

Abstract
Interleukin 5 (IL‐5) and eosinophils are thought to play an important role in the pathology of asthma. This study evaluated the pharmacokinetics, pharmacodynamics, safety, and tolerability of mepolizumab, a humanized anti‐IL5 IgG1 monoclonal antibody, in development for the treatment of severe eosinophilic asthma. This single‐blind study randomized 35 healthy Japanese male subjects (3:1) to receive either a single mepolizumab intravenous dose (10, 75, 250, or 750 mg) or placebo. Subjects were observed for up to 151 days postdose, depending on the dose administered. Blood samples were collected to measure mepolizumab concentrations, blood eosinophils, IL‐5, and antibodies to mepolizumab. Mepolizumab exhibited dose‐proportional pharmacokinetics. The terminal phase half‐life was 19.7–34.6 days, independent of dose. Higher mepolizumab plasma concentrations were associated with lower blood eosinophil counts. Mepolizumab 75–750 mg reduced blood eosinophils for ≥3 months postdose. Mepolizumab demonstrated a favorable safety profile: of 41 reported adverse events, most were mild in severity and none were serious. No neutralizing antibodies to mepolizumab were detected. Sustained reduction in blood eosinophils after single intravenous mepolizumab doses ≥ 75 mg, along with mepolizumab pharmacokinetics and a favorable tolerability profile in healthy Japanese subjects, provides a solid foundation for future studies with mepolizumab in Japanese patients with asthma.
Keywords: mepolizumab, anti–interleukin 5, blood eosinophils, pharmacodynamics, pharmacokinetics
Interleukin 5 (IL‐5) is produced by a number of cell types and is responsible for the maturation and release of eosinophils from the bone marrow.1
Mepolizumab is a humanized monoclonal antibody that recognizes human IL‐5 with high affinity and specificity, thereby inhibiting binding of IL‐5 to IL‐5 receptors. Mepolizumab has been reported to consistently and significantly reduce peripheral and tissue eosinophils in patients with asthma and in healthy volunteers.2, 3, 4
Asthma is characterized by chronic airway inflammation, bronchial hyperreactivity, and airflow obstruction.4 Eosinophils play a prominent role in airway inflammation in asthma, and are considered a central effector cell in asthma pathogenesis.4 Patients with asthma show increased expression of IL‐5 in bronchoalveolar lavage (BAL) fluid and bronchial biopsy tissue5; the level of IL‐5 in BAL fluid and the bronchial mucosa correlates with disease severity.5, 6, 7 This is of particular interest because IL‐5 promotes the differentiation, recruitment, and survival of eosinophils.6, 8
A treatment strategy that blocks IL‐5, thereby suppressing eosinophilic inflammation, has been shown to have a therapeutic benefit in asthma.2 In a published study in Western patients with refractory eosinophilic asthma and a history of recurrent severe exacerbations, mepolizumab intravenous doses of 75, 250, and 750 mg reduced the number of exacerbations per patient per year by 48% (95%CI, 31%–61%); P < .0001), 39% (95%CI, 19%–54%; P =.0005), and 52% (95%CI, 36%–64%; P < .0001), respectively, compared with placebo.2
The purpose of the current study was to evaluate the pharmacokinetics, pharmacodynamics, safety, and tolerability of single ascending mepolizumab doses administered intravenously to healthy Japanese male subjects.
Methods
Study Subjects
Eligible candidates were healthy Japanese men 20–55 years of age with a body weight of more than 45.0 kg and a body mass index (BMI) between 18.5 and 29.0 kg/m2, with no clinically relevant abnormalities as determined from medical history, physical examination, vital signs, and laboratory tests. This study was conducted in compliance with the Declaration of Helsinki and Good Clinical Practice after obtaining approval from the institutional review board. Written informed consent was obtained from each subject.
Study Design and Treatment
This was a single‐blind, placebo‐controlled, parallel‐group, single ascending dose study in healthy Japanese male subjects (GlaxoSmithKline study number MEA115705; clinicaltrial.gov identifier NCT01471327). The study comprised 4 groups (group 1, 10 mg; group 2, 75 mg; group 3, 250 mg; group 4, 750 mg), with 8 subjects per group. Within each group, subjects were randomized to active drug or placebo in a 3:1 ratio. Intravenous doses were administered as an infusion over approximately 30 minutes. Subjects remained in the clinical research unit for 24 hours after dosing and returned for outpatient visits throughout the study. Subjects were assigned to 1 of the 4 possible groups in accordance with the randomization schedule generated prior to the start of the study using validated internal software (RANDALL). This study was conducted at GSK Medicines Research Unit, Prince of Wales Hospital, Randwick , Australia, in compliance with the Declaration of Helsinki and Good Clinical Practice after obtaining ethics approval from the institutional review board (Bellberry Human Research Ethics Committee, Dulwich, South Australia). Written informed consent was obtained from each subject.
Safety Assessments
Safety assessments included monitoring for all adverse events (AEs), examination of vital signs, electrocardiogram (ECG), and clinical laboratory tests. AE and serious AE (SAE) data were collected from the start of study treatment until the end of follow‐up.
Pharmacokinetic Sampling and Bioanalysis
For the measurement of mepolizumab concentration in plasma, blood samples were taken prior to administration and 0.25, 0.5, 1, 2, 4, 8, 12, and 24 hours after administration on day 1. Additional blood samples were taken on days 4, 6, 8, 15, 29, 43, 57, 85, 121 (groups 3 and 4 only), and 151 (group 4 only).
Blood samples (approximately 2 mL) for determination of plasma concentrations of mepolizumab were collected in plastic lithium heparin tubes and immediately placed on ice. The plasma was separated by refrigerated centrifugation at 4°C (20 minutes at approximately 1500g). Plasma samples (at least 0.5 mL) were transferred to appropriately labeled polypropylene tubes, frozen on solid carbon dioxide (dry ice), and stored at the investigator site at approximately −20°C until shipment for analysis at Alliance Pharma Inc. (Malvern, Pennsylvnia). If samples could not be frozen in solid carbon dioxide, they were placed directly in a −20°C freezer.
Plasma concentrations of mepolizumab were determined using an enzyme‐linked immunosorbent assay method with a lower limit of quantification (LLQ) of 50 ng/mL (data on file). The upper limit of quantification was 5000 ng/mL. A 96‐well plate was coated with mouse idiotypic antibody against mepolizumab overnight. Mepolizumab in calibration standards, quality controls (QCs), and human plasma samples were bound to the capture antibody on the plate. Bound mepolizumab further bound to mouse anti‐human IgG1 (Fc specific) antibody labeled with horseradish peroxidase (HRP), catalyzing the HRP substrate to generate luminescence. The luminescent intensity was proportional to the amount of mepolizumab in standards, QCs, and samples.
The between‐run precision (%CV) was ≤8.1%, and the between‐run accuracy (% bias) ranged from ‐5.6% to ‐1.1%. Plasma mepolizumab concentration–time data were analyzed by noncompartmental methods with WinNonlin Professional version 4.1 (Pharsight Corporation, Mountain View, California) software. Calculations were based on the actual sampling times recorded during the study. From the plasma concentration–time data, the following pharmacokinetic parameters were determined and summarized, as data permitted: maximum observed plasma concentration (Cmax), time to Cmax (tmax), area under the plasma concentration–time curve (AUC0–t and AUC0– ∞), terminal phase half‐life (t1/2), time of last observed quantifiable concentration (tlast), volume of distribution determined by terminal phase (Vz) and at steady state (Vss), and plasma clearance (CL). Exploratory assessment of dose proportionality was evaluated using a power model for AUC0–∞ and Cmax.
Pharmacodynamic Assessment
Blood eosinophil count was measured as part of the standard hematological assessment at the following times: predose; 12 and 24 hours after administration on day 1; and on days 6, 15, 29, 43, 57, 85, 121 (groups 3 and 4 only), and 151 (group 4 only).
Free and total IL‐5 were measured at the following times: prior to the administration of mepolizumab; 1, 12, and 24 hours after administration on day 1; and on days 4, 8, 15, 29, 43, 57, 85, 121 (groups 3 and 4 only), and 151 (group 4 only).
Serum free and total IL‐5 were both measured by validated methods using an electrochemiluminescence (ECL)–based immunoassay on the Meso‐Scale Discovery (MSD) platform as described in detail below.
Serum samples were collected in serum separator tubes (SSTs; no anticoagulant).
The total IL‐5 assay used an anti‐IL‐5 monoclonal antibody for capture and a biotinylated anti‐IL‐5 monoclonal antibody for detection and was designed to provide IL‐5 quantification in the presence of excess mepolizumab. Serum total IL‐5 levels were back‐calculated against a calibration curve of GSK‐provided IL‐5 (data on file).
The free IL‐5 assay used mepolizumab for capture and a rat anti‐IL‐5/Sulfo‐TAG labeled goat anti‐rat antibody combination for detection. The assay was designed to capture IL‐5 that was not complexed with mepolizumab (data on file).
Validation parameters included evaluation of precision, accuracy/matrix, specificity, linearity, and analyte stability. All evaluated parameters were within acceptance criteria of ≤25% CVs and ≤25% REs.
For total IL‐5 and free IL‐5, any data values lower than the LLQ were imputed as zero. The LLQ values for total IL‐5 and free IL‐5 assays were 7.81 and 3.91 pg/mL, respectively.
Immunogenicity Assessment
Samples were assessed for antimepolizumab antibodies, and any positive samples were further tested for antimepolizumab neutralizing antibodies by ECL analysis based on a ligand‐binding immunoassay using the MSD platform as described in detail below. Serum samples were obtained prior to mepolizumab administration and on days 8, 29, 85, 121 (groups 3 and 4 only), and 151 (group 4 only) after administration.
Serum samples were analyzed for the presence of antimepolizumab antibodies and neutralizing antibodies. Blood samples (7 mL) were collected in SSTs (no anticoagulant).
For the validation of the antidrug antibody (ADA) assay for the presence of antimepolizumab antibody analysis, the following parameters were evaluated using purified rabbit antidrug antibodies: assay precision (<25% CV), screening and confirmation cut points, specificity, drug interference, sensitivity (1.03 ng/mL), and stability. For the detection, test samples were mixed with an anti‐IL‐5 blocking antibody and then overnight with biotin and Sulfo‐TAG drug conjugates to allow bridge formation. ADA binding to both conjugates and the streptavidin‐coated MSD plate resulted in an ECL signal directly proportional to the amount of ADAs in the sample.
Statistical Methods
No formal statistical sample size calculation was performed. The sample size was based on feasibility, and a sufficient number of subjects were enrolled into the study to provide 32 evaluable subjects for analysis. Safety and tolerability data were reviewed by the study team and the investigator at the end of each treatment period before progressing to the next dose. In addition, an interim analysis was performed when all subjects at all dose levels had completed the visit on day 57 and most subjects had completed the visit on day 85 to optimize the later development plan. The interim analysis was performed on the safety and pharmacokinetic data. The final analysis was performed after all subjects had completed the study. Placebo data from each cohort were pooled for the purpose of summary statistics. For exploratory assessment of dose proportionality, we applied the power model to log‐transformed AUC0–∞ and log‐transformed Cmax. For each of these parameters, the mixed‐effects model was fitted with loge (dose) as a fixed effect and individual subject intercept fitted as a random effect. Estimates of the mean slopes of loge (dose) were reported along with corresponding 90%CIs. The “all‐subjects population” was defined as all subjects who received a dose of study medication. The “PK population” was defined as subjects in the “all‐subjects population” from whom a PK sample was obtained and analyzed. The “PD population” was defined as all subjects in the “all‐subjects population” who had a baseline pharmacodynamic measurement and at least 1 posttreatment PD measurement.
Results
Study Population
Thirty‐five healthy Japanese male volunteers were enrolled in this study (Supplementary Figure 1) and randomized to treatment groups. Thirty‐three subjects completed the study. Two subjects (1 in the placebo group and 1 in the mepolizumab 250 mg group) withdrew consent. There were no subjects for whom the treatment blind (to subjects, single blind) was broken during the study. Twenty‐six subjects who were randomized to 10, 75, 250, or 750 mg of mepolizumab were included in the PK population. All 35 randomized subjects were included in the PD population.
Demographic characteristics were comparable with respect to age, BMI, weight, and height in the 5 treatment groups (Supplementary Table 1). Subjects were aged 24.5 to 33.7 years, whereas mean body weight ranged from 60.55 to 70.33 kg, and BMI ranged from 20.63 to 23.42 kg/m2 across the treatment groups.
Pharmacokinetics
The mean and individual plasma mepolizumab concentrations decreased in a biexponential manner following a 30‐minute continuous infusion with mepolizumab (Figure 1). Median values for tmax ranged from 0.5 to 2.5 hours (Table 1). Cmax and AUC0–∞ increased with dose after administration of mepolizumab with an intersubject variability (%CV) ranging from 9.4% to 14.6% and 9.0% to 22.5%, respectively. Mean values for Vz, Vss, and CL were similar across the dose range studied, 4.38–7.85 L, 4.40–6.52 L, and 6.19–7.87 mL/h, respectively. The %CV ranged from 10.7% to 36.7% for Vz, 10.8% to 23.9% for Vss, and 8.6% to 21.3% for CL.
Figure 1.
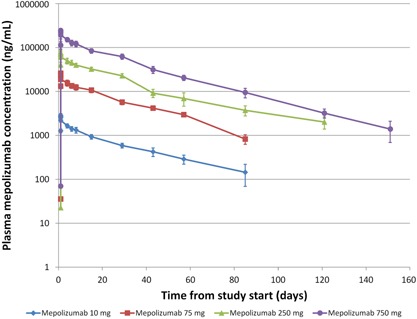
Mean (± SD) plasma mepolizumab concentration–time profile following single intravenous dose of mepolizumab. SD, standard deviation.
Table 1.
Summary of Selected Plasma Mepolizumab Pharmacokinetic Parameters Following Single‐Dose Administration
| Mepolizumab | ||||
|---|---|---|---|---|
| Parameter (unit) | 10 mg (n = 6) | 75 mg (n = 6) | 250 mg (n = 7) | 750 mg (n = 7) |
| AUC0–∞ (μg · day/mL) | 54.63 (12.27) | 493.36 (41.07) | 1698.66 (172.17) | 4495.64 (413.79) |
| AUC0–t (μg · day/mL) | 47.99 (6.47) | 469.14 (42.77) | 1460.74 (270.47) | 4448.41 (400.21) |
| Cmax (μg/mL) | 2.87 (0.27) | 26.46 (1.81) | 79.26 (11.60) | 253.65 (28.28) |
| tmax (day) | 0.042 (1 a ) (0.02–0.04) | 0.104 (2.5 a ) (0.04–0.17) | 0.042 (1 a ) (0.02–0.08) | 0.021 (0.5 a ) (0.02–0.33) |
| tlast (day) | 84.0 (83.0–84.0) | 84.0 (84.0–84.2) | 119.0 (23.1–121.1) | 150.0 (143.0–150.1) |
| t1/2 (day) | 27.43 (10.36) | 19.80 (2.42) | 36.14 (11.30) | 22.65 (2.32) |
| Vz (L) | 7.04 (1.08) | 4.38 (0.82) | 7.85 (2.88) | 5.47 (0.58) |
| Vss (L) | 6.52 (0.77) | 4.40 (0.69) | 5.65 (1.35) | 4.98 (0.54) |
| CL (mL/h) | 7.87 (1.68) | 6.37 (0.55) | 6.19 (0.63) | 7.01 (0.74) |
All parameters are indicated as mean (standard deviation) except for tmax and tlast, which are indicated as median (range).
Percent extrapolated for AUC0–∞ was less than 20%.
The units in parentheses are hours.
The mean t1/2 ranged from 19.8 to 36.1 days across the doses investigated, with a %CV ranging from 10.3% to 37.8%.
An exploratory analysis of the dose proportionality for AUC0–∞ and Cmax was conducted. The slope (90%CI) for loge‐transformed AUC0–∞ was 1.0284 (0.9970–1.0599) and for Cmax was 1.0279 (1.0014–1.0543). The lower limit of the 90%CI was slightly greater than 1 for Cmax.
Pharmacodynamics
Blood eosinophil counts
Mean blood eosinophil counts decreased to approximately half of baseline by 24 hours postdose in the mepolizumab treatment groups. There was a dose‐related decrease in the postbaseline/baseline ratio for blood eosinophils, with the 10‐mg dose showing lower reduction in blood eosinophils compared with the other doses at day 29 (Table 2). There was no reduction in the blood eosinophil postbaseline/baseline ratio in the placebo group during the study. Maximum decrease in the geometric mean of the blood eosinophil postbaseline/baseline ratio was achieved on day 6 for mepolizumab 10 mg, on day 29 for mepolizumab 75 and 250 mg and on day 43 for mepolizumab 750 mg. We noted a dose‐dependent duration for maximal blood eosinophil postbaseline/baseline ratio reduction; higher doses were associated with later return toward baseline, and only the 10‐mg group had reached baseline by day 85. In all the other groups (75, 250, and 750 mg), mean blood eosinophil count had not returned to baseline by the time of the last measurement (days 85, 121, and 151, respectively). Following administration of mepolizumab 10 mg, the maximum reduction (65%) in mean blood eosinophil count was reached by day 6, after which values gradually returned to slightly above baseline on day 85. Following administration of mepolizumab 75 and 250 mg, the maximum reduction (55%–75%) in mean blood eosinophil count was reached by day 6 and sustained up to day 57, after which values gradually returned toward baseline. Following administration of mepolizumab 750 mg, the maximum reduction (80%–85%) in mean blood eosinophil count was reached by day 6 and sustained up to day 85, after which values gradually returned toward baseline.
Table 2.
Mean (SD) Profile of the Postbaseline/Baseline Ratio in Blood Eosinophil Data
| Mepolizumab | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 10 mg | 75 mg | 250 mg | 750 mg | Placebo | ||||||
| n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | |
| Day 1 (12 h) | 6 | 1.05852 (0.533547) | 6 | 1.4518 (1.837609) | 7 | 1.00341 (0.21429) | 7 | 0.86561 (0.159091) | 9 | 1.34826 (0.479147) |
| Day 1 (24 h) | 6 | 0.5505 (0.157661) | 6 | 0.52597 (0.115643) | 7 | 0.57413 (0.16816) | 7 | 0.43741 (0.044098) | 9 | 1.10145 (0.26215) |
| Day 6 (120 h) | 6 | 0.37973 (0.124193) | 6 | 0.36625 (0.173148) | 7 | 0.43853 (0.174446) | 7 | 0.2035 (0.112465) | 9 | 1.01413 (0.413268) |
| Day 15 (336 h) | 6 | 0.45033 (0.190448) | 6 | 0.26483 0.098609) | 7 | 0.35593 (0.208822) | 7 | 0.19301 (0.116505) | 9 | 1.08557 (0.240293) |
| Day 29 (672 h) | 6 | 0.53628 (0.218912) | 6 | 0.27947 (0.177709) | 6 | 0.26021 (0.118443) | 7 | 0.18892 (0.130089) | 9 | 1.13588 (0.272369) |
| Day 43 (1008 h) | 5 | 0.80807 (0.214577) | 6 | 0.34136 (0.169534) | 6 | 0.26285 (0.143955) | 7 | 0.14869 (0.129909) | 8 | 0.94097 (0.282451) |
| Day 57 (1344 h) | 6 | 1.0012 (0.309113) | 6 | 0.29961 (0.157891) | 6 | 0.29338 (0.161407) | 7 | 0.14372 (0.091606) | 8 | 0.96401 (0.314589) |
| Day 85 (2016 h) | 6 | 1.07912 (0.43946) | 6 | 0.45509 (0.143149) | 6 | 0.37915 (0.18329) | 7 | 0.17998 (0.108718) | 8 | 0.89772 (0.341836) |
| Day 121 (2880 h) | 6 | 0.59373 (0.098405) | 7 | 0.2993 (0.156555) | 4 | 0.79153 (0.172963) | ||||
| Day 151 (3600 h) | 7 | 0.51711 (0.295935) | 2 | 1.09524 (0.673435) | ||||||
SD, standard deviation.
Free and total IL‐5
At baseline, serum total IL‐5 concentrations (free IL‐5 and IL‐5 bound to mepolizumab) were below the LLQ in all but 3 subjects. In the placebo group, serum total IL‐5 concentrations remained essentially unchanged during the study. In contrast, at all doses of mepolizumab, mean serum total IL‐5 concentrations increased from baseline postdosing in a dose‐dependent manner (Figure 2).
Figure 2.
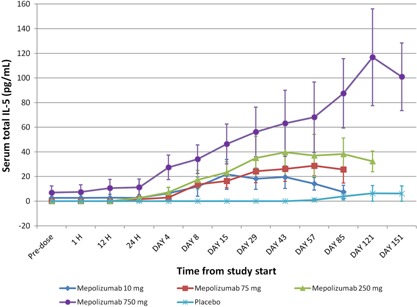
Mean (± SE) profile of total IL‐5 data. SE, standard error.
At baseline, serum‐free IL‐5 concentrations were below the LLQ in all but 1 subject. In the placebo group, serum‐free IL‐5 concentrations remained below the LLQ in all subjects during the study. In the 750‐mg group (n = 7), serum‐free IL‐5 concentrations were measurable in 1 subject on day 1 to day 8, in 2 subjects on days 29, 43, and 57, in 3 subjects on day 85, and in 5 subjects on days 121 and 151. For all groups and all times taken together, the serum‐free IL‐5 concentrations ranged from LLQ to 123.86 pg/mL. Serum‐free IL‐5 concentrations were measurable in most of the subjects at the later sampling points in the 750‐mg group, but remained below the LLQ in almost all subjects in the 10‐, 75‐, and 250‐mg groups during the study.
Immunogenicity
In the 10‐mg group, ADAs were detected in 3 subjects on day 8. One of these 3 subjects also tested positive for ADAs on day 29. No ADAs were detected after day 29. One subject in the 75‐mg group and 1 subject in the 250‐mg group tested positive for ADAs on day 8; both subjects tested negative at the later times. No ADAs were detected in any subjects in the 750‐mg or placebo group up to day 151 postdose. None of the samples that tested ADA positive tested positive for neutralizing activity.
Safety and Tolerability
Adverse events
No deaths or other SAEs were reported during the study. A total of 41 AEs were reported in 20 of the 35 subjects during the study. The most frequently reported AEs across the treatment groups, irrespective of causality, were headache and upper respiratory tract infection. The majority of AEs were reported as mild in severity.
The overall incidence of AEs was similar between the mepolizumab 750‐mg and placebo groups (71% and 67%, respectively) and slightly lower in the mepolizumab 10‐, 75‐, and 250‐mg groups (50%, 50%, and 43%, respectively).
Sixteen drug‐related AEs were reported in 13 subjects across the treatment groups. The most common drug‐related AE was headache. All drug‐related AEs were mild in severity, and all subjects recovered during the study.
Laboratory parameters, vital signs, and electrocardiograms
No consistent trends, changes, or differences between placebo and treatment groups were noted in clinical laboratory values (ie, hematology, chemistry, and urinalysis) except for eosinophil count. No hematology, chemistry, or urinalysis findings were associated by the investigator to an AE. There were no clinically relevant trends in vital signs during the study, and all ECG parameters remained within the normal range.
Discussion
In this study conducted in healthy Japanese male subjects, we administered single intravenous doses of mepolizumab to characterize the drug's PK, PD, safety, and tolerability and to support the identification of an effective dose for Japanese patients with severe asthma. The PK results showed dose‐proportional increases in AUC and Cmax following single intravenous infusions of mepolizumab over the range of 10–750 mg. Mean values for Vz, Vss, and CL were similar across the dose range, suggesting mepolizumab PK was linear in healthy Japanese subjects. In a previous phase 1 study conducted in healthy non‐Japanese volunteers,3 mean AUC0–∞ and Cmax were similar to our findings following administration of a single intravenous dose of mepolizumab 250 mg. The mean t1/2 values in the 75‐ and 750‐mg groups in this study were relatively similar (20 and 23 days, respectively) to the previous phase 1 study,3 but the mean t1/2 (36 days) following administration of mepolizumab 250 mg was longer than in the previous phase 1 study (18.5 days). 3 The t1/2 variability was higher at 250 mg than at 75 and 750 mg in this study. The reason for the longer t1/2 in the 250‐mg group is not fully understood, although the greater variability in this cohort of 7 subjects is likely a contributing factor. Our results indicate that there are no apparent differences in the PKs of mepolizumab between Japanese and Western individuals.
In line with the established mechanism of action of mepolizumab, a reduction in mean blood eosinophil counts to approximately half of baseline values by 24 hours was observed postdose in the mepolizumab treatment groups, and approximately maximum reduction was achieved by day 6. We found that the time to return toward baseline for the mean blood eosinophil count was dose related. The 10‐mg group differentiated from the other dose groups by returning toward baseline as early as day15 and was the only group to have returned to baseline by day 85. A decrease (at least 50% from baseline) in blood eosinophil counts following mepolizumab intravenous administration of 250 mg was also observed in healthy Western individuals.3 These results suggest that a single dose of mepolizumab in the range of 75–750 mg is able to sustain a maximum reduction in blood eosinophils in healthy volunteers for at least 2 months after a single administration. This sustained duration in maximum reduction was not achieved with mepolizumab 10 mg.
Serum‐free IL‐5 concentrations were not measurable at baseline in most subjects and was therefore not a useful PD end point for providing evidence of pharmacology in healthy volunteers. By contrast, and as expected, the mean serum total IL‐5 concentrations increased in a dose‐dependent manner from baseline after mepolizumab dosing. As the total IL‐5 assay detects both free IL‐5 and mepolizumab bound to IL‐5, the increase in serum total IL‐5 most likely reflects the binding of mepolizumab to IL‐5 in circulation. This would reduce IL‐5 clearance through the IL‐5 endogenous receptors by blocking receptor‐mediated endocytosis.9, 10
No deaths or other SAEs were reported after administration of single intravenous doses of mepolizumab up to 750 mg. Of 41 reported AEs, most were mild in severity. The observed AE profile was consistent with results from the overall clinical development experience.2, 11
We found a low incidence (18%, or 6 of 33) and low titers of ADAs after a single intravenous infusion of mepolizumab in this population. There was no correlation between the presence of ADAs and clinical events. Furthermore, all samples were negative for neutralizing antibodies. It is important to highlight that this observation was limited to a single dose.
These findings warrant further investigation of mepolizumab in a population of Japanese patients with asthma.
In summary, the PK profile suggests a dose‐proportional increase and linearity following a single intravenous infusion over the range of 10–750 mg in healthy Japanese subjects. There were no apparent differences in the PKs of mepolizumab between Japanese and Western individuals, on comparison with previous studies. Blood eosinophil reduction was observed and provided evidence of pharmacology following mepolizumab administration. Mepolizumab demonstrated a favorable safety profile in healthy Japanese male subjects.
Funding
This study was funded by GSK (MEA115705).
Declaration of Conflicting Interests
N. Tsukamoto, N. Takahashi, H. Itoh, and I. Pouliquen are employees of GlaxoSmithKline; N. Tsukamoto and I. Pouliquen hold stock in GlaxoSmithKline. Editorial support for preparation of the initial manuscript draft was provided by Asklep and was funded by GlaxoSmithKline. Editorial support in the form of copyediting was provided by Makoto Hirai, PhD, at Asklep (Tokyo, Japan) and Ian Grieve, PhD, at Gardiner‐Caldwell Communications (Macclesfield, United Kingdom) and was funded by GlaxoSmithKline.
Supporting information
Supplementary Figure 1. Study subject disposition.
Supplementary Table S1.
Acknowledgments
This study was funded by GlaxoSmithKline (GSK study number MEA115705). Employees of the sponsor participated in the conception and design of the study and the analysis and interpretation of the data. The authors acknowledge the following individuals for their contributions to this study and/or critical review during the development of this article: Shigeru Nohda, Toshiyasu Hirama, Hector Ortega, Steven Yancey, Deborah Templeton, Thomas Lee, Shbana Ahmad, Aimee Lomas, Simon Cozens, Amy Loercher, Jennifer Yuan, Paul Thomas, Kylie Riddell, Cheryl Friend, and Franziska Loehrer.
References
- 1. Greenfeder S, Umland SP, Cuss FM, Chapman RW, Egan RW. Th2 cytokines and asthma–the role of interleukin‐5 in allergic eosinophilic disease. Respir Res. 2001; 2(2):71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pavord ID, Korn S, Howarth P, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double‐blind, placebo‐controlled trial. Lancet. 2012; 380(9842):651–659. [DOI] [PubMed] [Google Scholar]
- 3. Smith DA, Minthorn EA, Beerahee M. Pharmacokinetics and pharmacodynamics of mepolizumab, an anti‐interleukin‐5 monoclonal antibody. Clin Pharmacokinet. 2011; 50(4):215–227. [DOI] [PubMed] [Google Scholar]
- 4. Wardlaw AJ, Brightling C, Green R, Woltmann G, Pavord I . Eosinophils in asthma and other allergic diseases. Br Med Bull. 2000; 56(4):985–1003. [DOI] [PubMed] [Google Scholar]
- 5. Hamid Q, Azzawi M, Ying S, et al. Expression of mRNA for interleukin‐5 in mucosal bronchial biopsies from asthma. J Clin Invest. 1991; 87(5):1541–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clutterbuck EJ, Hirst EM, Sanderson CJ. Human interleukin‐5 (IL‐5) regulates the production of eosinophils in human bone marrow cultures: comparison and interaction with IL‐1, IL‐3, IL‐6, and GMCSF. Blood. 1989; 73(6):1504–1512. [PubMed] [Google Scholar]
- 7. Humbert M, Corrigan CJ, Kimmitt P, Till SJ, Kay AB, Durham SR. Relationship between IL‐4 and IL‐5 mRNA expression and disease severity in atopic asthma. Am J Respir Crit Care Med. 1997; 156(3 Pt 1):704–708. [DOI] [PubMed] [Google Scholar]
- 8. Wang JM, Rambaldi A, Biondi A, Chen ZG, Sanderson CJ, Mantovani A. Recombinant human interleukin 5 is a selective eosinophil chemoattractant. Eur J Immunol. 1989; 19(4):701–705. [DOI] [PubMed] [Google Scholar]
- 9. Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008; 84(5):548–558. [DOI] [PubMed] [Google Scholar]
- 10. Tabrizi MA, Tseng CML, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today. 2006; 11(1–2):81–88. [DOI] [PubMed] [Google Scholar]
- 11. Ortega H, Yancey S, Cozens S. Pharmacokinetics and absolute bioavailability of mepolizumab following administration at subcutaneous and intramuscular sites. Clin Pharmacol Drug Dev. 2014; 3(1):57–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Study subject disposition.
Supplementary Table S1.