplice,28 segemehl,22 find_circ,13 and CIRI,26 to individually detect circRNA candidates in HeLa cells with three different RNA‐library treatments (Table 2): rRNA depletion (rRNA−), rRNA‐depleted RNAs with RNase R treatment (rRNA−&RNase R+), and rRNA‐depleted RNAs with poly(A) depletion (rRNA−&polyA−). Of note, as TopHat‐Fusion, MapSplice, and segemehl can also detect intergenic NCL events, we only considered the intragenic NCL events detected by these three methods. We showed that a considerable proportion (23–85%) of the detected circRNA candidates is dependent on the individual RNA‐library treatment (Figure 5(a)). Such proportions vary among the methods used (Figure 5(a)), which also reflect the discrepancies among different identification results. This result thus indicates that genome‐wide analysis of circRNAs may be biased by both the method and RNA‐library treatment. Moreover, we find that as much as 31–76% of the intragenic NCL events detected in rRNA− data are absent from both poly(A)‐depleted data and RNase R‐selected data (Figure 5(a)). As circRNAs tend to be non‐polyadenylated10, 19, 23 or RNase R‐resistant,11, 13, 43 such intragenic NCL events that are dependent on rRNA‐depleted data may not arise from backsplicing, suggesting that these circRNA candidates should be further curated.
Table 2.
HeLa Cell Transcriptome Data Derived from Different RNA‐library Treatments Used in This Study. All Data are Paired‐end RNA Sequencing Data
| RNA‐library Treatment | Sequencing Platform | Read Length | NCBI SRA ID (Read Number) |
|---|---|---|---|
| rRNA− | Illumina Hiseq 2000 | 101 bp |
SRR1637089 (44,933,450) SRR1637090 (35,685,310) |
| rRNA−&RNase R+ | Illumina Hiseq 2000 | 101 bp |
SRR1636985 (13,309,745) SRR1636986 (23,505,713) |
| rRNA−&polyA− | Illumnia GA II | 76 bp | SRR317048 (70,788,979) |
Figure 5.
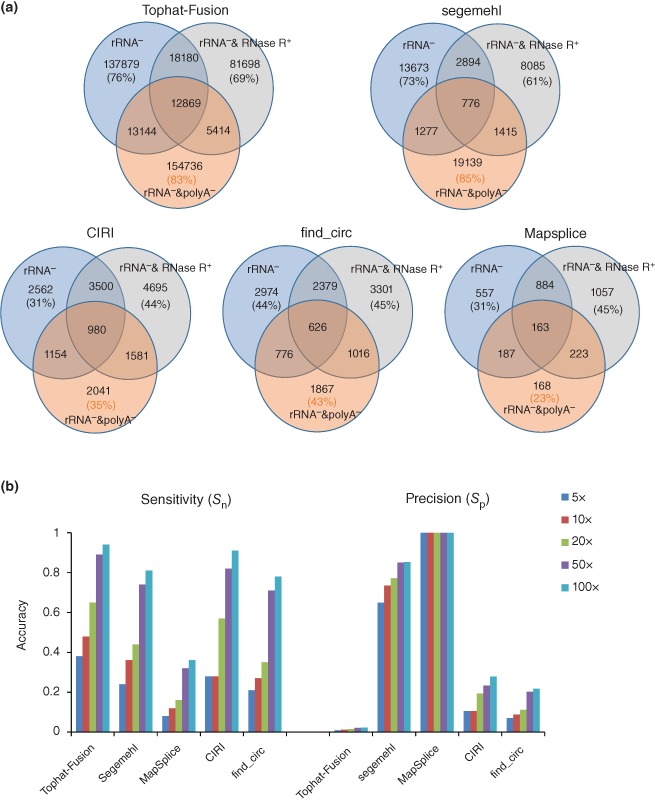
Comparison of identified ecircRNAs based on different bioinformatics methods or the use of sequencing data from different treatments. (a) Venn diagram of identified circRNAs based on HeLa cell transcriptome data with different RNA‐library treatments for each individual algorithm. The percentage of ecircRNA events identified from each RNA‐library treatment is showed in parentheses. (b) Evaluation of sensitivity (S n) and precision (S p) of five ecircRNA‐detecting algorithms, based on simulated datasets of different expression levels of NCL transcripts. S n and S p, both of which range from 0 to 1, are defined as TP/(TP+FN) and TP/(TP+FP), respectively. TP (true positive), FP (false positive), and FN (false negative) represent the number of correctly identified events, the number of incorrectly identified events, and the number of missing events, respectively.
Evaluation of Sensitivity and Precision of circRNA‐Detecting Methods
To evaluate the sensitivity (S n) and precision (S p) of different circRNA‐detecting methods, we utilized Mason57 to generate paired‐end reads (with read length 2 × 100 nt) from 100 simulated intragenic NCL transcripts with different expression levels (5‐ to 100‐fold), and then mixed these simulated data with the same background dataset generated from the GENCODE‐annotated (version 19) co‐linear transcripts. The simulated NCL transcripts must not be derived from pseudogenes or mitochondrial or ribosomal genes,58 and their junction sites were randomly generated and located at the boundaries of annotated exons. The above‐mentioned circRNA‐detecting methods (i.e., TopHat‐Fusion, MapSplice, segemehl, find_circ, and CIRI) were then applied to the simulated datasets. Our results revealed that the S n and S p values were both positively correlated with the expression levels of circRNAs (Figure 5(b)). When examining the tested dataset at all simulated expression levels, TopHat‐Fusion exhibited the highest S n values but the lowest S p values (all S p < 0.1), whereas the opposite was observed for MapSplice (Figure 5(b)). It was notable that although MapSplice achieved 100% precision (all S p = 1) under all simulated conditions, it had very poor sensitivity (all S n < 0.4) (Figure 5(b)). This reveals that certain methods achieve better precision by sacrificing sensitivity, highlighting the difficulty in reaching a balance between sensitivity and precision. Overall, segemehl, find_circ, and CIRI exhibited similar levels of sensitivity, but find_circ and CIRI demonstrated relatively lower precision (all S p < 0.3) than segemehl (all S p > 0.6) (Figure 5(b)). Therefore, here segemehl seemed to achieve a better balance between sensitivity and precision than the other methods examined. Of note, here all tools for evaluation were used with default parameters. In fact, different stringency levels of parameter settings (e.g., the number of RNA‐seq reads supporting the NCL junctions, the alignment quality of both ends of mapped RNA‐seq reads, the control of canonical splice signals encompassing the NCL junctions, etc.) may significantly affect the number of identified candidate circles for the same tool, and thereby affect the accuracy. Generally, circRNA‐detecting tools with low‐stringency parameters could achieve better sensitivity but worse precision than those with high‐stringency ones.
GLOBAL PROPERTIES OF EXONIC CIRCULAR RNAS
Flanking Introns of ecircRNAs
In addition to the aforementioned excess of RCSs in ecircRNA‐flanking introns, various independent studies reached the following conclusion: ecircRNAs tend to have longer flanking introns than expected (comparing to the average or control sets) in diverse species (e.g., humans, flies, and nematodes).10, 11, 15, 24, 37 In human, the ecircRNA‐flanking introns are three‐ to fivefold longer than randomly selected introns.11, 15 In Drosophila, upstream and downstream flanking introns of ecircRNAS have median lengths of 4662 and 2962 nt, respectively, both of which are much longer than the median length of all introns (94 nt).24 In addition, C. elegans ecircRNAs were observed to have 10‐fold longer flanking introns than the median length of all the introns.37 Although longer flanking introns seemed to promote ecircRNA formation, statistical analysis indicated that long flanking introns were not necessary for ecircRNA formation in humans.19 A later study revealed that a longer flanking intron itself does not cause ecircRNA formation; instead, the longer the intron, the greater the possibility that it contains more cis elements (e.g., inverted Alu elements) that promote ecircRNA formation.15 However, no specific motifs or structures have been found in flanking intron pairs so far.24 It is known that different species exhibit remarkable variations in intron length. Whether such a fundamental difference may have influenced ecircRNA biogenesis awaits further elucidation.
Sequence Context of ecircRNAs
Recent transcriptome‐wide analyses have revealed several common features in animal ecircRNAs. First, ecircRNAs may consist of a single exon or multiple exons (see also Figure 4(b)). Many of ecircRNAs encircle the second exon10 or the exon(s) near the 5′ end24 of the corresponding co‐linear counterpart. Typically, one gene contains one circular form, but some genes can form multiple circular products. Most human ecircRNAs contain less than 5 exons with a median length of 547 nt.23 Only a few ecircRNAs are smaller than 80 nt in length.26 Long exons tend to be enclosed within the circles.11 For the case of circRNAs with a single exon, the encircled exons are longer than overall expressed exons.11, 15 Second, GT‐AG canonical splice sites are usually required for circRNA formation, although cryptic splice sites are sometimes used instead.30 Splice sites used for co‐linear cis‐splicing10, 11, 24 or NCL trans‐splicing41 may also be used to form ecircRNAs. Nevertheless, there is no special global pattern associated with splice sites for circRNA biogenesis.30 Third, introns between the encircled exons are usually excised, but are retained in some rare cases (Figure 4(b)).11, 23, 24 Noncoding RNAs, intergeneric regions, or antisense regions are possibly (if not seldom) encompassed in ecircRNAs.23, 24, 26 Fourth, with the exception of the splice site sequences, RNA motifs are generally not present within the circles shared by most ecircRNAs. Only a handful of ecircRNAs were observed to contain MBL motifs21 and miRNA binding sites.13, 14
Conservation of ecircRNAs among Species
Several studies have shown that some ecircRNAs are evolutionarily conserved between three Drosophila species24 or between humans and mice,11, 13, 23, 37 implying circular forms are not by‐products of splicing or randomly misspliced products. The conservation of splicing regulatory elements in host genes may be responsible for the conservation of circRNA generation between species. A recent study revealed that the ecircRNAs expressed in both human and mouse have a higher probability of forming circles by base‐pairing of RCSs in the flanking introns as compared with those expressed only in one species or exons with bracketing intron length‐matched controls,37 suggesting that the human–mouse orthologous ecircRNAs are also conserved in terms of their exon circularization between these two species. However, exons within the human–mouse orthologous circles do not exhibit greater sequence conservation than their neighboring linear exons.23 The correlation between retention of ecircRNA orthologues across evolutionarily distant species and biological significance demands further investigation.
Abundance and Tissue‐specific Accumulation
A large amount of ecircRNAs have been identified in various cell types and eukaryotes (Table 1). A prominent website, circBase,59 continues to collect identified circRNAs. In human, as high as ∼100,000 ecircRNAs have been identified (Table 1), although the number of human ecircRNAs should be estimated more conservatively because of the possibility of false positives arising from high‐throughput sequencing or identification processes (as described above). It was suggested that circRNAs comprise 1 to >10% of all transcripts in human cells.10, 19, 23 No specific pattern of association between the circular forms and their corresponding linear transcripts has been observed. Most ecircRNAs are expressed at a very low level (0.1–1% of the expression levels of their co‐linear counterparts), but a few cases were more abundant than their co‐linear isoforms.11 When it comes to tissue‐specificity, most circRNAs were detected in only a few tissues/cell types.13, 19, 24, 26, 30 A study used 15 human cell types to show that widely expressed circRNAs exhibited significantly higher expression than narrowly expressed ones.26 Interestingly, in flies, ecircRNAs tended to arise from neural‐related genes and had a higher expression level in neural tissues.24 Similarly, ecircRNAs were reported to be enriched in mouse brain.60
Subcellular Localization
Several studies have shown that ecircRNAs are enriched in cytoplasmic samples.10, 11, 13, 23 As ecircRNAs are generated by the spliceosomal machinery in the nucleus and can be found in chromatin‐bound RNA pools, they are likely to be transported by the nuclear export system, or escape from nuclei during cell division. Cytosolic localization may also support the post‐transcriptional function of ecircRNAs. The most representative examples are CDR1as/ciRS‐7 and circRNA Sry, which are predominantly localized in the cytoplasm and function as miRNA sponges when the specific miRNA (miR‐7 for CDR1as/ciRS‐7; and miR138 for circRNA Sry) is present.13, 14 However, a very recent study showed that some exonic circles with intronic segments retained between exons were predominantly located in the nucleus, where they cis‐regulated their parent genes through specific RNA–RNA interactions.61 These observations indicate that different ecircRNAs may differ in their preferred subcellular localizations, suggesting they may also possess varied functions.
Translation Potential
As most ecircRNAs carry open reading frames, one may speculate that they may be translated into peptides. It was shown that peptides can be translated from ecircRNAs in vitro 62 or in vivo,63 as initiated from viral internal ribosome entry sites (IRESs)62 or from prokaryotic ribosome‐binding sites.63 Translation of ecircRNAs produced from backsplicing in human cells transfected with vectors has recently been demonstrated.31 Nevertheless, there is no evidence that spliceosome‐generated ecircRNAs can serve as mRNAs. Analyses based on mass spectrometry data,19, 60 ribosome profiling,23, 60 and polysome profiling8, 11, 60 have indicated that ecircRNAs tend to be untranslatable.
FUNCTIONS OF EXONIC CIRCULAR RNAS
miRNA Sponge
The ecircRNAs from human/mouse CDR1as/ciRS‐7 and mouse Sry have been experimentally validated to be highly associated with the miRNA effector protein Argonaute in the presence of miR‐7 and miR‐138, respectively.13, 14 CDR1as/ciRS‐7 contains 74 miR‐7 binding sites, while circRNA Sry contains 16 miR‐138 binding sites. The miRNA binding does not destabilize these two ecircRNAs; instead it competes with the binding between the miRNA and its target coding genes, and thereby reduces the effect of miRNA‐mediated posttranscriptional repression. It has been conclusively demonstrated that over‐expressing circRNAs of CDR1as/ciRS‐7 or Sry increases the expression of miRNA target reporter constructs, while knockdown of these ecircRNAs has the opposite effect.14 Downregulation of miR‐7 targets was also observed in CDR1as/ciRS‐7 knockdown human cells.13 Therefore, these two ecircRNAs are believed to serve as miRNA sponges13, 14 or miRNA reservoirs64 to attenuate miRNA‐mediated responses. In other words, with the same specific miRNA binding sites, ecircRNAs may play a regulatory role for competing endogenous RNA activities, which act as miRNA decoys to regulate the miRNA effects on their coding RNA targets by competing for miRNA binding. Expression of the corresponding coding RNA targets could be elevated by increasing the expression of such competing endogenous RNAs (or ‘ceRNAs’). An in vivo experiment in zebrafish further demonstrated that injection of human or mouse CDR1as/ciRS‐7 could lead to reduced midbrain sizes, similar to the phenotype of miR‐7 knockdown.13 In addition to miR‐7 miRNA sites, CDR1as/ciRS‐7 carries one miR‐671 site, which triggers its own linearization and destruction.65 Moreover, ecircRNAs from the human C2H2 zinc finger gene family were also predicted to function as miRNA sponges.23 A bioinformatics study showed that the miRNA sites in circRNAs are depleted of polymorphisms, suggesting the important role of circRNAs in regulating miRNA activities.66 Although these results indicated that some circRNAs indeed function as miRNA sponges, several studies suggested that most circRNAs do not act as miRNA‐sponges, as a large majority do not have more miRNA binding sites than co‐linear mRNAs.23, 60
Regulation of Parental Gene Transcription
Since ecircRNAs can be considered to be one type of alternative splicing isoform, they may play a role in regulating gene expression at the level of alternative splicing. There are two possible scenarios: (1) they form multiple mRNA isoforms, of which some are translated to functional proteins; and (2) they reduce the pool of canonically spliced transcripts which can be translated into functional proteins. The former is less probable, because no evidence supports the translational potential of ecircRNAs at present (as stated above); whereas the latter seems to be more likely. Some intron‐retained circRNAs were demonstrated (1) to be associated with human RNA polymerase II, and (2) to tend to be localized in the nucleus, suggesting that they might regulate gene expression.61 Knockdown of circRNA EIF3J could cause a significant decrease of EIF3J.61 The similar trend was also observed in circRNA PAIP2 and its parental gene.61 Further experiments revealed that these circRNAs might interact with U1 snRNP, and thereby upregulate their parental genes in cis.61 Although circRNAs and their corresponding co‐linear forms may compete with each other for biogenesis during splicing,21 the generated circles may promote both circRNA and mRNA expression in some cases.61
Regulatory Role of ecircRNAs during Development and Cell Proliferation
It was observed that ecircRNAs tended to be tissue specific and enriched in brain.12, 24, 60 Although the reason may be that most the parent genes of ecircRNAs were also enriched in brain, the relative contribution of ecircRNA to the total transcriptional output of the same gene was remarkably higher in brain than in other tested tissues.60 Compared with the corresponding co‐linear isoforms, ecircRNAs increase during aging of the central nervous system24 and decrease during cell proliferation.25 The reason for the negative correlation of global ecircRNA abundance with proliferation may be that ecircRNAs are more stable than their co‐linear isoforms, and prefer to accumulate in cells with a slower division rate.25 A similar phenomenon was observed in fission yeast (Schizosaccharomyces pombe): after starvation, decreased cell proliferation was accompanied by an increase in ecircRNA abundance.67 In addition, accumulation of ecircRNAs is decreased in various cancer cells and idiopathic pulmonary fibrosis, as compared with that in normal tissues.25 Expression profiles of ecircRNAs were more diverse among cancer cell lines than among noncancer cells, whereas the opposite trend was observed for their corresponding co‐linear isoforms.26 These observations revealed that changes in the physical condition of tissues or cells through developmental processes, aging, or disease can affect ecircRNA accumulation, suggesting that ecircRNAs might be an important biomarker for monitoring such changes. Whether (and perhaps, how) the change in ecircRNA abundance itself causes the change in cellular physical condition awaits further investigation.
Interactions with RNA‐binding Proteins
It has been shown that RNA‐binding proteins (RBPs), such as Argonaute,13, 14 RNA polymerase II,20 and MBL,21 can bind to ecircRNAs. The protein binding capacity of ecircRNAs is likely to be more complex than previously thought. Some ecircRNAs can store, sort, or localize RBPs, and probably regulate the function of RBPs by acting as competing elements, in the same way as they modulate miRNA activity.13, 68 The unique tertiary structure of ecircRNAs may also play an important role in the assembly of RNA or RBP complexes (Box 1).
BOX 1. MAMMALIAN CIRCULAR INTRONIC RNAs.
Through the spliceosomal machinery, intron lariats can escape the usual intron debranching and degeneration processes, and thus form stable circular intronic RNAs (ciRNAs).20 The formation process was suggested to rely on a 7 nt GU‐rich motif near the 5′ splice site and a 11 nt C‐rich motif near the branchpoint site.20 Although ciRNAs were first observed in mammalian cells over two decades ago,69, 70 the function of ciRNAs is understudied. Recently, a comprehensive analysis of RNA‐seq data in human cells provided several clues to their function.20 First, ciRNAs tend to be enriched in the nucleus and do not exhibit an excess of microRNA target sites. Second, some ciRNAs (e.g., ci‐ankrd52, ci‐mcm5, and ci‐sirt7) can enhance expression of their parent mRNAs. Third, ciRNAs can interact with RNA polymerase II and regulate polymerase II transcription. Fourth, in some cases, the expression of ciRNAs is positively correlated with that of their parent genes. Fifth, ciRNAs tend to exhibit relatively little evolutionary conservation between human and mouse. These observations indicate that ciRNA may possess a biological role distinct from that of ecircRNAs. In addition, ciRNA may associate with the polymerase II elongation machinery and upregulate their corresponding parent genes.20 The low evolutionary conservation of intronic sequences further suggests that ciRNAs increase transcriptome complexity between species.
CONCLUSION
It is now generally believed that eukaryotic spliceosomes exhibit a certain degree of flexibility in splice site choice, resulting in widespread alternatively spliced isoforms, including circRNAs, in transcriptomes. Although the functions of ecircRNAs are mostly unknown, circRNAs tend to exhibit tissue/cell type‐specific accumulation, and some of them are conserved among species. In addition to their well‐documented role as microRNA sponges, some evidence shows that they may regulate their parental gene transcription and cell proliferation. Thus, their existence cannot be simply explained as a consequence of missplicing or an inconsequential by‐product of pre‐mRNA splicing. Some ecircRNAs are indeed functional. However, a lot of questions regarding ecircRNA biogenesis and degradation await answers. For example, base‐pairing of matched RCSs in the flanking introns of circles is important, but not necessary, for enhancing ecircRNA formation.30, 31 It is worth asking whether this property of RCSs is common to all species, and whether ecircRNA biogenesis varies among species. In addition, ecircRNAs tend to have longer flanking introns,11, 15, 24, 37 but there is no information about how longer flanking introns are related to ecircRNA formation. Moreover, binding between splicing factor MBL proteins and MBL motifs located in flanking introns may promote ecircRNA formation from MBL genes.21 This finding has drawn great attention as MBL is known to be important in tissue‐specific alternative splicing.71 Besides MBL, however, no other trans‐splicing element affecting backsplicing has been found. To date, studies of circRNAs have mainly focused on ecircRNAs formed by spliceosomal machinery. Little is known about circRNAs generated from non‐spliceosomal mechanisms. Their prevalence and whether they carry functions similar to those formed by spliceosomes are still waiting further investigation. In addition, whether circRNAs are generated co‐transcriptionally or post‐transcriptionally remains a matter of debate. While co‐transcriptional generation of ecircRNAs is supported by fly nascent RNA‐seq data,21 (1) the requirement for a downstream functional 3′ processing signal and (2) the collaboration between intronic repeats and exonic sequences in circularization suggest a posttranscriptional model.32
In term of ecircRNA identification, RNA‐seq data continue to be important sources, as the accessibility of such data increases exponentially with the use of different experimental designs and variously processed biosamples. The three major hurdles to ecircRNA detection from RNA‐seq data are as follows: (1) discrimination between ecircRNAs, trans‐spliced RNAs, and genetic rearrangements; (2) removal of sequencing errors, alignment errors, and in vitro artifacts; and (3) biased identification results due to the use of different bioinformatics methods or sequencing data derived from different treatments. These difficulties may introduce severe bias into the trends of ecircRNA analysis. To date, there is no systematic method to effectively distinguish between different types of NCL events (i.e., circRNAs, trans‐spliced RNAs, and genetic rearrangements). In addition, with currently available bioinformatics algorithms, it remains a considerable challenge to effectively eliminate false calls from sequencing/alignment errors without losing sensitivity. Our results showed that the identification results vary dramatically among different ecircRNA‐detecting tools and among different RNA‐treatment data (Figure 5(a)); in particular, the sensitivity varies considerably for lowly expressed circRNAs (Figure 5(b)). Our evaluations of the accuracy for five well‐known circRNA‐detecting methods (TopHat‐Fusion, MapSplice, segemehl, find_circ, and CIRI) revealed that TopHap‐Fusion yielded the best sensitivity but the worst precision, whereas MapSplice demonstrated the opposite trend (Figure 5(b)). Of these five methods, segemehl seemed to achieve the greatest balance between sensitivity and precision (Figure 5(b)). Our observation reveals that there remains a need for a robust pipeline capable of identifying ecircRNAs with better balance between sensitivity and precision. In addition, the frequent occurrence of template switching in cDNA products presents another challenge to the accurate identification of ecircRNAs. Currently, no systematic approach is available for identifying ecircRNAs in the human transcriptome while using control experiments to remove potential template switching events. In summary, there are still a lot of unanswered questions for this important but largely uncharted class of transcripts, including unknown factors relating to ecircRNA biogenesis, function, and identification (Figure 6). The world of the transcriptome may be more complicated than we previously thought.
Figure 6.
Summary of selected unanswered questions regarding ecircRNA biogenesis, function, and identification.
ACKNOWLEDGMENTS
We thank Li‐Yuan Hung for comments. This work was supported by the Genomics Research Center, Academia Sinica, Taiwan; and the Ministry of Science and Technology (MOST), Taiwan (under the contracts MOST 103‐2628‐B‐001‐001‐MY4 and MOST 104‐2911‐I‐001‐502).
Conflict of interest: The authors have declared no conflicts of interest for this article.
References
FURTHER READING
- CircBase, a database for circular RNAs, http://circbase.org/. [DOI] [PMC free article] [PubMed]
- circ2Traits, a database of human circular RNAs associated with disease or traits, http://gyanxet‐beta.com/circdb/.
- TopHat‐Fusion (version 2.0.13), an algorithm for detecting both intergenic and intragenic NCL events, http://ccb.jhu.edu/software/tophat/fusion_index.html.
- MapSplice (version 2.17), an algorithm for detecting both intergenic and intragenic NCL events, http://www.netlab.uky.edu/p/bioinfo/MapSplice2.
- segemenhl (version 0.2.0), an algorithm for detecting both intergenic and intragenic NCL events (filterjunctions.py was applied to extracting circRNA candidates), http://www.bioinf.uni‐leipzig.de/Software/segemehl/.
- find_circ (version 1.0), an algorithm for detecting circRNAs, https://github.com/bioxfu/circRNAFinder/blob/master/src/find_circ.py.
- CIRI (version 1.1), an algorithm for detecting circRNAs, http://sourceforge.net/projects/ciri/.
- Simulated datasets, the simulated datasets that were generated in this study for evaluation of the sensitivity and precision of the circRNA‐detecting tools examined, ftp://treeslab1.genomics.sinica.edu.tw/simulated_expression/.
REFERENCES
- 1. Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK. Viroids are single‐stranded covalently closed circular RNA molecules existing as highly base‐paired rod‐like structures. Proc Natl Acad Sci USA 1976, 73:3852–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Taylor J, Pelchat M. Origin of hepatitis delta virus. Future Microbiol 2010, 5:393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grabowski PJ, Zaug AJ, Cech TR. The intervening sequence of the ribosomal RNA precursor is converted to a circular RNA in isolated nuclei of Tetrahymena. Cell 1981, 23:467–476. [DOI] [PubMed] [Google Scholar]
- 4. Lehmann K, Schmidt U. Group II introns: structure and catalytic versatility of large natural ribozymes. Crit Rev Biochem Mol Biol 2003, 38:249–303. [DOI] [PubMed] [Google Scholar]
- 5. Dalgaard JZ, Garrett RA. Protein‐coding introns from the 23S rRNA‐encoding gene form stable circles in the hyperthermophilic archaeon Pyrobaculum organotrophum. Gene 1992, 121:103–110. [DOI] [PubMed] [Google Scholar]
- 6. Danan M, Schwartz S, Edelheit S, Sorek R. Transcriptome‐wide discovery of circular RNAs in Archaea. Nucleic Acids Res 2012, 40:3131–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert JM, Oliner JD, Kinzler KW, Vogelstein B. Scrambled exons. Cell 1991, 64:607–613. [DOI] [PubMed] [Google Scholar]
- 8. Capel B, Swain A, Nicolis S, Hacker A, Walter M, Koopman P, Goodfellow P, Lovell‐Badge R. Circular transcripts of the testis‐determining gene Sry in adult mouse testis. Cell 1993, 73:1019–1030. [DOI] [PubMed] [Google Scholar]
- 9. Cocquerelle C, Mascrez B, Hetuin D, Bailleul B. Mis‐splicing yields circular RNA molecules. FASEB J 1993, 7:155–160. [DOI] [PubMed] [Google Scholar]
- 10. Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 2012, 7:e30733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19:141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rybak‐Wolf A, Stottmeister C, Glazar P, Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal‐Fluss R, et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell 2015, 58:870–885. [DOI] [PubMed] [Google Scholar]
- 13. Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495:333–338. [DOI] [PubMed] [Google Scholar]
- 14. Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495:384–388. [DOI] [PubMed] [Google Scholar]
- 15. Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. Complementary sequence‐mediated exon circularization. Cell 2014, 159:134–147. [DOI] [PubMed] [Google Scholar]
- 16. Gualandi F, Trabanelli C, Rimessi P, Calzolari E, Toffolatti L, Patarnello T, Kunz G, Muntoni F, Ferlini A. Multiple exon skipping and RNA circularisation contribute to the severe phenotypic expression of exon 5 dystrophin deletion. J Med Genet 2003, 40:e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koh W, Pan W, Gawad C, Fan HC, Kerchner GA, Wyss‐Coray T, Blumenfeld YJ, El‐Sayed YY, Quake SR. Noninvasive in vivo monitoring of tissue‐specific global gene expression in humans. Proc Natl Acad Sci USA 2014, 111:7361–7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bahn JH, Zhang Q, Li F, Chan TM, Lin X, Kim Y, Wong DT, Xiao X. The Landscape of microRNA, Piwi‐Interacting RNA, and circular RNA in human saliva. Clin Chem 2015, 61:221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. Cell‐type specific features of circular RNA expression. PLoS Genet 2013, 9:e1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, Zhu S, Yang L, Chen LL. Circular intronic long noncoding RNAs. Mol Cell 2013, 51:792–806. [DOI] [PubMed] [Google Scholar]
- 21. Ashwal‐Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, Kadener S. circRNA biogenesis competes with pre‐mRNA splicing. Mol Cell 2014, 56:55–66. [DOI] [PubMed] [Google Scholar]
- 22. Hoffmann S, Otto C, Doose G, Tanzer A, Langenberger D, Christ S, Kunz M, Holdt LM, Teupser D, Hackermuller J, et al. A multi‐split mapping algorithm for circular RNA, splicing, trans‐splicing and fusion detection. Genome Biol 2014, 15:R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guo JU, Agarwal V, Guo H, Bartel DP. Expanded identification and characterization of mammalian circular RNAs. Genome Biol 2014, 15:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Westholm Jakub O, Miura P, Olson S, Shenker S, Joseph B, Sanfilippo P, Celniker Susan E, Graveley Brenton R, Lai EC. Genome‐wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age‐dependent neural accumulation. Cell Rep 2014, 9:1966–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bachmayr‐Heyda A, Reiner AT, Auer K, Sukhbaatar N, Aust S, Bachleitner‐Hofmann T, Mesteri I, Grunt TW, Zeillinger R, Pils D. Correlation of circular RNA abundance with proliferation – exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Sci Rep 2015, 5:8057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gao Y, Wang J, Zhao F. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol 2015, 16:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim D, Salzberg SL. TopHat‐Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol 2011, 12:R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, et al. MapSplice: accurate mapping of RNA‐seq reads for splice junction discovery. Nucleic Acids Res 2010, 38:e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol 2014, 15:108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Starke S, Jost I, Rossbach O, Schneider T, Schreiner S, Hung L, Bindereif A. Exon circularization requires canonical splice signals. Cell Rep 2015, 10:1–9. [DOI] [PubMed] [Google Scholar]
- 31. Wang Y, Wang Z. Efficient backsplicing produces translatable circular mRNAs. RNA 2015, 21:172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liang D, Wilusz JE. Short intronic repeat sequences facilitate circular RNA production. Genes Dev 2014, 28:2233–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zaphiropoulos PG. Circular RNAs from transcripts of the rat cytochrome P450 2C24 gene: correlation with exon skipping. Proc Natl Acad Sci USA 1996, 93:6536–6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Surono A, Takeshima Y, Wibawa T, Ikezawa M, Nonaka I, Matsuo M. Circular dystrophin RNAs consisting of exons that were skipped by alternative splicing. Hum Mol Genet 1999, 8:493–500. [DOI] [PubMed] [Google Scholar]
- 35. Kameyama T, Suzuki H, Mayeda A. Re‐splicing of mature mRNA in cancer cells promotes activation of distant weak alternative splice sites. Nucleic Acids Res 2012, 40:7896–7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dubin RA, Kazmi MA, Ostrer H. Inverted repeats are necessary for circularization of the mouse testis Sry transcript. Gene 1995, 167:245–248. [DOI] [PubMed] [Google Scholar]
- 37. Ivanov A, Memczak S, Wyler E, Torti F, Porath Hagit T, Orejuela Marta R, Piechotta M, Levanon Erez Y, Landthaler M, Dieterich C, et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep 2015, 10:1–8. [DOI] [PubMed] [Google Scholar]
- 38. Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, Roslan S, Schreiber AW, Gregory PA, Goodall GJ. The RNA binding protein quaking regulates formation of circRNAs. Cell 2015, 160:1125–1134. [DOI] [PubMed] [Google Scholar]
- 39. Al‐Balool HH, Weber D, Liu Y, Wade M, Guleria K, Nam PL, Clayton J, Rowe W, Coxhead J, Irving J, et al. Post‐transcriptional exon shuffling events in humans can be evolutionarily conserved and abundant. Genome Res 2011, 21:1788–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu CS, Yu CY, Chuang CY, Hsiao M, Kao CF, Kuo HC, Chuang TJ. Integrative transcriptome sequencing identifies trans‐splicing events with important roles in human embryonic stem cell pluripotency. Genome Res 2014, 24:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu CY, Liu HJ, Hung LY, Kuo HC, Chuang TJ. Is an observed non‐co‐linear RNA product spliced in trans, in cis or just in vitro? Nucleic Acids Res 2014, 42:9410–9423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim P, Yoon S, Kim N, Lee S, Ko M, Lee H, Kang H, Kim J, Lee S. ChimerDB 2.0 – a knowledgebase for fusion genes updated. Nucleic Acids Res 2010, 38:D81–D85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Suzuki H, Tsukahara T. A view of pre‐mRNA splicing from RNase R resistant RNAs. Int J Mol Sci 2014, 15:9331–9342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Marzluff WF, Wagner EJ, Duronio RJ. Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat Rev Genet 2008, 9:843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang L, Duff MO, Graveley BR, Carmichael GG, Chen LL. Genomewide characterization of non‐polyadenylated RNAs. Genome Biol 2011, 12:R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol 2014, 32:453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McPherson A, Wu C, Hajirasouliha I, Hormozdiari F, Hach F, Lapuk A, Volik S, Shah S, Collins C, Sahinalp SC. Comrad: detection of expressed rearrangements by integrated analysis of RNA‐Seq and low coverage genome sequence data. Bioinformatics 2011, 27:1481–1488. [DOI] [PubMed] [Google Scholar]
- 48. Wang Q, Xia J, Jia P, Pao W, Zhao Z. Application of next generation sequencing to human gene fusion detection: computational tools, features and perspectives. Brief Bioinform 2013, 14:506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McPherson A, Wu C, Wyatt AW, Shah S, Collins C, Sahinalp SC. nFuse: discovery of complex genomic rearrangements in cancer using high‐throughput sequencing. Genome Res 2012, 22:2250–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Annala MJ, Parker BC, Zhang W, Nykter M. Fusion genes and their discovery using high throughput sequencing. Cancer Lett 2013, 340:192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Houseley J, Tollervey D. Apparent non‐canonical trans‐splicing is generated by reverse transcriptase in vitro. PLoS One 2010, 5:e12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McManus CJ, Duff MO, Eipper‐Mains J, Graveley BR. Global analysis of trans‐splicing in Drosophila. Proc Natl Acad Sci USA 2010, 107:12975–12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cocquet J, Chong A, Zhang G, Veitia RA. Reverse transcriptase template switching and false alternative transcripts. Genomics 2006, 88:127–131. [DOI] [PubMed] [Google Scholar]
- 54. Ouhammouch M, Brody EN. Temperature‐dependent template switching during in vitro cDNA synthesis by the AMV‐reverse transcriptase. Nucleic Acids Res 1992, 20:5443–5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Djebali S, Lagarde J, Kapranov P, Lacroix V, Borel C, Mudge JM, Howald C, Foissac S, Ucla C, Chrast J, et al. Evidence for transcript networks composed of chimeric RNAs in human cells. PLoS One 2012, 7:e28213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lasda E, Parker R. Circular RNAs: diversity of form and function. RNA 2014, 20:1829–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Holtgrewe M, Emde AK, Weese D, Reinert K. A novel and well‐defined benchmarking method for second generation read mapping. BMC Bioinformatics 2011, 12:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ge H, Liu K, Juan T, Fang F, Newman M, Hoeck W. FusionMap: detecting fusion genes from next‐generation sequencing data at base‐pair resolution. Bioinformatics 2011, 27:1922–1928. [DOI] [PubMed] [Google Scholar]
- 59. Glazar P, Papavasileiou P, Rajewsky N. circBase: a database for circular RNAs. RNA 2014, 20:1666–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. You X, Vlatkovic I, Babic A, Will T, Epstein I, Tushev G, Akbalik G, Wang M, Glock C, Quedenau C, et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci 2015, 18:603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li Z, Huang C, Bao C, Chen L, Lin M, Wang X, Zhong G, Yu B, Hu W, Dai L, et al. Exon‐intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol 2015, 22:256–264. [DOI] [PubMed] [Google Scholar]
- 62. Chen CY, Sarnow P. Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science 1995, 268:415–417. [DOI] [PubMed] [Google Scholar]
- 63. Perriman R, Ares M Jr. Circular mRNA can direct translation of extremely long repeating‐sequence proteins in vivo. RNA 1998, 4:1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hansen TB, Kjems J, Damgaard CK. Circular RNA and miR‐7 in cancer. Cancer Res 2013, 73:5609–5612. [DOI] [PubMed] [Google Scholar]
- 65. Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, Kjems J. miRNA‐dependent gene silencing involving Ago2‐mediated cleavage of a circular antisense RNA. EMBO J 2011, 30:4414–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thomas LF, Saetrom P. Circular RNAs are depleted of polymorphisms at microRNA binding sites. Bioinformatics 2014, 30:2243–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang PL, Bao Y, Yee MC, Barrett SP, Hogan GJ, Olsen MN, Dinneny JR, Brown PO, Salzman J. Circular RNA is expressed across the eukaryotic tree of life. PLoS One 2014, 9:e90859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hentze MW, Preiss T. Circular RNAs: splicing's enigma variations. EMBO J 2013, 32:923–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Qian L, Vu MN, Carter M, Wilkinson MF. A spliced intron accumulates as a lariat in the nucleus of T cells. Nucleic Acids Res 1992, 20:5345–5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kopczynski CC, Muskavitch MA. Introns excised from the delta primary transcript are localized near sites of Delta transcription. J Cell Biol 1992, 119:503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Konieczny P, Stepniak‐Konieczna E, Sobczak K. MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res 2014, 42:10873–10887. [DOI] [PMC free article] [PubMed] [Google Scholar]