

Abstract
Background
This study aimed to explore the preventive effects of gallic acid (GA) on the toxicity induced by NiSO4 in Beas-2B cells.
Material/Methods
Beas-2B cell viability was measured by MTT assay. The degree of oxidative stress was detected by measuring the levels of reactive oxygen species (ROS) and lipid peroxide (LPO). The rate of apoptosis was measured by flow cytometry. Ras/ERK-related protein levels were analyzed by Western blot analysis, which including Ras, ERK, c-Myc, PARP, and PARP cleavage.
Results
MTT assay showed that NiSO4 induced cytotoxicity, while GA had a protective role against toxicity. Additionally, GA could reduce the apoptotic cell number and the level of ROS in Beas-2B cells induced by NiSO4. Western blot analysis demonstrated that NiSO4 could up-regulate the related protein in the Ras/ERK signaling pathway. Furthermore, we observed that GA could alleviate the toxicity of NiSO4 through regulating protein changes in the Ras/ERK signaling pathway.
Conclusions
Preventive effects of GA on NiSO4-induced cytotoxicity in Beas-2B cells may be through the Ras/ERK signaling pathways.
MeSH Keywords: Gallic Acid, Nickel, Preventive Medicine
Background
Nickel compounds are well-known carcinogens that have been identified in industrial environments, such as plating, aluminum coloring, battery production, catalyst, etc. Nickel compounds can enter the body through inhalation, ingestion, and dermal absorption, and inhalation is a main route to cause respiratory disease [1]. Both in vitro and in vivo experiments have demonstrated that respiratory distress and lung and nasal cancer are the common adverse health effects of exposure to nickel compounds [2–4]. A large body of evidence indicates that soluble nickel compounds such as nickel sulfate (NiSO4) and nickel chloride (NiCl2) may cause the cellular production of reactive oxygen species (ROS) and lipid peroxide (LPO) [5–7]. Therefore, the potential harm of nickel compounds for occupational workers cannot be neglected. However, the molecular mechanism and critical signaling pathways by which nickel compounds induce cytotoxicity, oxidative damage, and apoptosis still remain elusive.
Gallic acid (3,4,5-trihydroxy benzoic acid, GA) is a kind of polyphenol compound in nature with high medicinal value, which widely exists in grapes, pomegranates, black tea, and traditional Chinese medicine, such as Moutan, dogwood, gallnut, saxifrage, etc. [8,9]. GA is known to have multiple pharmacological functions, such as anti-carcinogenic, anti-hepatitis B virus, anti-HIV, trypanocidal activity, anti-inflammatory, antibacterial, and antiviral [10–12]. However, the potential protective effect of GA against nickel compounds’ toxicity has not been investigated. This study was conducted to explore cytotoxicity, oxidative stress, and apoptosis induced by NiSO4 in vitro culture of human bronchial epithelial Beas-2B cells. The potential preventive effect of GA against NiSO4-induced toxicity was further researched. In the present study, we chose human bronchial epithelial Beas-2B cells. Since the bronchial epithelium is an important protective barrier against inhalable particle matter, and inhalation is the main way for humans to be exposed to nickel compounds, the Beas-2B cell line is a suitable model to research the toxic effect of nickel compounds.
The Ras/ERK signaling pathway family includes Ras, MEK, ERK, c-Myc, etc. c-Myc is a member of the Myc family of transcription factors, and it is frequently overexpressed in abnormal cells [13]. By modifying the expression of target gene, c-Myc leads to a variety of biological effects, including regulation of cell growth, promotion of cell proliferation, apoptosis, stem cell self-renewal, and DNA damage response [14]. PARP is a chromatin-associated enzyme that is very important for the stability and survival of cells. PARP cleavage is thought to be an important marker of apoptosis, and it is also generally considered to be a marker of caspase-3 activation [3]. Therefore, we chose Ras, ERK, c-Myc, PARP, and PARP cleavage in the Ras/ERK pathway to carry out the experiment.
As far as we know, this study is the first to report the potential protective effect of GA against NiSO4-induced cell injury through the Ras/ERK signaling pathway.
Material and Methods
Reagents and antibodies
Nickel sulfate (NiSO4) (Product No.: 851028, purity≥98%) was obtained from Qingong Chemical Plant in Shanghai, China, and gallic acid (GA) (Lot No.: M0116A, purity >98%) was bought from Meilun Biological Company in Dalian, China. The reagent kits for ROS, lactate dehydrogenase (LDH), malondialdehyde (MDA), and glutathione (GSH) were purchased from Jiancheng Biosciences, China. Annexin V-FITC Apoptosis Detection Kit I was purchased from BD Biosciences, USA. The following antibodies were used: Ras, phosphor-ERK1/2, ERK1/2, phosphor-c-Myc, c-Myc, PARP, PARP cleavage antibody, β-actin antibody, and secondary antibody (Cell Signaling Technology, USA). Manumycin A (Ras inhibitor) was from Abcam, USA, and PD98059 (ERK inhibitor) was from Tocris Bioscience, UK. PhosSTOP was from Roche Bioscience, Germany. All other reagents were purchased from Sigma Chemical Company in the USA unless otherwise specified.
Cell culture conditions
Human bronchial epithelial Beas-2B cells were obtained from the Cell Bank of the Chinese Academy of Sciences. Beas-2B cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Corning). The culture medium contained 10% fetal bovine serum (FBS, Gibco) and 1% penicillin/streptomycin. Cells were maintained at 37°C as monolayers in a humidified atmosphere containing 5% CO2.
MTT assay
The viability of Beas-2B cells was measured by MTT assay as described by Ahamed et al. with some specific modification [15].
Cellular reactive oxygen species content determination
The determination of cellular ROS content was strictly carried out according to the kit instructions. In summary, Beas-2B cells were seeded into 24-well cell culture plates at a concentration of 5×104 cells/well. After treatment, cells were collected and incubated with 1 mL of DCFH-DA at 37°C for 20 min. In addition, this experiment needed to set a positive control. The cells were washed 3 times with DMEM and observed under laser scanning confocal microscope at once (Leica TCS SP5, Germany). The chemical fluorescence method was used to detect the activity of ROS in different concentrations of GA for 24 h.
Cellular glutathione, lactate dehydrogenase, and malondialdehyde content determination
The determination of cellular LDH, MDA, and GSH content was strictly carried out according to the kit instructions. After treatment, cells were evaluated by Microplate Reader (Thermo Fisher Scientific, USA).
Flow cytometry to examine the apoptosis
The apoptosis rate was detected by Annexin V-FITC Apoptosis Detection Kit I. In summary, after treatment, Beas-2B cells were washed twice with pre-cooling PBS and added to a flow cytometry tube with 500 μL of Annexin binding buffer; then 5 μL of Annexin-V-FITC and 5 μL of PI were added to each tube. Finally, cells were gently mixed with avoidance of light at room temperature for 15 min. The apoptosis rate was examined using FACSCalibur (BD Biosciences, USA).
Western blot analysis
The extraction method for total protein and Western blot were introduced in a previous study. In summary, after treatment, cells were washed twice with PBS and split in a cell lysis buffer containing 1 mM PMSF and 1 mM PhosSTOP on ice for 50 min. Then, the cell suspension was centrifuged for 10 min at 12,000 rpm at 4°C, and supernatant was collected. The total protein concentration was determined by using the BCA Protein Assay kit. Protein (20 μg-40 μg) of each group lysate was separated using 8–10% (w/v) SDS-polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membrane. The membranes were blocked with 5% non-fat milk in tris-buffered saline with Tween 20 (TBST) for 30 min at 37°C, and incubated with rabbit-polyclonal primary antibodies against RAS (1: 1000), p-ERK1/2 (1: 1000), ERK1/2 (1: 1000), p-c-Myc (1: 1000), c-Myc (1: 1000), PARP (1: 1000), PARP cleavage (1: 1000), and β-actin (1: 1000) overnight at 4°C. In the next step, the blots were washed three times with TBST, followed by 30 min of incubation with appropriate goat anti-rabbit alkaline phosphatase (AP) conjugated secondary antibody (1: 2000). The antibody-reactive bands were monitored by stabilized substrate for alkaline phosphatase and its densitometry was quantified using ChemiAnalysis image analysis software (Clinx Science Instruments, China).
Statistical analysis
Each experiment was performed at least three times, and representative data are shown. Statistical significance was determined by one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test, and all statistical analysis was carried out using GraphPad Prism Software. P<0.05 was considered to be significant.
Results
Effect of GA on cell viability
Figure 1 shows the cell viability of Beas-2B cells exposed to GA (10 μM-200 μM) for 24 h. Results suggested that GA in the concentration range of 10 μM-150 μM did not decrease significant numbers of living cells (P>0.05). A higher concentration of GA (200 μM) induced 23.58% reduction in cell viability (P<0.05). The safe doses of GA (10–150 μM) were further used to research its protective effect.
Figure 1.
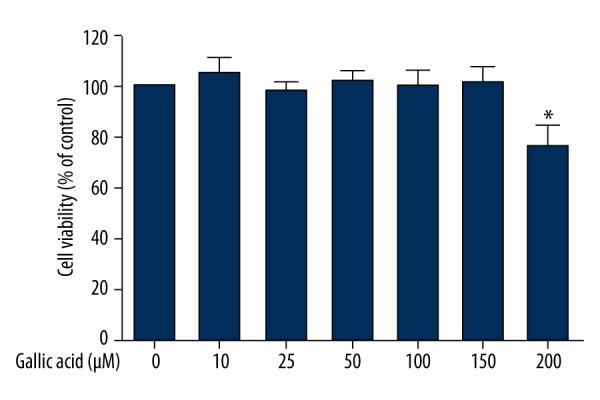
Effect of GA on cell viability. Beas-2B cells were exposed to different concentrations of GA for 24 h, and cell viability was determined by MTT assay as described in the Material and Methods section. Data represent mean ±SD of three independent experiments made in three replicates. * P<0.05, compared with control group.
Effect of GA on NiSO4-induced apoptosis
The potential of GA to prevent the apoptosis induced by NiSO4 in Beas-2B cells was examined. Cells were exposed to NiSO4 at the concentration of 500 μM for 24 h in the presence of GA (10–150 μM). As shown in Figure 2, flow cytometry analysis indicated that co-exposure to GA (10–150 μM) had a significant preventive effect on apoptosis induced by NiSO4.
Figure 2.

Effect of GA on NiSO4-induced apoptosis. Beas-2B cells were exposed to 500 μM NiSO4 for 24 h in the presence or advance of GA (0–150 μM), and apoptosis was determined by flow cytometry analysis as described in the Material and Methods section. (A) Control group. (B) 500 μM NiSO4. (C) 10 μM GA + 500 μM NiSO4. (D) 25 μM GA + 500 μM NiSO4. (E) 50 μM GA + 500 μM NiSO4. (F) 100 μM GA + 500 μM NiSO4. (G) 150 μM GA + 500 μM NiSO4. Data represent mean ±SD of three independent experiments made in three replicates. * Significant preventive effect of different concentrations of GA on reduction of oxidative stress caused by NiSO4 (P<0.05).
Effect of GA on NiSO4-induced oxidative stress
To investigate the protective effect of GA on NiSO4-induced oxidative stress, Beas-2B cells were first treated with GA (10 μM-150 μM) for 24 h and then incubated with 500 μM NiSO4 for 24 h. We observed that co-exposure to GA significantly inhibited the oxidant (ROS, LDH, and MDA) induction and antioxidant (GSH) reduction in Beas-2B cells exposed to NiSO4 (P<0.05) (Figure 3A–3D). The fluorescence intensity of Beas-2B cells treated with GA (10 μM, 50 μM, and 150 μM) for 24 h before NiSO4 infection decreased compared with the only 500 μM NiSO4-infected group (Figure 4).
Figure 3.

Effect of GA on NiSO4-induced oxidative stress. Beas-2B cells were exposed to 500 μM NiSO4 for 24 h in the presence or advance of GA (0–150 μM), and the cells’ oxidative stress parameters were determined according to the kit instructions as described in the Material and Methods section. (A) ROS, (B) LDH, (C) MDA, and (D) GSH. Data represent mean ±SD of three independent experiments made in three replicates. * Significant preventive effect of different concentrations of GA on reduction of oxidative stress caused by NiSO4 (P<0.05).
Figure 4.

Effect of GA on NiSO4-induced reactive oxygen species fluorescence intensity. The expression of reactive oxygen species fluorescence intensity was observed by laser confocal microscopy. (A) Control group. (B) 500 μM NiSO4. (C) 10 μM GA+500 μM NiSO4. (D) 50 μM GA+500 μM NiSO4. (E) 150 μM GA+500 μM NiSO4. The original magnification is 20×.
Effect of GA on NiSO4-induced Ras/ERK signaling pathway protein expressions
Western blot analysis showed that the Ras protein level was increased by NiSO4 in Beas-2B cells after 24 h of exposure. Further, along with the concentration of Ras increasing, the levels of p-ERK1/2, c-Myc, and PARP cleavage also increased while PARP showed the opposite expression. We also observed that PARP cleavage, a marker of cell apoptosis, was increased by NiSO4 (Figure 5A). Additionally, to research whether the increased c-Myc protein was regulated by the Ras/ERK pathway, cells were treated with certain inhibitors for 1 h prior to the time that cells were exposed to NiSO4 (500 μM) for 24 h. The results showed that both Ras and ERK inhibitors decreased NiSO4-induced c-Myc protein (Figure 5B).
Figure 5.

Effect of GA on NiSO4-induced Ras/ERK signaling pathway protein expressions. Shown are expression levels of Ras/ERK signaling pathway proteins after Beas-2B cells were treated with NiSO4 alone or NiSO4 + GA. (A) NiSO4 regulated Ras/ERK signaling protein expressions in Beas-2B cells. Cells were exposed to different concentrations of NiSO4 for 24 h; effect of GA on NiSO4-induced Ras/ERK signaling pathway protein expressions. Cells were exposed to 500 μM NiSO4 for 24 h in the presence or advance of GA (0–150 μM). (B) Beas-2B cells were not treated or treated with Ras inhibitor manumycin A (10 μM) or ERK inhibitor PD98059 (20 μM) for 1 h, and then with 500 μM NiSO4 for 24 h. The protein expression levels were determined by Western blotting analysis as described in the Material and Methods section.
GA’s depressive effect on NiSO4-induced Ras, p-ERK1/2, c-Myc, and PARP cleavage protein up-regulation was observed in the groups co-exposed to GA (10 μM-150 μM), especially in the 100 μM and 150 μM co-exposure GA groups (P<0.05) (Figure 5A).
Discussion
The present study aimed to identify the molecular mechanism and critical signaling pathways by which GA produces a potential protective effect against NiSO4 toxicity. The results from our study suggest that NiSO4 induced cytotoxicity, oxidative stress, and apoptosis through the Ras/ERK pathway in human bronchial epithelial Beas-2B cells. Addition of GA at the certain safe doses obviously inhibited NiSO4-induced cell injury and also reduced the expression of Ras/ERK signaling protein, particularly in the 100 μM and 150 μM co-exposure GA groups.
It is well known that the body is in the equilibrium state of oxidation and antioxidation under normal physiological conditions. Studies in recent years, however, have shown that nickel ions through the Fenton reaction cause an oxidative stress reaction in the body, generating ROS and producing adverse biological effects in cells, including cell membrane damage and apoptosis [16]. LPOs, including MDA, LDH, and the others, can reflect the change of oxidative stress. By measuring these oxidative stress parameters, we found that the level of GSH decreased with the increase of ROS and LPO levels after NiSO4 exposure. By adjusting the LPO index changes in Beas-2B cells, GA, as a kind of antioxidant extracted from plants, effectively reduced the oxidative stress effect caused by NiSO4, thus reducing the damage. Our research found that GA can be a contributing factor to lipid peroxidation, evidenced by MDA and LDH levels that were significantly decreased in the co-exposure GA groups when compared to the NiSO4 treatment group. As an antioxidant, GA widely exists in grapes, pomegranates, black tea, and traditional Chinese medicine. Studies have confirmed that GA through the Fenton reaction produces a hydroxyl radical and xanthine oxidase free-radical system, resulting in superoxide anion free-radical scavenging, and that GA has an inhibitory effect on human liver microsomal cytochrome P450 3A (CYP3A) mediated oxidation, to reduce the tissue accumulation of ROS. In the induction of apoptosis, GA mainly had a pro-oxidative effect. We also demonstrated that GA significantly protected against the oxidative stress induced by NiSO4 in Beas-2B cells.
Apoptosis is a physiological mechanism that plays a very important role in maintaining the stability of the organismic internal environment, but when apoptosis is too high or too low, it will have adverse effects on the body. A large number of studies have shown that oxidative stress may induce apoptosis [17,18]. Our observation showed that NiSO4 induced apoptosis accompanied by LPO level changes (see Supplementary Figures 1–3). ROS can initiate a chain reaction, which is easy to react with various kinds of unsaturated fatty acids and cholesterol in the cell membrane. Previous studies have demonstrated that nickel ion-induced oxidative stress leads to apoptosis [19–23]. There is a close link between the development of oxidative stress and apoptosis [24]. Before NiSO4 infected Beas-2B cells, GA intervention could significantly inhibit apoptosis caused by NiSO4. By reducing the increased level of ROS and apoptosis rate, GA inhibited the cell injury, and therefore has respiratory protective actions after NiSO4 exposure.
In order to investigate the specific molecular mechanism of the protective effect on human bronchial epithelial Beas-2B cells apoptosis induced by GA, we further detected the expression levels of Ras/ERK signaling pathway proteins, including Ras, ERK, c-Myc, etc. The Myc oncogene family is considered to be related to the occurrence of many kinds of malignant diseases [25], in which the c-Myc gene encoding protein is a transcription factor, which is expressed in most human diseases [13]. Previous studies also have shown that there were several potential mechanisms of c-Myc-induced apoptosis, including the increase of DNA strand breaks and the decrease of bcl-2 expression [26,27]. In recent years, particular attention has been paid to studies on the multiple pathways that control c-Myc, such as NF-κB [24], Ras/JNK [28], and Ras/ERK [29]. Li et al. reported that nickel ions induced c-Myc in Beas-2B cells by the Ras/ERK pathway [3]. Western blot analysis results indicated that 250~1000 μM NiSO4 significantly increased the protein levels of Ras, p-ERK, and c-Myc (Figure 5). Ras/ERK signaling pathway inhibitors were used to confirm whether the Ras/ERK pathway may be related to NiSO4-induced c-Myc. The results showed that both Ras and ERK inhibitors reduced NiSO4-induced c-Myc protein. Meanwhile, 500 μM-1000 μM NiSO4 significantly increased PARP cleavage, which represented a marker of apoptosis. After GA intervention, the expression levels of c-Myc–related proteins were suppressed. Especially in the 100–150 μM GA co-exposure group, GA down-regulated the expression levels of c-Myc and PARP cleavage. These findings demonstrate that GA has a protective effect against NiSO4-induced apoptosis via inhibiting the Ras/ERK signaling pathway.
To sum up, the results of our study suggested the safe dose of GA in Beas-2B cells is 10–150 μM. The protective effect of GA against NiSO4-induced cell toxicity in Beas-2B cells might be through the Ras/ERK pathway, which indicates that GA has an effect on preventing cell injury.
Conclusions
In this study, we found that GA could reduce NiSO4-induced toxicity through down-regulation of the Ras/ERK signaling pathway in Beas-2B cells. The limitation is that the protective effect of GA on NiSO4-induced cell toxicity was only evaluated by in vitro experiments; however, there have been relatively few systematic in vivo studies involving the mechanism of the protective effect of GA. Thus, it has not been possible to identify whether the activation of Ras/ERK signaling pathway is the direct manifestation of toxicity or whether it is the protective response of the cells toward toxic substances. In order to provide a sufficient basis for identification of the mechanism of the protective effect of GA when exposed to toxic substances, further studies are needed to elucidate this relationship.
Supplementary materials
NiSO4 induced cytotoxicity in Beas-2B cells. Cells were exposed to different concentrations of NiSO4 for 24 h, and cell viability was determined by MTT assay as described in the Material and Methods section. Data represent mean ±SD of three independent experiments made in three replicates. * P<0.05, compared with control group.
NiSO4 induced cytotoxicity in Beas-2B cells. Nuclear staining of Beas-2B cells was with Hoechst 33258. (A) Control group. (B) 250 μM NiSO4. (C) 500 μM NiSO4. (D) 750 μM NiSO4. (E) 1000 μM NiSO4. The original magnification is 40
NiSO4 induced oxidative stress in Beas-2B cells. Cells were exposed to different concentrations of NiSO4 for 24 h, and cells’ oxidative stress parameters were determined according to the kit instructions as described in the Material and Methods section. (A) ROS, (B) LDH, (C) MDA, (D) GSH. Data represent mean ±SD of three independent experiments made in three replicates. * P<0.05, compared with control group.
Footnotes
Conflicts of interest
The authors declare that there are no conflicts of interest.
Source of support: This work was supported by the National Natural Science Foundation of China (No. 81372965)
References
- 1.Grandjean P. Human exposure to nickel. IARC Sci Publ. 1984;53:469–85. [PubMed] [Google Scholar]
- 2.Ma L, Bai YN, Pu HQ, et al. A retrospective cohort mortality study in Jinchang, the largest nickel production enterprise in China. Biomed Environ Sci. 2014;27:567–71. doi: 10.3967/bes2014.088. [DOI] [PubMed] [Google Scholar]
- 3.Li Q, Suen TC, Sun H, et al. Nickel compounds induce apoptosis in human bronchial epithelial Beas-2B cells by activation of c-Myc through ERK pathway. Toxical Appl Pharmacol. 2009;235:191–98. doi: 10.1016/j.taap.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doll R, Morgan LG, Speizer FE. Cancers of the lung and nasal sinuses in nickel workers. Br J Cancer. 1970;24:623–32. doi: 10.1038/bjc.1970.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chakrabati SK, Bai C, Subramanian KS. DNA-protein crosslinks induced by nickel compounds in isolated rat lymphocytes: Role of reactive oxygen species and specific amino acids. Tixicol Appl Pharmacol. 2001;170:153–65. doi: 10.1006/taap.2000.9097. [DOI] [PubMed] [Google Scholar]
- 6.Cavallo D, Ursini CL, Setini A, et al. Evaluation of oxidative damage and inhibition of DNA repair in an in vitro study of nickel exposure. Toxicol In Vitro. 2003;17:603–7. doi: 10.1016/s0887-2333(03)00138-3. [DOI] [PubMed] [Google Scholar]
- 7.Chakrabarti SK, Bai C. Role of oxidative stress in nickel chloride-induced cell injury in rat renal cortical slices. Biochem Pharmacol. 1999;58:1501–10. doi: 10.1016/s0006-2952(99)00232-4. [DOI] [PubMed] [Google Scholar]
- 8.Manach C, Williamson G, Morand C, et al. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am J Clin Nutr. 2005;81:230S–42S. doi: 10.1093/ajcn/81.1.230S. [DOI] [PubMed] [Google Scholar]
- 9.Liu H, Zheng A, Liu H, et al. Identification of three novel polyphenolic compounds, origanine A-C, with unique skeleton from Origanum vulgare L. using the hyphenated LC-DAD-SPE-NMR/MS methods. J Agric Food Chem. 2012;60:129–35. doi: 10.1021/jf204406u. [DOI] [PubMed] [Google Scholar]
- 10.Kawada M, Ohno Y, Ri Y, et al. Anti-tumor effect of gallic acid on LL-2 lung cancer cells transplanted in mice. Anticancer Drugs. 2001;12:847–52. doi: 10.1097/00001813-200111000-00009. [DOI] [PubMed] [Google Scholar]
- 11.Ohno T, Inoue M, Ogihara Y. Cytotoxic activity of gallic acid against liver metastasis of mastocytoma cells P-815. Anticancer Res. 2001;21:3875–80. [PubMed] [Google Scholar]
- 12.Nose M, Koide T, Morikawa K, et al. Formation of reactive oxygen intermediates might be involved in the trypanocidal activity of gallic acid. Biol Pharm Bull. 1998;21:583–87. doi: 10.1248/bpb.21.583. [DOI] [PubMed] [Google Scholar]
- 13.Hann SR, Eisenman RN. Proteins encoded by the human c-myc oncogene: Differential expression in neoplastic cells. Mol Cell Biol. 1984;4:2486–97. doi: 10.1128/mcb.4.11.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lüscher B, Larsson LG. The world according to MYC. Conference on MYC and the transcriptional control of proliferation and oncogenesis. EMBO Rep. 2007;8:1110–14. doi: 10.1038/sj.embor.7401121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahamed M, Akhtar MJ, Siddiqui MA, et al. Oxidative stress mediated apoptosis induced by nickel ferrite nanoparticles in cultured A549 cells. Toxicology. 2011;283:101–8. doi: 10.1016/j.tox.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 16.Lu S, Zhang W, Zhang R, et al. Comparison of cellular toxicity caused by ambient ultrafine particles and engineered metal oxide nanoparticles. Part Fibre Toxicol. 2015;12:1–12. doi: 10.1186/s12989-015-0082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murakawa M, Jung SK, Iijima K, et al. Apoptosis-inducing protein, AIP, from parasite-infected fish induces apoptosis in mammalian cells by two different molecular mechanisms. Cell Death Differ. 2001;8:298–307. doi: 10.1038/sj.cdd.4400811. [DOI] [PubMed] [Google Scholar]
- 18.Schmitt CA, Lowe SW. Apoptosis and therapy. J Pathol. 1999;187:127–37. doi: 10.1002/(SICI)1096-9896(199901)187:1<127::AID-PATH251>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 19.Wu B, Cui H, Peng X, et al. Dietary nickel chloride induces oxidative stress, apoptosis and alters Bax/Bcl-2 and caspase-3 mRNA expression in the cecal tonsil of broilers. Food Chem Toxicol. 2014;63:18–29. doi: 10.1016/j.fct.2013.10.033. [DOI] [PubMed] [Google Scholar]
- 20.Freitas M, Barcellos-de-Souza P, Barja-Fidalgo C, et al. Nickel induces apoptosis in human neutrophils. Biometals. 2013;26:13–21. doi: 10.1007/s10534-012-9590-2. [DOI] [PubMed] [Google Scholar]
- 21.Doreswamy K, Shrilatha B, Rajeshkumar T, et al. Nickel-induced oxidative stress in testis of mice: Evidence of DNA damage and genotoxic effects. J Androl. 2004;25:996–1003. doi: 10.1002/j.1939-4640.2004.tb03173.x. [DOI] [PubMed] [Google Scholar]
- 22.Huang J, Cui H, Peng X, et al. The association between splenocyte apoptosis and alterations of Bax, Bcl-2 and caspase-3 mRNA expression, and oxidative stress induced by dietary nickel chloride in broilers. Int J Environ Res Public Health. 2013;10:7310–26. doi: 10.3390/ijerph10127310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valeeva ET, Galimova RR, Karimova LK, et al. [Cases of nickel carbonyl acute poisoning at major petrochemical enterprises]. Med Tr Prom Ekol. 2009;11:17–19. [in Russian] [PubMed] [Google Scholar]
- 24.Ding J, Zhang X, Li J, et al. Nickel compounds render anti-apoptotic effect to human bronchial epithelial Beas-2B cells by induction of cyclooxygenase-2 through an IKKbeta/p65-dependent and IKKalpha- and p50-independent pathway. J Biol Chem. 2006;281:39022–32. doi: 10.1074/jbc.M604798200. [DOI] [PubMed] [Google Scholar]
- 25.Simkó M, Mattsson MO. Extremely low frequency electromagnetic fields as effectors of cellular responses in vitro: Possible immune cell activation. J Cell Biochem. 2004;93:83–92. doi: 10.1002/jcb.20198. [DOI] [PubMed] [Google Scholar]
- 26.Vafa O, Wade M, Kern S, et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9:1031–44. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- 27.Guan F, Zhang D, Wang X, et al. Nitric oxide and bcl-2 mediated the apoptosis induced by nickel(II) in human T hybridoma cells. Toxicol Appl Pharmacol. 2007;221:86–94. doi: 10.1016/j.taap.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 28.Ke Q, Li Q, Ellen TP, et al. Nickel compounds induce phosphorylation of histone H3 at serine 10 by activating JNK-MAPK pathway. Carcinogenesis. 2008;29:1276–81. doi: 10.1093/carcin/bgn084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vafa O, Wade M, Kern S, et al. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9:1031–44. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
NiSO4 induced cytotoxicity in Beas-2B cells. Cells were exposed to different concentrations of NiSO4 for 24 h, and cell viability was determined by MTT assay as described in the Material and Methods section. Data represent mean ±SD of three independent experiments made in three replicates. * P<0.05, compared with control group.
NiSO4 induced cytotoxicity in Beas-2B cells. Nuclear staining of Beas-2B cells was with Hoechst 33258. (A) Control group. (B) 250 μM NiSO4. (C) 500 μM NiSO4. (D) 750 μM NiSO4. (E) 1000 μM NiSO4. The original magnification is 40
NiSO4 induced oxidative stress in Beas-2B cells. Cells were exposed to different concentrations of NiSO4 for 24 h, and cells’ oxidative stress parameters were determined according to the kit instructions as described in the Material and Methods section. (A) ROS, (B) LDH, (C) MDA, (D) GSH. Data represent mean ±SD of three independent experiments made in three replicates. * P<0.05, compared with control group.