

Summary
Accurate analysis and quantification of different testicular cell populations are of central importance in studies of male reproductive biology. The traditional histomorphometric and immunohistochemical methods remain the gold standard in studying the complex dynamics of the testicular tissue. Through past years advances have been made in the application of flow cytometry for the rapid analysis of testicular cell populations. Detection of DNA content and of surface antigens and fluorescent reporters have been widely used to analyze and sort cells. Detection of intracellular antigens can broaden the possibilities of applying flow cytometry in studies of male reproduction. Here, we report a detailed protocol for the preparation of rat testicular tissue for detection of intracellular antigens by flow cytometry, and a pipeline for subsequent data analysis and troubleshooting. Rat testicular ontogenesis was chosen as the experimental model to validate the performance of the assay using vimentin and γH2AX as intracellular markers for the somatic and spermatogenic cells, respectively. The results show that the assay is reproducible and recapitulates the rat testis ontogenesis.
Keywords: development, flow cytometry, spermatogenesis, testis
Introduction
Flow cytometry is widely used for the rapid analysis of various cellular features both in vitro and in vivo. In recent years, flow cytometry has emerged as a standard way to analyze also solid organs after tissue dissociation in addition to the traditional applications for cell lines and hematological cells (Tuomela et al., 2010). Flow cytometry is an attractive application to study the behavior of the different testicular cells, since it can considerably accelerate experimental workflows. Situations warranting a method for quick analysis are for example screening for suitable dose and time windows in toxicological studies, and the analysis of testicular phenotypes of transgenic animals.
The dynamics of the different testicular cell populations have traditionally been analyzed with histomorphometric methods. Despite being accurate and offering valuable information on the localization the cells and tissue architecture, they are time‐consuming and laborious. Automated image analysis is often not applicable to testicular samples because of their complexity. Flow cytometry has been used as a more data‐intensive method to study the behavior of the different testicular cell populations. A golden standard for flow cytometry analysis of testicular cells has been the use of stoichiometric DNA stains to study the different germ cell populations based on their DNA content (Toppari et al., 1985, 1986, 1988; Nurmio et al., 2007; Michaelis et al., 2013).
More recently, flow cytometry has emerged as a method to sort specific cell populations from fresh testicular cell preparations. Both antibodies against cell surface markers and fluorescence reporter animal models have been used to enrich specific cell populations and to detect malignant cell contamination (Hou et al., 2011; Dovey et al., 2013; Van Saen et al., 2013; Wakeling et al., 2013; Valli et al., 2014). While fresh cell suspension and surface antigens are necessary for some applications, they have some shortcomings in others. First, the requirement for fresh samples for each experiment limits the applicability when rarely available material is used. Second, sortable cell surface markers are required and especially for somatic cells, suitable antibodies are hard to find. In addition, this method does not enable the study of intracellular signaling cascades.
Intracellular flow cytometry has been extensively used to study various cell and tissue types (Strobl & Knapp, 2004; Turac et al., 2013). Advantages of intracellular flow cytometry include a broader pool of potential target antigens, elimination of the need to generate animal models or cell lines with cell type‐specific fluorescent reporters, and the possibility to store and transfer the samples for later analysis. An obvious limitation of intracellular flow cytometry is that it does not provide viable sorted cells for downstream analyses as the cell are fixed and permeabilized during the sample preparation. In the context of testicular research, intracellular flow cytometry has been previously used to study proliferating cell populations in the fetal and adult mouse testes (Elzeinova et al., 2013; Ferguson et al., 2013; Wakeling et al., 2013).
In this report, we describe a detailed flow cytometry protocol to study intracellular antigens in testicular cells. We have validated the protocol in ontogenical series of rat testicular tissue by monitoring the dynamics of the somatic and germ cell populations during testis development.
Materials and Methods
Protocol
Here, we describe a detailed protocol for testicular tissue dissociation, subsequent fixation and permeabilization, and detection of the different testicular cell populations in rat testis (Fig. 1). Different marker antibodies can be used in conjunction with this protocol depending on the cell type of interest. We recommend the use of antibodies that have been developed for the detection of intracellular antigens by flow cytometry. Testicular ontogenesis offers a powerful tool to assess antibody specificity.
Figure 1.
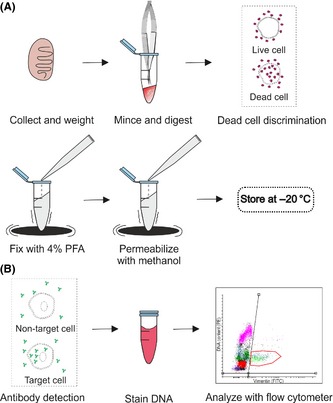
Workflow. (A) Sample preparation. Ten milligram of tissue is minced and digested with enzymes. The solution is filtered to obtain a single cell suspension and optional fixable dead cell dye is added. Cells are fixed with 4% paraformaldehyde (PFA) and permeabilized with methanol while gently vortexing. Samples are stored in −20 °C. (B) Staining procedure. After washing and blocking, a fluorescently labeled antibody is added to the cells. This is followed by DNA staining with propidium iodide and an analysis with a flow cytometer.
Materials required:
McPherson–Vannas scissors
Polystyrene Eppendorf tubes
Low affinity pipette tips
FACS tubes with 35 μm mesh caps
Use the hard clear plastic (such as polystyrene) Eppendorf tubes and low binding affinity pipette tips throughout the experiment.
- Tissue dissociation
-
1.1Prepare 1 mL digestion medium per sample with 1 mg/mL collagenase/dispase (cat. # 10269638001; Roche, Basel, Switzerland), 1 mg/mL hyaluronidase (cat. # H3506; Sigma, St. Louis, MO, USA), 1 mg/mL DNAse I (cat. # DN‐25; Sigma, St. Louis, MO, USA) in DMEM/F12.
-
1.2Collect the testes in ice‐cold phosphate‐buffered saline (PBS) and remove the tunica albuginea carefully. Aliquot approximately 10 mg of tissue per each sample using the McPherson–Vannas scissors. Record the exact weights of the tissue aliquots.
-
1.3Mince the testicular tissue with the McPherson–Vannas scissors in 200 μL of the digestion medium until all seminiferous tubule pieces are ≤1 mm long. Add the remaining 800 μL of the digestion medium to each sample.
-
1.4Incubate for 25 min at 37 °C with slow continuous rotation. Perform an additional mechanical disruption every 5 min by carefully pipetting the solution five times back and forth using 1000 μL Maxymym Recovery tips (Axygen, Corning Inc., Corning, NY, USA). Performing this step according to instructions is critical to ensure an optimal recovery of the fragile spermatocytes and to minimize debris.
-
1.5Filter the cell suspension using a 35 μm pore size to achieve single cell suspension. A flow cytometry tube cell strainer cap (cat. # 352235; BD Falcon, BD Biosciences, Franklin Lakes, NJ, USA) attached to an Eppendorf tube functions well for the filtration. Pipette the cell suspension slowly through the mesh to avoid clogging and cell damage. Pellet the cells by centrifugation (400 g, 10 min at +4 °C), discard the supernatant and resuspend the pellet in 1 mL of PBS. If preparing samples without dead cell discrimination, continue to step 3.1.
-
1.1
- Dead cell discrimination (optional)A fixable dead cell stain can be added to the cell suspension. We used LIVE/DEAD Near‐IR (Invitrogen, Thermo Fischer Scientific, Waltham, MA, USA), which binds to amines on cell surface, but also intracellularly when the integrity of the cell is compromised. Intracellular amine staining yields an intense fluorescent signal in flow cytometry and allows the exclusion of dead cells from the downstream analyses.
-
2.1Add the LIVE/DEAD Near‐IR stain reconstituted in DMSO to the cells at a final concentration of 1 : 10 000. Incubate for 30 min on ice and pellet by centrifugation (400 g, 10 min at +4 °C) and resuspend in 1 mL of PBS.
-
2.1
- Fixation and permeabilizationTo allow the detection of intracellular antigens by specific antibodies and to enable long‐term storage of the samples, the testicular cells are fixed using 4% paraformaldehyde (PFA) and permeabilized with 90% methanol (an adaptation of the protocol from Cell Signaling Technology Inc., Danvers, MA, USA).
-
3.1Add 32% PFA (cat. # 15714; Electron Microscopy Sciences, Hatfield, PA, USA) to a final concentration of 4% to the cell suspension while vortexing the solution slowly. This prevents aggregation of the cells during fixation. For fixation, incubate the samples for 10 min at 37 °C with gentle rotation. Chill the samples on ice for 1 min.
-
3.2Centrifuge the cells at 1200 g, 7 min at 4 °C to remove the fixative. Discard the supernatant and resuspend the pellet in 100 μL of PBS. Permeabilize the cells by adding 900 μL of ice‐cold 100% methanol in a drop‐wise fashion while gently vortexing the cells. Incubate for 30 min on ice. After this step, the samples can be stored in −20 °C.
-
3.1
- Detection of intracellular antigens and DNA content of the testicular cells
-
4.1Remove methanol solution from the samples by centrifugation at 1200 g, 7 min at 4 °C. Discard the supernatant. For washing resuspend the pellet in with 1 mL of incubation buffer (0.5% sera of choice in PBS). If using a directly fluorochrome‐conjugated primary antibody, use the sera of the species in which the antibody was made. In the case of using an unconjugated primary antibody together with a fluorochrome‐conjugated secondary antibody, choose the sera based on the secondary antibody. Centrifuge at 1200 g, 7 min at 4 °C and remove supernatant.
-
4.2Resuspend the pellet in 100 μL of incubation buffer per assay tube and block for 10 min at room temperature (RT). Add the primary antibody at an assay‐dependent concentration and incubate for 1 h at RT. Remember to include an unstained control (with no antibody, LIVE/DEAD cell stain or DNA stain) and controls with each fluorochrome (only antibody, LIVE/CELL stain or DNA stain) alone. Move to step 4.4 if using a secondary antibody.
-
4.3Wash the samples by adding 700 μL of incubation buffer and centrifuge at 1200 g, 7 min at 4 °C. Discard supernatant. Resuspend in 1 mL of PBS and centrifuge at 1200 g, 7 min at 4 °C. (Move to step 4.5 if secondary antibody is not used.)
-
4.4Secondary antibody incubation (Optional)
-
4.5When using an unconjugated primary antibody, the signal is detected with a fluorochrome‐conjugated secondary antibody.
-
4.6Wash once by adding 700 μL of incubation buffer and centrifuge at 1200 g, 7 min at 4 °C. Discard the supernatant and resuspend the pellet in 100 μL of incubation buffer. Add the secondary antibody at assay‐dependent concentration and incubate for 1 h at RT. Perform washes the same way as in step 4.3 and proceed to the DNA staining.
-
4.7For DNA staining with propidium iodide (PI), resuspend the cells in 250 μL of 0.2% BSA, 5 μg/mL RNase in PBS and incubate the samples for 15 min at 37 °C in gentle rotation. Add 250 μL of PBS followed by PI (100 μg/mL). Filter the samples prior to analysis.
-
4.1
Analyze with a flow cytometer. Keep the samples on ice and protected from light.
Animals
Male Sprague–Dawley rats of different ages (5, 10, 16, 24, and 60‐day‐old) were chosen to target the major developmental time windows of rat testis differentiation in the ontogenic study. The animals were sacrificed by CO2 inhalation followed by cervical dislocation. Animals were housed under environmentally controlled conditions (12 h light/12 h darkness; temperature 21 ± 1 °C) in the animal facility of the University of Turku. All procedures were carried out according to the institutional and ethical policies of the University of Turku and approved by the local ethics committee on animal experimentation. For each time‐point, five animals from different litters were used for the analyses.
Detection of vimentin‐ and γH2AX‐Ser139 positive cells
The flow cytometry testis samples from 5, 10, 16, and 24‐day‐old and adult rats were prepared as described above with the exception that no dead cell stain was added to the samples. Three 10 mg samples were taken from different parts of each analyzed testis from five different animals. From the juvenile animals with small testes, both testes of each animal were pooled as one sample and the three parallel samples represent different animals from the same litter and the n refers to litter. Rabbit anti‐vimentin conjugated to Alexa488 (cat. # 9854; Cell Signaling Technologies) was used at a concentration of 1/50 to detect vimentin‐positive cells and rabbit sera were used in the incubation buffer with this antibody.
For the detection and quantitation of γH2AX‐Ser139 positive cells by flow cytometry, mouse anti‐γH2AX‐Ser139 antibody (cat. # 05‐636; EMD Millipore Billerica, MA, USA) was used at a concentration of 1/100 on the 16‐day‐old and adult rat testis samples. About 0.5% normal donkey serum was used as the incubation buffer. After washing, the cells were incubated for 1 h at RT with a donkey anti‐mouse‐Alexa488 secondary antibody (Invitrogen) diluted 1/500. For the γH2AX‐assay, FxCycle Far‐Red DNA stain (cat. # F‐10348; Life Technologies Ltd, Paisley, UK) was used as a DNA counterstain. FxCycle was reconstituted in DMSO according to the manufacturer's instructions. After the antibody staining and washes, the samples were resuspended in 250 μL of 0.2% BSA, 5 μg/mL RNase in PBS, incubated for 15 min at 37 °C in gentle rotation, and 250 μL of PBS was added. Then 7.5 μL of FxCycle DNA stain was pipetted to the samples and incubated for 30 min at RT.
Data acquisition and gating strategy
Data acquisition
BD LSRII (Becton Dickinson, Franklin Lakes, NJ, USA) was used in sample analysis. The LSRII allows user to set the acquired volume when running the sample in a 96‐well‐plate format. Analyzing a fixed volume enables cell‐count normalization. The suitable volume depends on the cell concentration, and 25 μL was the optimal volume in our experiments. Even though a set volume is required for the quantitative analysis, the populations can be visualized also without volume control by using flow cytometry cell counting beads (e.g., TruCount tubes by BD).
For Alexa488 and PI, a 488 nm laser was used for excitation and emissions were collected with 530/30 nm and 575/26 nm bandpass filters, respectively. FxCycle DNA stain was excited with 633 nm laser and emission was detected with a 660/20 nm bandpass filter. For the LIVE/DEAD Near‐IR kit dead cell marker, the 633 nm laser was used for excitation and the emission was detected with a 780/60 nm bandpass filter. In each run, an unstained control and a single‐stained control for each fluorochrome were used. Compensation was adjusted based on these controls, and the single‐stained controls also helped setting the positive signal thresholds when gating the different cell populations (Figure S1B). The controls were prepared from the same testicular samples that were analyzed, but in the case of very low‐abundancy target cells and precious samples, compensation beads can also be used. Because of the differences in fluorochromes and in flow cytometer laser and filter set specifications, each experiment should be carefully planned with a person with knowledge of the flow cytometer properties.
Gating
Flow cytometry data analysis was performed with the non‐commercial Flowing Software ver. 2.5 (Mr. Perttu Terho; Turku Centre for Biotechnology, Finland; www.flowingsoftware.com).
Gating of the fixed testis samples differs somewhat from gating for less complex in vitro samples from more uniform cell lines. The complexity of the cell morphology in the testis makes traditional approaches for doublet discrimination (such as using the FSC/SSC‐plot, or FSC‐Area/FSC‐Height plots) difficult to use in a reproducible manner between different samples (Fig. 2A; Figure S1A). Also, the proportion of the cells that could be confidently assigned as doublets is small. Therefore, we began the gating of the appropriate cell populations from the FSC/PI density plot, where debris (small particles), cell aggregates (high on FSC axis) and extremely bright particles (high on PI axis) were excluded from the analysis (Fig. 2C and D, solid black line). Next, the cell populations with different amounts of DNA (1C, 2C, 4C) were gated using the PI signal (Fig. 2B–D, black dotted areas).
Figure 2.
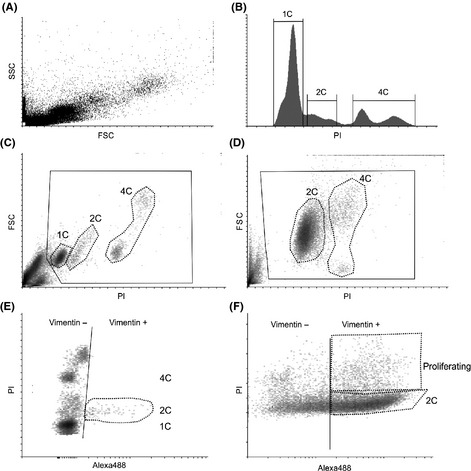
A multi‐step gating strategy to obtain information about the different cell populations. (A) A representative FSC/SSC dot plot of an adult rat testis. (B) A propidium iodide (PI) fluorescence histogram showing the cells clustered according to the DNA contents. 1C = haploid germ cells, 2C = diploid cells (spermatogonia and somatic cells), 4C = tetraploid cells (germ cells in meiosis I and cells in G2/M‐phase), proliferating = cells with DNA amount between 2C and 4C. (C) FSC/PI density plot of an adult rat testis showing gating of the cell populations corresponding to the histogram. Solid black line = gate excluding debris and cell aggregates, dotted black lines = cell populations according to DNA amount. (D) FSC/PI density plot of a 5‐day‐old rat testis. Note that no haploid cells are present at this time‐point. Solid black line = gate excluding debris and cell aggregates, dotted black lines = cell populations according to DNA amount. (E) PI/Alexa488 density plot of an adult rat testis showing vimentin‐positive cells as a part of the 2C population. (F) PI/Alexa488 density plot of a 5‐day‐old rat testis showing the vimentin‐positive cells in both the 2C and in the proliferating cell populations.
To analyze the number and DNA content of the vimentin‐positive cells, a PI/Alexa488 density plot was used. The threshold of the double‐negative population was set first using the unstained control (Figure S1B). Then, the threshold for vimentin‐positive and vimentin‐negative cells (Fig. 2E and F, solid black line) was defined by the single‐stained controls (Figure S1B). Knowledge of the normal dynamics of the different cell types during testicular development will aid the judgment whether an antibody gives a specific signal. It is known that in the adult rat testis, the vimentin‐positive somatic cells should all be diploid and non‐proliferative (Fig. 2E). In a 5‐day‐old juvenile testis, a considerable amount of the cells in the testis are vimentin positive, and a large proportion of them are proliferating (Fig. 2F).
A similar gating pipeline was applied to the γH2AX and LIVE/DEAD cell staining. The unstained control maximum intensity was used as a cutoff value for the dead cell population in the LIVE/DEAD cell staining and the positive control sample provided additional help in setting the correct threshold (Figure S2).
Calculations
The final cell counts were displayed as number of cells per milligram of tissue. The formula below was used to obtain these results:
V(total) is the total volume of the stained cell suspension (here 550 μL), V(run) is the analyzed sample volume (25 μL), and the number of events is the amount of cells in each population after the gating. Finally, the figures are normalized to the weight of tissue taken for the assay.
Analysis of cell damage
To analyze the level of cell damage caused by the initial single cell preparation procedure, samples were prepared from adult rat testis as described in the protocol including the dead cell detection with LIVE/DEAD Near‐IR dye and PI. For a positive control, a part of the same testis used for the assay was incubated at +42 °C for 6 h to induce cell death. The tissue was subsequently processed according to the sample preparation protocol.
Troubleshooting
A few approaches have proven useful to decrease unspecific background from an antibody staining.
Dead cell discrimination: Damaged cells often cause bright unspecific staining. Adding a stain for dead cell discrimination to the assay enables exclusion of dead cells from analysis. Also, gating out more debris using the FSC/SSC‐plot often clarifies the assay considerably.
Increased stringency of staining conditions: Antibody dilution optimization is one of the basic steps one should perform. One should opt for directly conjugated flow cytometry‐validated antibodies when possible. If unspecific staining persists, it is useful to increase the amount of serum used for blocking and to add a detergent such as 0.05% Triton‐X or Tween‐20 to the washing buffer.
Choice of fluorochromes: PI and Alexa488 can be a problematic pair in the case of very bright fluorescence, as the bleed through from one channel to another can distort the data. In such cases, changing the DNA stain should be considered. There are several DNA stains [e.g. DRAQ5 (Cell Signaling Technologies), DRAQ7 (Cell Signaling Technologies), FxCycle (Invitrogen)] available with different wavelength options and narrower emission profiles than PI. Further information can be found from several sources such as https://www.bdbiosciences.com/documents/Multicolor_AppNote.pdf.
Doublet discrimination: Some experimental set‐ups may require more rigorous doublet discrimination than used here. Such a need might arise from, e.g., difficulties in the tissue digestion because of fibrosis in some animal models or the need to quantify the 4C population more precisely. Doublet discrimination using FSC‐Area/FSC‐Width – plots was the most applicable method for doublet discrimination in the rat testis samples (Figure S1A).
Immunofluorescence
For additional validation of the target cells detected by the antibodies used for the flow cytometry assays, standard immunofluorescence was performed on PFA‐fixed paraffin‐embedded adult rat testis. Briefly, the tissue sections were deparaffinized and permeabilized in 10 mm sodium citrate buffer, pH 6 using a pressure cooker. 100 mM NH4Cl was used to quench autofluorescence and the samples were blocked with 5% normal rabbit sera (vimentin antibody) or normal donkey sera (γH2AX‐Ser139 antibody). The same primary antibodies as used for the flow cytometry (mouse anti‐γH2AX‐Ser139 antibody, cat. # 05‐636; Millipore) and anti‐vimentin conjugated to Alexa488 (cat. # 9854; Cell Signaling Technologies) were diluted at 1/100 in blocking solution and incubated overnight at +4 °C. The vimentin‐stained sections were directly counterstained with DAPI (4′,6‐diamidino‐2‐phenylindole) and mounted after washing off the unbound antibody. The γH2AX‐Ser139‐stained sections were washed and incubated with the secondary donkey anti‐mouse‐AlexaFluor546‐antibody diluted 1/500 in blocking solution and incubated 1 h at RT. After washing the sections were counterstained with DAPI and mounted with VectaShield Hard‐set mounting medium (cat. # H‐1400; Vector Laboratories, Burlingame, CA, USA). The samples were imaged using Axioimager M1 fluorescence microscope (Zeiss, Oberkochen, Germany).
Results
Cell populations revealed by the DNA stain
It was not feasible to separate distinctive testicular cell type populations based on cell size and granularity only (Fig. 2A). However, when PI was introduced, clear cell populations could be distinguished according to their DNA content (Fig. 2B–D). Here, for convenience, the term ploidy is used for the reference to the different cell populations with different amount of DNA, not for their chromosome number as in cytogenetics. In the adult testis, the haploid cells with the least DNA form the 1C peak in the histogram (Fig. 2B). Elongated spermatids, which do not bind DNA in a stoichiometric fashion, could be seen in the histogram as a less intense shoulder of the 1C peak (Fig. 2B, gate for 1C). In the FSC/PI density plot, they form a single population (Fig. 2C). The tetraploid (4C) cell population in both adult and juvenile testis is comprised of both spermatocytes in meiosis I (leptotene, zygotene and pachytene) and of the mitotically dividing cells that are in G2‐phase of the cell cycle (proliferating spermatogonia and somatic cells). Two populations containing different sized cells had a 4C DNA content. In the adult testis, the population with larger cells likely presented pachytene spermatocytes, while the population with smaller cells comprised all other 4C cells (Fig. 2C). A similar pattern of two 4C populations separated by size was observed in the 5‐day‐old rat testis, but there these populations represented mitotically proliferating germ and somatic cells in the G2/M‐phase of cell cycle (Fig. 2D). The diploid (2C) cell population is the most heterogeneous in the adult testis because this population includes not only spermatogonia, but also all the non‐proliferating somatic cells (major cell types are Sertoli cells, Leydig cells, peritubular myoid cells and macrophages). The cells that were found between the 2C and the 4C populations were regarded as proliferating cells at different stages of DNA synthesis. To assess how well the different cell populations were recovered during the sample preparation, PI staining of samples at different time points of rat testicular development was prepared (Fig. 3). The different populations were gated from the FSC/PI density plots and the results were displayed as cells/mg of testis. The assay recapitulated the relative behavior of the cell populations during rat testicular ontogenesis: as the animals age, the relative proportion of the 2C cells (spermatogonia and somatic cells) decreases as the amount of other germ cells increases when meiotic and haploid cells emerge (Fig. 3).
Figure 3.
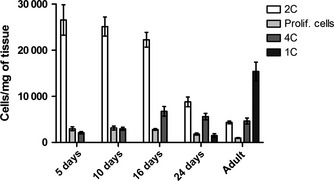
Distribution of cells according to DNA amount in the ontogenical series of rat testis flow cytometry. The population dynamics of cells treated according to the present protocol and analyzed with propidium iodide (PI) recapitulate rat testicular ontogenesis. Diploid (2C), tetraploid (4C) and haploid (1C) were gated using the FSC/PI density plot and PI histogram. N = 5/group.
Cell population analysis with vimentin
We chose to study the behavior of the somatic cell population during rat testis development as an experimental model to test the usefulness of flow cytometry. Vimentin was chosen as a somatic cell marker to dissect the identity of the diploid cell population. The vimentin antibody gave a strong signal in Sertoli cells in the adult testis and a fainter signal was detected in the interstitial cell compartment (Fig. 4A). We labeled the fixed testicular cells with anti‐vimentin antibody and performed subsequently the PI staining (Fig. 4B–D). A large relative amount of vimentin‐positive cells, together with active proliferation was observed in the testis before puberty, and once active spermatogenesis commenced in puberty, the relative amount of somatic cells and their proliferation decreased by the time the animals reached adulthood (Fig. 4C and D). Simultaneously, the relative proportion of the vimentin‐positive cells of all the analyzed testicular cells decreased as the germ cell population expanded when spermatogenesis progressed (Fig. 4B and C).
Figure 4.
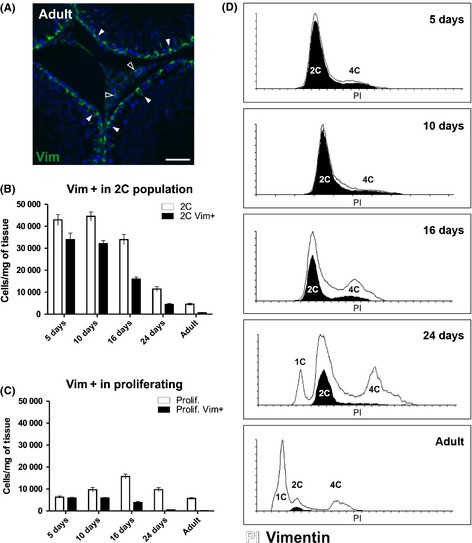
Flow cytometry analysis using vimentin antibody recapitulates somatic cell dynamics during testis development in the rat. (A) Immunofluorescence on paraformaldehyde‐fixed paraffin‐embedded adult rat testis using anti‐vimentin‐Alexa488 (green) and DAPI as a nuclear counter stain. Solid arrowheads = Sertoli cells, open arrowheads = interstitial cells. Scale bar represents 50 μm. (B) Vimentin‐positive cells (black) clustering within the 2C cell population (white) in 5, 10, 16, 24 days and adult rat testis. N = 5/group. C. Vimentin‐positive cells (black) clustering within the proliferating cell population (white) in 5, 10, 16, 24 days and adult rat testis. (D) Representative histograms of propidium iodide and vimentin staining from 5‐, 10‐, 16‐, 24‐day‐old and adult. The proportion of proliferating vimentin‐positive cells (black histogram) was high in 5‐day‐old testis (C and D), but their proportion gradually decreased so that they are absent from adult testes. The majority of the observed 2C cells in the 5‐day‐old testis are vimentin‐ positive(B and D). As spermatogenesis progresses and the number of spermatogonia increases, the relative amount of vimentin‐positive cells in the 2C population decreases (B and D).
γH2AX‐positive testicular cell populations
Phoshorylated γH2AX‐histone protein is associated with both DNA double strand breaks and sex vesicle formation in the testis (Hamer et al., 2003). It is expressed in a subset of the diploid spermatogonia, step 11–14 haploid spermatids and tetraploid spermatocytes (Fig. 5A) (Hamer et al., 2003; Meyer‐Ficca et al., 2005). FxCycle DNA stain was used to detect the DNA content of the cells in the γH2AX‐S139 assay. Most of the 4C cells were positive for γH2AX‐S139 in the adult testis (Fig. 5C and E), while in the 16‐day‐old testis there were more γH2AX‐S139‐negative 4C cells corresponding to the higher relative amount proliferating somatic cells and differentiating spermatogonia (Fig. 5B and C). Some γH2AX‐S139‐positive cells clustered also within the 2C cell population in both time points (Fig. 5B–D). Another major γH2AX‐S139‐positive cell population clustered within the 1C haploid cells in the adult testis, while this signal was absent from the 16‐day‐old testis (Fig. 5B, C and F).
Figure 5.
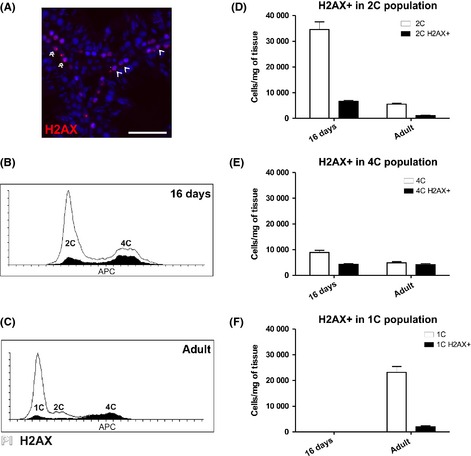
γH2AX‐S139 can be used as a marker for specific germ cell subpopulations in the flow cytometry assay. (A) Immunofluorescence on paraformaldehyde‐fixed paraffin‐embedded 16dpp and adult rat testis using anti‐γH2AX‐S139 (red) and DAPI as a nuclear counter stain. Double arrowhead = 4C cells with γH2AX‐positive sex body. Open arrowhead = γH2AX ‐positive spermatogonia. Scale bar represents 50 μm. (B–D) In the adult testis, γH2AX‐S139‐positive cell are detected in the subpopulations expected based on literature: Fraction of 1C and 2C cells and the majority of 4C cells. No γH2AX‐S139‐positive cells are observed at 16 dpp in the 1C cells population. (B) Representative histogram of 16 dpp rat testis stained with γH2AX‐S139‐antibody (black) and DNA stain (FxCycle, white). (C) Representative histogram of adult rat testis stained with γH2AX‐S139‐antibody (black) and DNA stain (FxCycle, white). (D) γH2AX‐S139‐positive cells (black) clustering within the 2C cell population (white) in 16‐day‐old and adult rat testis. N = 5/group. (E) γH2AX‐S139‐positive cells (black) clustering within the 4C cell population (white) in 16‐day‐old and adult rat testis. N = 5/group. (F) γH2AX‐S139‐positive cells (black) clustering within the 1C cell population (white) in 16‐day‐old and adult rat testis. N = 5/group.
Assay‐induced cell damage and assay reproducibility
To assess the extent of cell death induced by single cell solution preparation, a dead cell dye (LIVE/DEAD Near‐IR) was introduced to the assay. The cells that lost their membrane integrity acquired a bright far‐red fluorescence. Over half of the cells in the heat‐treated positive control samples were positive for the dead cell marker (Figure S2). However, testis treated according to our assay protocol without heat treatment showed only low levels of cell death after the single cell preparation (Figure S2) and, importantly, cell death affected all cell populations quite equally.
The variation between different animals in the data sets was relatively low at all time points (Fig. 6). There was a low intra‐animal variation in both 5‐day‐old and adult animals indicating that the assay is reproducible irrespective to the structure of the tissue (Fig. 6).
Figure 6.
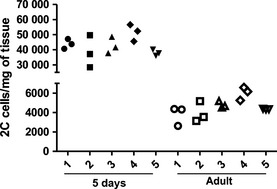
Intra‐ and inter‐animal variation in the 2C population in 5‐day‐old and adult rat is very similar. The number of cells in the 2C population in the parallel samples was plotted for five different animals in both the 5‐day‐old and the adult rats.
Discussion
In this study, we have described a detailed protocol for the rapid analysis of testicular cell populations. This flow cytometry method is readily applicable for several experimental settings within the field of testicular research. It can be used for quick screening of testicular phenotypes of transgenic animals, testicular effects of in vivo drug exposure, behavior of cell dynamics in in vitro cultures of testicular tissue, and in the future for the analysis of human testicular biopsies. Adding to the speed of the analysis is that proliferation and apoptosis of many different testicular cell types can be analyzed simultaneously, which is useful especially when dealing with limited sample amounts. Flow cytometry does not replace the golden‐standard histology and immunohistochemical methods that allow analyzing the cell–cell associations and localization, but can be applied for instance to screen time points for a more targeted analysis. Obtaining histological sections for analysis can take weeks depending on the laboratory, while this flow cytometry‐based assay can be performed within one working day.
The novelty of the present method arises from the use of intracellular staining instead of membrane labeling, and a rigorous validation of the sample preparation, antibody staining and data analysis pipeline using testicular ontogenesis as an experimental setting. A special emphasis was laid on the sample preparation procedure optimization to obtain representative cell suspensions, and to avoid the loss of the more fragile spermatocytes. We tested the effect of different enzyme cocktails and different intensities of mechanical disruption on the extent of cell debris and cell type composition of the samples (data not shown). A well‐known pitfall in analyzing the relative representation of cell populations with flow cytometry arises from changes in cellularity (Elzeinova et al., 2013; Ferguson et al., 2013). When the amount of the input material (weight of tissue) and the volume analyzed (cell concentration) are known, absolute cell numbers per tissue weight can be obtained. In addition, we show a detailed data analysis and troubleshooting pipeline, making the application of this method by others more attainable.
The ratios of the different cell populations during development mirrored the dynamics assessed previously using histomorphometric methods (Clermont & Perey, 1957; Roosen‐Runge & Leik, 1968). With even PI alone, the emergence of the meiotic cells from post‐natal day (PND)16 onwards and appearance of the first haploid germ cells at PND24 in the rat testis could be assessed. However, the major step forward here is the possibility to distinguish somatic cells and germ cells with 2C amount of DNA from each other.
To validate the protocol for intracellular staining in the testis samples, we chose to couple detection of vimentin‐positive cells to the DNA stain assay. Sertoli cells are the major cell type expressing vimentin in the rat testis, but it is also expressed in the interstitial cell compartment in the rat testis (our results, Alam & Kurohmaru, 2014; Kopecky et al., 2005). More precise dissection of the somatic cell population can be achieved by coupling additional marker antibodies (such as Sox9 for Sertoli cells or 3βHSD for Leydig cells) to the assay.
The assay was able to recapitulate the somatic cell dynamics in the developing rat testis. In the post‐natal rat testis, Sertoli cells proliferate until the PND18 (Clermont & Perey, 1957) while Leydig cell proliferation can still be detected at the age of 63 days in the rat (Rijntjes et al., 2009). The germ cell/somatic cell ratio followed the expected trend: at the age of 5 days, a majority of the cells in the testis are somatic cells, while spermatogonia form a small fraction of cells. By the time adult spermatogenesis is fully established, the amount of vimentin‐positive cells in relation to germ cells in the tissue sample was very low as expected. As the Sertoli cells are the major cell type with a strong vimentin signal, it can be concluded that the flow cytometry assay could recapitulate Sertoli cell ontogenesis relatively well despite some interfering signal from the interstitial vimentin‐positive cells. Thus, the assay can be used as a proxy to study Sertoli cell dynamics during testis development.
Phoshorylated γH2AX was chosen to detect different germ cell subtypes in the rat testis samples. A γH2AX‐Ser139 signal can be detected in intermediate and B spermatogonia, zygotene and pachytene spermatocytes and step 11–14 elongated spermatids (Hamer et al., 2003; Meyer‐Ficca et al., 2005). In the adult rat testis, the γH2AX‐positive cell clustered as expected based on the immunohistochemical staining and literature. Strongest γH2AX‐signal was detected in the 4C cells and over 80% of the cells in this population expressed γH2AX corresponding to the expected behavior of the meiotic 4C cells. The small population of γH2AX‐positive 2C cells represented the differentiating spermatogonia. Five percent of all 1C cells were positive for γH2AX showing that even the faint signal from step 11–14 spermatid nuclei could be detected using flow cytometry. In the 16‐day‐old rat testis, there are no spermatids and as expected there were no γH2AX‐positive 1C cells either. A larger proportion of the 2C cells were positive for γH2AX in the 16‐day‐old than in the adult testis which corresponded to the amplification of the intermediate and B spermatogonia at this time‐point (Clermont & Perey, 1957). These results show that the γH2AX assay functions in the rat testis and it could be applied in the future to detect the different testicular germ cell populations.
The flow cytometry‐based assay does not aim to replicate the results from the traditional histomorphometric methods, but provides an additional time‐saving method to analyze testicular tissue. There are several issues contributing to the discrepancy between relative cell counts obtained by flow cytometry vs. histology. Undigested cells are lost from the analysis which can cause biased interpretation of the results. The expected ratio between 4C spermatocytes and Sertoli cells in adult rat testis would be 4.71 spermatocytes for each Sertoli cell (Wing & Christensen, 1982). The 4C to vimentin‐positive cell ratio obtained in this study from the adult rat testis was 7.6, indicating that a portion of the adult Sertoli cells are lost during the sample preparation because of incomplete digestion as expected. A similar phenomenon was observed in the 5‐ and 10‐day‐old testes when compared to reference values from the literature (Clermont & Perey, 1957). Age, species, and testicular fibrosis can contribute to an incomplete digestion and care should be taken when applying this method to experimental groups with very different cell composition. Nevertheless, the assay performed uniformly in testes from very different ages: despite a very different tissue composition and structure, the intra‐ and inter‐animal variations were at a similar level in both 5‐day‐old and adult rats.
We present here a readily applicable protocol for intracellular flow cytometry on testicular tissue. We hope that this protocol and its modifications will accelerate research in the field of male reproduction.
Disclosures
The authors have nothing to disclose.
Author contributions
ER and MN contributed to experimental design, data analysis, and wrote manuscript. SCM conducted majority of the experiments, data analysis, and wrote the manuscript. KJ was involved in experimental design and data analysis. JS provided data acquisition and analysis. JT was involved in experimental design, data analysis, wrote the manuscript and funded the research.
Supporting information
Figure S1. (A) Doublet discrimination gating strategy using FSC‐Area/FSC‐Width dot plots. Black rectangle = cells that are possibly doublets. (B) Setting negative/positive thresholds for gating using unstained and single fluorochrome‐stained controls. Ab = antibody.
Figure S2. The sample preparation protocol induces only little cell death. The amount of cell damage during sample preparation was assessed with the LIVE/DEAD Near‐IR dye. (A) In the positive control (heat treatment‐induced apoptosis) 59.6% of cells were apoptotic (black histogram). White histogram shows the unstained control. (B) An overlay histogram of an adult rat testis sample displaying the DNA staining (PI) profile (white) and the proportion of the damaged cells (black).
Acknowledgments
We wish to thank Ketlin Adel for skillful technical assistance with flow cytometry data acquisition. This work has been funded by the Academy of Finland, the Sigrid Jusélius Foundation and the Turku University Hospital special research grants.
Emmi Rotgers and Sheyla Cisneros‐Montalvo contributed equally to this study.
References
- Alam MS & Kurohmaru M. (2014) Disruption of sertoli cell vimentin filaments in prepubertal rats: an acute effect of butylparaben in vivo and in vitro. Acta Histochem 116, 682–687. doi:10.1016/j.acthis.2013.12.006. [DOI] [PubMed] [Google Scholar]
- Clermont Y & Perey B. (1957) Quantitative study of the cell population of the seminiferous tubules in immature rats. Am J Anat 100, 241–267. doi:10.1002/aja.1001000205. [DOI] [PubMed] [Google Scholar]
- Dovey SL, Valli H, Hermann BP, Sukhwani M, Donohue J, Castro CA, Chu T, Sanfilippo JS, Orwig KE. (2013) Eliminating malignant contamination from therapeutic human spermatogonial stem cells. J Clin Invest 123, 1833–1843. doi:10.1172/JCI65822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elzeinova F, Peknicova J, Ded L, Kubatova A, Margaryan H, Dorosh A, Makovický P, Rajmon R. (2013) Adverse effect of tetracycline and doxycycline on testicular tissue and sperm parameters in CD1 outbred mice. Exp Toxicol Pathol 65, 911–917. doi:10.1016/j.etp.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Ferguson L, How JJ & Agoulnik AI. (2013) The fate of spermatogonial stem cells in the cryptorchid testes of RXFP2 deficient mice. PLoS ONE 8, e77351. doi:10.1371/journal.pone.0077351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamer G, Roepers‐Gajadien HL, van Duyn‐Goedhart A, Gademan IS, Kal HB, van Buul PP & de Rooij DG. (2003) DNA double‐strand breaks and gamma‐H2AX signaling in the testis. Biol Reprod 68, 628–634. [DOI] [PubMed] [Google Scholar]
- Hou M, Stukenborg JB, Nurmio M, Andersson M, Toppari J, Soder O & Jahnukainen K. (2011) Ontogenesis of ap‐2gamma expression in rat testes. Sex Dev 5, 188–196. doi:10.1159/000328822. [DOI] [PubMed] [Google Scholar]
- Kopecky M, Semecky V & Nachtigal P. (2005) Vimentin expression during altered spermatogenesis in rats. Acta Histochem 107, 279–289. [DOI] [PubMed] [Google Scholar]
- Meyer‐Ficca ML, Meyer RG, Jacobson EL & Jacobson MK. (2005) Poly(ADP‐ribose) polymerases: managing genome stability. Int J Biochem Cell Biol 37, 920–926. [DOI] [PubMed] [Google Scholar]
- Michaelis M, Langhammer M, Hoeflich A, Reinsch N, Schoen J & Weitzel JM. (2013) Initial characterization of an outbreed mouse model for male factor (in)fertility. Andrology 1, 772–778. doi:10.1111/j.2047‐2927.2013.00108.x. [DOI] [PubMed] [Google Scholar]
- Nurmio M, Toppari J, Zaman F, Andersson AM, Paranko J, Soder O & Jahnukainen K. (2007) Inhibition of tyrosine kinases PDGFR and C‐kit by imatinib mesylate interferes with postnatal testicular development in the rat. Int J Androl 30, 366–376; discussion 376. doi:10.1111/j.1365‐2605.2007.00755.x. [DOI] [PubMed] [Google Scholar]
- Rijntjes E, Swarts HJ, Anand‐Ivell R & Teerds KJ. (2009) Prenatal induced chronic dietary hypothyroidism delays but does not block adult‐type leydig cell development. Am J Physiol Endocrinol Metab 296, E305–E314. doi:10.1152/ajpendo.90750.2008. [DOI] [PubMed] [Google Scholar]
- Roosen‐Runge EC & Leik J. (1968) Gonocyte degeneration in the postnatal male rat. Am J Anat 122, 275–299. doi:10.1002/aja.1001220208. [DOI] [PubMed] [Google Scholar]
- Strobl H & Knapp W. (2004) Myeloid cell‐associated lysosomal proteins as flow cytometry markers for leukocyte lineage classification. J Biol Regul Homeost Agents 18, 335–339. [PubMed] [Google Scholar]
- Toppari J, Eerola E & Parvinen M. (1985) Flow cytometric DNA analysis of defined stages of rat seminiferous epithelial cycle during in vitro differentiation. J Androl 6, 325–333. [DOI] [PubMed] [Google Scholar]
- Toppari J, Mali P & Eerola E. (1986) Rat spermatogenesis in vitro traced by quantitative flow cytometry. J Histochem Cytochem 34, 1029–1035. [DOI] [PubMed] [Google Scholar]
- Toppari J, Bishop PC, Parker JW & diZerega GS. (1988) DNA flow cytometric analysis of mouse seminiferous epithelium. Cytometry 9, 456–462. doi:10.1002/cyto.990090509. [DOI] [PubMed] [Google Scholar]
- Tuomela J, Gronroos TJ, Valta MP, Sandholm J, Schrey A, Seppanen J, Marjamäki P, Forsback S, Kinnunen I, Solin O, Minn H, Harkonen PL. (2010) Fast growth associated with aberrant vasculature and hypoxia in fibroblast growth factor 8b (FGF8b) over‐expressing PC‐3 prostate tumour xenografts. BMC Cancer 10, 596. doi:10.1186/1471‐2407‐10‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turac G, Hindley CJ, Thomas R, Davis JA, Deleidi M, Gasser T, Karaöz E, Pruszak J. (2013) Combined flow cytometric analysis of surface and intracellular antigens reveals surface molecule markers of human neuropoiesis. PLoS ONE 8, e68519. doi:10.1371/journal.pone.0068519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valli H, Sukhwani M, Dovey SL, Peters KA, Donohue J, Castro CA, Chu T, Marshall GR, Orwig KE. (2014). Fluorescence‐ and magnetic‐activated cell sorting strategies to isolate and enrich human spermatogonial stem cells. Fertil Steril, 102, 566–580. e7. doi: 10.1016/j.fertnstert.2014.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Saen D, Goossens E, Aerts JL, Haentjens P & Tournaye H (2013). Does early cell death cause germ cell loss after intratesticular tissue grafting? Fertil Steril, 99, 1264–1272. e1. doi: 10.1016/j.fertnstert.2012.12.019. [DOI] [PubMed] [Google Scholar]
- Wakeling SI, Miles DC & Western PS. (2013) Identifying disruptors of male germ cell development by small molecule screening in ex vivo gonad cultures. BMC Res Notes 6, 168. doi:10.1186/1756‐0500‐6‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing TY & Christensen AK. (1982) Morphometric studies on rat seminiferous tubules. Am J Anat 165, 13–25. doi:10.1002/aja.1001650103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (A) Doublet discrimination gating strategy using FSC‐Area/FSC‐Width dot plots. Black rectangle = cells that are possibly doublets. (B) Setting negative/positive thresholds for gating using unstained and single fluorochrome‐stained controls. Ab = antibody.
Figure S2. The sample preparation protocol induces only little cell death. The amount of cell damage during sample preparation was assessed with the LIVE/DEAD Near‐IR dye. (A) In the positive control (heat treatment‐induced apoptosis) 59.6% of cells were apoptotic (black histogram). White histogram shows the unstained control. (B) An overlay histogram of an adult rat testis sample displaying the DNA staining (PI) profile (white) and the proportion of the damaged cells (black).