

Abstract
Although epidemiological evidence suggests a human genetic basis of pulmonary tuberculosis (PTB) susceptibility, the identification of specific genes and alleles influencing PTB risk has proven to be difficult. Previous genome-wide association (GWA) studies have identified only three novel loci with modest effect sizes in sub-Saharan African and Russian populations. We performed a GWA study of 550,352 autosomal SNPs in a family-based discovery Moroccan sample (on the full population and on the subset with PTB diagnosis at <25 years), which identified 143 SNPs with p < 1 × 10−4. The replication study in an independent case/control sample identified four SNPs displaying a p < 0.01 implicating the same risk allele. In the combined sample including 556 PTB subjects and 650 controls these four SNPs showed suggestive association (2 × 10−6 < p < 4 × 10−5): rs358793 and rs17590261 were intergenic, while rs6786408 and rs916943 were located in introns of FOXP1 and AGMO, respectively. Both genes are involved in the function of macrophages, which are the site of latency and reactivation of Mycobacterium tuberculosis. The most significant finding (p = 2 × 10−6) was obtained for the AGMO SNP in an early (<25 years) age-at-onset subset, confirming the importance of considering age-at-onset to decipher the genetic basis of PTB. Although only suggestive, these findings highlight several avenues for future research in the human genetics of PTB.
Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis, results in 8.6 million new cases and 1.3 million deaths each year (World Health Organization). With the rise of multi-drug resistant M. tuberculosis strains and in the absence of an efficacious vaccine, TB remains a major public-health problem worldwide (Kaufmann et al. 2014). One-third of the world’s population is exposed to M. tuberculosis, and after exposure, most, individuals become infected, with around 10 % of infected individuals who develop clinical disease (Casanova and Abel 2002; Ernst 2012). Around half of these patients, particularly young children, develop “primary” TB, often associated with extra-pulmonary disease, within 2 years of infection (Casanova and Abel 2002). The other half of affected individuals develop clinical TB later in life, typically pulmonary TB (PTB) due to reactivation of the original infection, or in some cases, a novel infectious episode (Casanova and Abel 2002; Ernst 2012; O’Garra et al. 2013). In addition to environmental (e.g., microbial) and nongenetic (e.g., acquired immunodeficiency) host factors, there is emerging molecular evidence suggesting that human genetics contributes to the variability in response to M. tuberculosis (Abel et al. 2014b). This is the case for the control of TB infection (Sepulveda et al. 1994; Jepson et al. 2001; Cobat et al. 2009, 2010, 2012, 2014b) and of childhood TB with the discovery of singlegene inborn errors of IFN-γ immunity (Casanova and Abel 2002; Alcaïs et al. 2005; Boisson-Dupuis et al. 2011, 2015). The study of genetic susceptibility to PTB has proven more difficult, although there is considerable evidence, particularly from twin studies (Puffer 1944), to support a major role for human genetic factors in the development of PTB (Abel et al. 2014b; Meyer and Thye 2014). Candidate gene association studies investigating PTB resulted in the identification of a number of common associated variants (Möller and Hoal 2010; Azad et al. 2012; Abel et al. 2014b). Although few have been satisfactorily replicated, several alleles within NRAMP1 have been identified across populations and clinical phenotypes (Vidal et al. 1993; Bellamy et al. 1998; Malik et al. 2005; Abel et al. 2014b). An expression study comparing PTB patients with healthy controls (Berry et al. 2010), and an ex vivo cell stimulation eQTL study (Barreiro et al. 2012) have provided new candidate genes and pathways to be tested.
Genetic markers investigated in the context of genome-wide (GW) association (GWA) studies comprise common variants representing the whole human genome in a hypothesis-generating approach that has been applied to infectious diseases such as leprosy and malaria (Abel et al. 2014a), and there have been five PTB studies reported to date (Meyer and Thye 2014). A sub-Saharan African study from Ghana and the Gambia and another from Russia have led to the identification of only three signals that reached the GW threshold for statistical significance (5 × 10−8). One of the two variants identified in the African GWA studies, rs4331426, is located in a gene desert on chromosome 18q11.2 (Thye et al. 2010) and the other, rs2057178, is near WT1 on chromosome 11p13 (Thye et al. 2012), and both displayed modest effect sizes (OR = 1.19 and 0.77, respectively). The chromosome 11 signal was replicated in populations from Indonesia and Russia as reported in the original study (Thye et al. 2012), and in an independent GWA study conducted in an admixed South African population (Chimusa et al. 2014). Replication of the chromosome 18q11.2 signal in two independent Chinese study populations provided conflicting results (Dai et al. 2011; Wang et al. 2013). The Russian study identified a cluster of intronic ASAP1 variants in a study population of over 15,000 subjects, also with a weak effect size (OR = 0.84 for the most significant SNP rs4733781), with possible functional involvement in dendritic cell mobility (Curtis et al. 2015). Another GWA study combining Japanese and Thai samples reported a variant with a modest effect on chromosome 20q12 among PTB subjects with age of onset <45 years (Mahasirimongkol et al. 2012). Finally, a three-stage GWA study among Indonesians and Russian Caucasians led to suggestive findings (Png et al. 2012).
The screening phases of previous PTB GWA studies have been conducted in study populations from sub-Saharan Africa, Asia, and Europe, but none from North Africa. We previously mapped a major susceptibility locus to chromosomal region 8q12–13 (Baghdadi et al. 2006) in a Moroccan study population which led to the identification of a cluster of susceptibility variants near TOX for early age-at-onset (<25 years) PTB (Grant et al. 2013). A subsequent candidate pathway approach focusing on genes of the IL12/IFN-γ circuit identified susceptibility variants in the promoter region of STAT4, again with stronger evidence of association for early age-at-onset PTB (Sabri et al. 2014). Our PTB work in Morocco points to age as another source of genetic heterogeneity in terms of genetic effect, with earlier age-at-onset consistently displaying greater risk of disease (Grant et al. 2013). Here, we conducted a GWA study in a primary family-based population and a replication case-control population, both from Morocco. Although the combined analysis of both samples did not lead to signals significant at the GW level, the strongest combined signal (2 × 10−6) was identified on chromosome 7p21 in the younger subset (age at onset <25 years).
Methods
Recruitment of PTB subjects and healthy controls
Study subjects were recruited from hospital Mohammed V of Rabat and TB diagnostic centers located in endemic areas of Casablanca and Salé (Morocco), where the annual incidence of TB is estimated at approximately 150 cases/100,000 inhabitants (World Health Organization 2011, 2013). In the present study, affected individuals presenting with PTB only were enrolled. These participants were enrolled in the primary family-based sample if their two parents or any number of unaffected siblings were also willing to participate, and otherwise were enrolled in the case–control replication study. Thus for both the family-based and the case–control studies, among subjects given a diagnosis of PTB on the basis of clinical symptoms and pathologic findings on chest radiographs, only those with positive sputum smear microscopy results (Ziehl–Neelsen staining) and/or positive sputum culture on Lowenstein-Jensen medium identified as M. tuberculosis, were recruited as cases. Parents and siblings were classified as TB cases using the same criteria, and for those who had TB in the past using medical records providing the same information. Parents and siblings were considered to be unaffected on the basis of normal findings of clinical examination, normal findings on chest radiographs, and an absence of a history of TB. Controls were recruited from among healthy blood donors, and only those with a normal clinical exam and without any history of TB or pulmonary disease were retained as previously described (Grant et al. 2013). This resulted in the recruitment of a total of 558 individuals into the family-based study who comprised 252 parents, 239 PTB affected offspring and 67 unaffected offspring. These individuals were also part of the previously described primary family-based screen (Grant et al. 2013) that included 706 individuals, and corresponded to the samples with enough available DNA for the present GWAS. In addition, 317 independent cases and 657 independent controls were recruited for the replication population, and this case/control sample is the same as that used in our previous study (Grant et al. 2013). The sampling of all the cohorts was approved by the appropriate ethical committees and/or institutional review boards, and written informed consent was obtained from all adults and from the parents of minors.
Genotyping and quality control of genotype data
All DNA samples from the discovery cohort were genotyped using the Illumina Human610-Quad BeadChip (Illumina, San Diego, CA) at the Centre National de Génotypage (Evry, France). After per-SNP quality control steps, 550,352 autosomal SNPs were retained (See Online Resource 1). Genotypes of the 252 founders were analyzed in a principal components analysis (see Online resource 1), and considered in a graphical display, also including populations from the 1000 genomes project (Online Resource 2). A SEQUENOM custom panel was designed comprising 143 SNPs selected for replication in the case–control population. A total of 317 cases and 650 controls were retained for analyses following quality control filtering (Online Resource 1, Table 1). The combined sample of the 239 affected offspring from the family-based study and the 317 cases from the replication study consisted of 66.1 % males with a mean (SD) age of PTB onset of 26 (10.2) years.
Table 1.
Clinical characteristics of study populations from the Moroccan family-based and case–control studies
| Characteristic | Family-based
|
Case–control
|
||
|---|---|---|---|---|
| Founders | Offspring | Cases | Controlsa | |
| N | 252 | 306 | 317 | 650 |
| N by status (affected; unaffected) | 42; 210 | 239; 67 | 317; 0 | 0; 650 |
| Age in years: mean (SD, range) by status | ||||
| Affectedb | 41.4 (12.9, 16–70) | 21.3 (7.8, 1–48) | 29.3 (10.8, 1–69) | – |
| Unaffected | 51.1 (9.4, 26–73) | 25.9 (9.5, 5–50) | – | 31.5 (9.3, 16–68) |
| % Males by status | ||||
| Affected | 55 | 56.2 | 71.9 | – |
| Unaffected | 38.4 | 38.8 | – | 57.9 |
Controls were recruited from among healthy blood donors. Although we cannot exclude the possibility that some of these controls may develop PTB later in life (in any case no more than 5%, the expected proportion of infected individuals who develop PTB after infection by M. tuberculosis), the level of misclassification should be quasi-negligible (<5%) as the control population is older than the cases and do not have any history of TB
Age at PTB onset for affected individual
Analysis strategy
A full GWA scan was conducted across all 550,352 SNPs under the additive, recessive and dominant models using three sets of configurations of the family-based study population. The 558 samples included 135 nuclear families with 187 affected PTB offspring informative for family-based association test (FBAT) analysis (Online Resource 3) following quality control measures (See Online Resource 1). First, the full family-based study population was evaluated using FBAT (Horvath et al. 2001) on all informative families from among the 558 genotyped individuals (Online Resource 3; see Supplemental Statistical methods in Online Resource 1 for further details including power calculations). Second, we restricted the FBAT analysis to the sub-sample of 134 offspring with PTB onset <25 years. Third, the set of SNPs displaying an FBAT p < 0.01 in the first full family-based population scan were analyzed using an augmented family-based population by means of the conditional logistic regression framework. A selection of 246 unrelated individuals from among the founder population and among the individuals found to be uninformative for FBAT were grouped together in a stratum for analysis in combination with the strata consisting of the affected offspring and her/his pseudosiblings as previously described (Grant et al. 2013). The 246 individuals correspond to: a single affected offspring from among 64 families uninformative for FBAT, 56 single parents and 63 parental pairs making up a total of 107 PTB affected cases and 139 unaffected controls. We previously demonstrated that the parents in a family-based study may serve as an independent replication study population (unpublished data). Unobserved genotypes were imputed in the family-based study leading to 7,467,568 combined genotyped and imputed SNPs across autosomes, and 5,390,648 autosomal SNPs retained after quality control filtering (Online Resource 1).
The case-control replication study was conducted on SNPs selected according to the results of the primary family-based study: all SNPs displaying a p < 1 × 10−4 in any of the three GW analyses were selected for genotyping. In the replication study, we tested for association under the additive, dominant and recessive models using a one-sided test for the risk genotype, allowing for a type I error rate of 0.01 by performing Chi-squared tests using PLINK software (Purcell et al. 2007) (see Online Resource 1 for power calculations). The conditional logistic regression framework was used to perform combined analyses of the family-based and case-control samples as described previously (Grant et al. 2013): all cases and controls from the case-control study were grouped together in one large stratum. The most significant model in the combined analysis was retained as the final model.
Results
We initially performed FBATs across 550,352 high-quality autosomal SNPs, and retained the minimum p value obtained across the additive, dominant and recessive models. No SNPs reached GW significance in the primary family-based study as illustrated in the Manhattan plot (Fig. 1), and the most significant p value was recorded for rs2339463 on chromosome 3 [under the additive model, based on the minor allele as the reference, OR (95 % confidence interval) = 0.39 (0.25–0.59); p = 2.2 × 10−6]. A Q-Q plot restricted to the additive model of FBAT analyses showed that the observed p values followed the diagonal representing the expected distribution under the null hypothesis of no association, with a corresponding genomic inflation factor of 1.012. A proportion of markers across the GW study displaying higher significance SNPs fell under the expected significance level probably reflecting the low sample size and limited power of the study (Online Resource 4; Statistical Methods in Online Resource 1). In selecting SNPs for genotyping in the replication phase among 317 cases and 650 controls from Morocco, we performed two more sets of analyses across all 550,352 autosomal SNPs. A second set of FBAT analyses was performed by restricting affected offspring to those with age-at-onset under 25 years. The most significant SNP was recorded for rs2884122 on chromosome 7 with p = 1.2 × 10−7 [under the additive model, OR = 0.48 (0.30–0.76)]. A third set of analyses was conducted in the conditional logistic regression framework incorporating the FBAT-informative families and adding in a single stratum those genotyped individuals not informative for the FBAT analyses (see Methods). The most significant association (under the additive model, OR = 0.51 [0.38–0.68]; p = 2.7 × 10−6) was observed at SNP rs1156325 on chromosome 7. A total of 143 SNPs reached the threshold of p = 1 × 10−4 in any of these GWA analyses and were selected for replication in the case/control population (Online Resource 5).
Fig. 1.
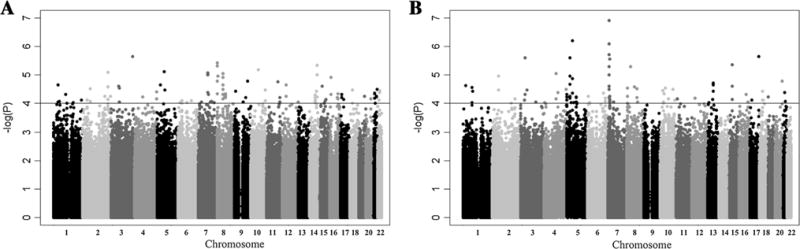
Manhattan plot displaying family-based GWA results for pulmonary tuberculosis using FBAT among 135 informative families from Morocco across 550,352 autosomal SNPs for: a the full study population and b the subset of families with 134 offspring having an age-at-onset at 25 years or younger. The −log 10 of the minimum p value obtained from the additive, dominant and recessive tests is displayed against chromosomal position going across autosomes, with a gray scale indicating chromosomes from 1 to 22. A horizontal line at a −log10 p value of 4 indicates the cut-off used for the selection of markers for replication in the case–control population
Genotyping was successful for 129 SNPs (while the process failed for 14 SNPs) (Online Resource 5). Only four SNPs displayed a p < 0.01 with the same risk allele as identified in the primary family-based study, in the replication sample comprising 317 cases and 650 controls. These four SNPs were further explored (Table 2). A combined analysis was performed in both the primary family sample and the replication case/control sample using the conditional logistic regression framework. The combined analysis model yielding the most significant p value was selected, and was additive for rs358793, rs916943, and rs6786408, and recessive for rsl7590261. Three SNPs, rs358793, rsl7590261 and rs6786408 provided a combined p value in the whole population between 5 × 10−4 and 10−5 (Table 2). SNP rs17590261 displayed a strong OR (6.24) with a large confidence interval (2.38–16.33) corresponding to a recessive model with a low number of individuals homozygous for the risk allele, including six affected offspring (from five informative families) who were homozygous in the family sample, and nine cases (out of 317) compared to six controls (out of 650) who were homozygous in the replication sample. The fourth SNP, rs916943, which had initially been identified as significant based on analysis of the <25 years stratum, also provided a lower p value (p = 2 × 10−6) in the analysis restricted to the early-onset PTB subjects. For this SNP, we found significant evidence (p = 0.012) for heterogeneity between the two age groups (<25 years or ≥25 years at PTB diagnosis) using the Chi-square test for heterogeneity (Cochran Q test) (Cochran 1954) as implemented in GWAMA (Mägi and Morris 2010). Thus, four signals with suggestive evidence of association with PTB were identified, although none of them reached GW statistical significance.
Table 2.
Results combining primary family-based and replication case–control studies in the conditional logistic regression framework
| SNP | Closest geneb | Locationc | M | M | MAF founders |
MAF cases | MAF controls |
Model | Discovery | Replication | Full sample
|
Age < 25 yearsa
|
||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pd | pd | OR (95 % CI)d | pd | OR (95 % CI)d | pd | |||||||||
| rs916943 | AGMO | 7 : 15593768 Intron 2 | T | C | 0.08 | 0.08 | 0.05 | ADD | 2.7 × 10−6 | 0.009 | 1.86 (1.33, 2.6) | 0.0002 | 2.73 (1.78, 4.18) | 2.0 × 10−6 |
| rs358793 | WNT5A | 3: 55361433 138.3 kb downstream | T | C | 0.49 | 0.48 | 0.57 | ADD | 9.7 × 10−5 | 0.0004 | 0.68 (0.57, 0.82) | 4.0 × 10−5 | 0.69 (0.53, 0.90) | 0.0057 |
| rs17590261 | PCDH10 | 4: 136564124 2.49 Mb upstream | C | T | 0.1 | 0.14 | 0.11 | REC | 3.5 × 10−5 | 0.003 | 6.24 (2.38, 16.33) | 2.2 × 10−5 | 5.47 (1.66, 18.05) | 0.0023 |
| rs6786408 | FOXP1 | 3: 71263218 Intron 5 | A | C | 0.48 | 0.54 | 0.45 | ADD | 2.8 × 10−5 | 0.0007 | 1.47 (1.23, 1.79) | 1.9 × 10−5 | 1.67 (1.28, 2.13) | 8.2 × 10−5 |
M major allele, m minor allele, MAF minor allele frequency
The number of affected individuals included in this analysis are 277 affected offspring and cases <25 years (or 50% of the total affected offspring and cases used in the full sample)
Gene in which the SNP is located, or closest gene
Chromosome with location in base pairs, and gene position or distance from the closest gene
Two-sided test based on the minor allele among founders
We searched for other SNPs in linkage disequilibrium (LD) with the SNPs representing the four signals rs358793, rs17590261, rs6786408 and rs916943 based on data from the 1000 Genomes Project (CEU population). There were 26 SNPs within 17 kb of rs358793 with an R2 > 0.8, and 65 SNPs within 74.5 kb of rs17590261 with an R2 > 0.8. Using the same criteria, there were no SNPs in LD with rs6786408 and rs916943 (R2 > 0.8). None of the 26 or 65 SNPs, all successfully imputed, displayed an association p value greater than 0.2 log units in significance than the originally identified genotyped SNP The full results from imputed association tests revealed one signal at p < 10−4 that was not in LD with a genotyped SNP identified for replication in the case-control study and displayed in Online Resource 5. The T allele at rs256659 on chromosome 12 (an IMPUTE2 information score of 0.96 was obtained for this SNP) was found to be over-transmitted under the additive model (p = 3.7 × 10−6). This SNP was then genotyped in the case control replication study, but the association was not replicated.
We also specifically tested the SNPs resulting from the two reported associations with PTB on chromosome 18 and chromosome 11 in GWA studies of populations from Ghana and The Gambia (Thye et al. 2010, 2012) in our Moroccan data (Table 3). These SNPs were not genotyped in our primary family-based GWA study, but were successfully imputed. The same risk alleles as those reported were identified for the two SNPs comprising the chromosome 11 signal, leading to one-sided p values of 0.07 for rs2057178 and 0.03 for rs11031731. These two SNPs were genotyped in the case/control replication study and were again associated with PTB with p = 0.043 and 0.037 for rs2057178 and rs11031731, respectively. Of note, these two SNPs are in very strong LD in the Moroccan population with an r2 estimated at 1 among the 650 controls. The results were more conflicting for the chromosome 18 signal for rs4331426, as the opposite allele to that identified in the published study was found to be over-transmitted to affected offspring. However, in the case–control replication population, we found a significant association of rs4331426 with PTB with the same direction of association as that previously reported. We finally examined the ASAP1 SNPs recently reported to be associated with PTB in the Russian GWA study (Curtis et al. 2015). The two main associated SNPs were successfully imputed (scores of 0.93 for rs4733781 and 0.99 rs10956514) in our family-based population. Minor alleles were the same as in Russia with close frequencies of 0.3 for rs4733781 (compared to 0.31 in Russian controls), and 0.46 for rs10956514 (compared to 0.36 in Russian controls). The same direction of association, although this time non significant (the one-sided test yielded p = 0.12 for rs4733781, estimated OR = 0.73, and p = 0.15 for rs10956514, estimated OR = 0.84), was obtained in our sample using FBATdosage (Cobat et al. 2014a) with the minor allele being under-transmitted to affected offspring.
Table 3.
Validation study exploring association with GWAS replicated SNPs from a study population including samples from Ghana and the Gambia in a case–control population from Morocco (additive model)
| SNP | M | M | Ghana/The Gambiaa
|
Morocco family-basedb
|
Morocco case–control
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MAF cases | MAF controls | MAF founders | Fdos-pc | MAF cases | MAF controls | OR (95 % CI)d | pd | ||||
| Chr11 | rs2057178 | A | G | 0.27 | 0.31 | 0.16 | 0.07 | 0.16 | 0.20 | 0.78 (0.59–1.00) | 0.043 |
| Chr11 | rs11031731 | A | G | 0.27 | 0.31 | 0.16 | 0.03 | 0.16 | 0.20 | 0.77 (0.59–1.00) | 0.037 |
| Chr18 | rs4331426 | G | A | 0.50 | 0.45 | 0.15 | >0.5 | 0.15 | 0.11 | 1.50 (1.09–2.14) | 0.009 |
M major allele, m minor allele, MAF minor allele frequency, the risk allele identified in the Ghana/The Gambia study population is underlined, Source Cobat et al. (2014a)
Source Thye et al. 2010
Thye T, Owusu-Dabo E, Vannberg FO, et al. Common variants at 11p13 are associated with susceptibility to tuberculosis. Nat. Genet. 2012; 44:257–259
Weighted averages were calculated based on published data on populations from Ghana and The Gambia
Genotypes were imputed using IMPUTE2 and information scores obtained were 0.79 for rs2057178, 0.88 for rs11031731 and 0.97 for rs4331426
One-sided test using FBAT-dosage on imputed probabilities of genotypes
One-sided test with respect to the minor allele
Discussion
In this GWA study of PTB conducted in a primary Moroccan family-based population followed by replication in an independent Moroccan case-control population, we identified four SNPs displaying suggestive association with PTB (2 × 10−6 < p < 4 × 10−5 in the combined analysis). Although these findings require validation in other study populations with similar (North African) or other ethnic backgrounds, two of the four SNPs are located within genes whose functions involve macrophages (FOXP1 and AGMO), which are the site of human infection, latency, and reactivation by M. tuberculosis. In humans, both FOXP1 and AGMO are widely expressed, in particular in hematopoietic cells (http://www.proteinatlas.org). SNP rs6786408 is located in the 5th intron of FOXP1 (628 kb including 21 exons) on chromosome 3. FOXP1 or forkhead box P1 is a transcriptional repressor. Transgenic mice overexpressing human FOXP1 in monocyte/macrophage lineage cells showed impaired macrophage functions including migratory capacity, cytokine production, phagocytosis and respiratory burst (Shi et al. 2008). In humans, de novo FOXP1 mutations were associated with mental retardation (Hamdan et al. 2010). The SNP associated with early age-at-onset PTB, rs916943, is located in AGMO (alkylglycerol monooxygenase). AGMO was found to play a role in modulating the production by macrophages of PAF (platelet activating factor) in mice, which impacts on changes to vascular permeability, oxidative burst, chemotaxis of leukocytes, and augmentation of arachidonic acid metabolism in phagocytes (Tokuoka et al. 2013). The two other identified SNPs, rs358793 and rs17590261, are located within intergenic regions. SNP rs358793 is situated 252.8 kb downstream of CACNA2D3 and 138.3 kb upstream of WNT5A, and is likely to affect binding of transcription factors (www.regulomedb.org) with no RNA genes in close proximity. No SNPs in high LD with rs358793 with available data displayed functional features. SNP rs17590261 is located on chromosome 4 in a region with no functional annotation. The nearest coding gene is PCDH10 at 2.49 Mb downstream, and the nearest RNA gene is long intergenic non-protein coding RNA 613 at 219.3 kb upstream. Among the 65 SNPs in high LD with rs17590261, 28 SNPs with data displayed minimal binding evidence (regulomedb.org).
Exploration of associations of SNPs previously identified in other GWA studies showed that the present Moroccan study replicated the association on chromosome 11 at rs2057178 and rs11031731, discovered in populations from The Gambia and Ghana (Thye et al. 2010, 2012). For SNP rs11031731, the association was significant both using the imputed genotype data in the primary family-based study (p = 0.03), and the genotyped data in the case-control study (p = 0.037). While the frequency of the protective allele A, was lower in Morocco (0.16) than in the initial African study (0.31), the corresponding OR of developing PTB was estimated in the case/control study at 0.77 (0.59–1.0) very close to that observed in the Gambian and Ghanaian populations. Interestingly, the chromosome 11 finding was also replicated in populations from Indonesia and Russia (Thye et al. 2012), and in an admixed South African Colored population (Chimusa et al. 2014). The differing linkage disequilibrium (LD) patterns across these populations combined with our results in the Moroccan study populations give credence to the likelihood that these identified SNPs on chromosome 11 near the WT1 locus (encoding Wilms tumor 1) include the causal variant(s) or are in high LD with the causal variants across ethnicities. Currently, the functional role played by these variants is not understood. For rs4331426 on chromosome 18, our results showed some evidence for association in the case–control study while the direction of association was opposite in the imputed data of the family-based sample. The chromosome 18 SNP was also not replicated in South African (Chimusa et al. 2014) nor in Chinese (Dai et al. 2011; Wang et al. 2013) populations, suggesting that this signal may be restricted to certain ethnicities. Finally, our Moroccan results were consistent with the recently reported Russian ASAP1 signal (Curtis et al. 2015), although they were not significant in our sample of smaller size.
The genetic basis of PTB has proven particularly challenging to characterize, first using the candidate gene approach and now the GWA study approach. Notably, no GWA study to date has identified nor replicated previously detected PTB susceptibility factors from candidate gene analyses. Several sources are likely to be responsible for the limited results observed overall: the small number of signals exceeding the GW threshold, the challenge in replicating findings across study populations, and the modest risk estimates. Genetic heterogeneity probably plays an important role, with different variants involved in different patients. Rare or even private causal variants are also likely to be implicated in driving disease risk. Finally, the phenotypic heterogeneity of TB-related traits is probably responsible for further difficulties in detecting underlying human genetic risk factors. In PTB, the most commonly used phenotype-defining characteristic is the presence of M. tuberculosis in the sputum of patients, regardless of the other clinical, microbiological and demographic criteria. This approach ignores the dynamic and idiosyncratic nature of PTB, the likelihood that different stages of this process result from different sets of genetic causal factors, and the possible impact of the M. tuberculosis strain and inoculum on clinical outcomes (Abel et al. 2014b). The extended natural history of M. tuberculosis infection and pathogenesis, involving a latency period that can last up to decades, complicates the interpretation of results, as PTB cases may either be reinfection or reactivation cases, such that in a given study population, pathogen variability over time as well as variability related to geography are considerations. The present Moroccan study population benefits from geographical and socio-economic homogeneity, although genetic variability of pathogen strains of M. tuberculosis may influence the results of GWA studies focusing on pulmonary TB in adults.
Another likely source of variability is age-at-onset, which may be related to differential human immune barrier functionality when faced with M. tuberculosis. In areas where M. tuberculosis is endemic and migration is negligible, age at first PTB onset is clearly correlated with the duration of the latency period. A variant associated with early-onset PTB, such as one belonging to the cluster of variants previously identified in TOX (Grant et al. 2013), is likely to play a role in prompting the critical transition from infection to active pulmonary disease. It is not yet possible to determine whether this effect may influence short (i.e., <2 years in the context of a primary infection or a reinfection), or long (i.e., >2 years in the context of reactivation) latency periods, or both. Such specificity may underlie the finding at the AGMO SNP rs916943 (p = 2.0 × 10−6 in the under 25 years group compared to p = 2.0 × 10−4 in the full population). Although our present findings require replication in other populations, our Moroccan GWA study has highlighted several avenues for future research of the genetic basis of PTB.
Supplementary Material
Acknowledgments
We thank all patients and family members from Morocco and all the healthy Moroccan subjects for their participation in this study. We thank the late Dr M. Chentoufi from the TB Diagnostic Center (CDST) of Hay Mohammadi. We are grateful to Fatima El Bordi, the nurse from the CDTMR of Salé and to Amal Rzioui of Pneumology department of Hospital Mohammed V. We thank the Centre National de Génotypage for carrying out the genotyping. This work was supported by grants from Institut National de la Santé et de la Recherche Médicale, University Paris Descartes, the European Research Council (grant n°ERC-2010-AdG-268777), the EU-grant HOMITB (HEALTH-F3-2008-200732), the French National Research Agency (ANR) under the “Investments for the future” program (grant n°ANR-10-IAHU-01), the St. Giles Foundation, the National Center for Research Resources and the National Center for Advancing Sciences (NCATS) of the National Institute of Health (grant n°8ULTR000043), the Rockefeller University, the National Institute of Allergy and Infectious Diseases (grant n°U01AI088685). AVG was supported by the Fondation pour la Recherche Médicale and Fondation BNP Paribas.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00439-016-1633-2) contains supplementary material, which is available to authorized users.
Ethical approval All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent Informed consent was obtained from all individual participants included in the study (as stated in supplemental methods).
Contributor Information
J. El Baghdadi, Email: baghdadijamila@yahoo.fr.
L. Abel, Email: laurent.abel@inserm.fr.
References
- Abel L, Alcaïs A, Schurr E. The dissection of complex susceptibility to infectious disease: bacterial, viral and parasitic infections. Curr Opin Immunol. 2014a;30C:72–78. doi: 10.1016/j.coi.2014.07.002. [DOI] [PubMed] [Google Scholar]
- Abel L, El-Baghdadi J, Bousfiha AA, et al. Human genetics of tuberculosis: a long and winding road. Philos Trans R Soc Lond B Biol Sci. 2014b;369:20130428. doi: 10.1098/rstb.2013.0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcaïs A, Fieschi C, Abel L, Casanova J-L. Tuberculosis in children and adults: two distinct genetic diseases. J Exp Med. 2005;202:1617–1621. doi: 10.1084/jem.20052302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad AK, Sadee W, Schlesinger LS. Innate immune gene polymorphisms in tuberculosis. Infect Immun. 2012;80:3343–3359. doi: 10.1128/IAI.00443-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baghdadi JE, Orlova M, Alter A, et al. An autosomal dominant major gene confers predisposition to pulmonary tuberculosis in adults. J Exp Med. 2006;203:1679–1684. doi: 10.1084/jem.20060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro LB, Tailleux L, Pai AA, et al. Deciphering the genetic architecture of variation in the immune response to Mycobacterium tuberculosis infection. Proc Natl Acad Sci USA. 2012;109:1204–1209. doi: 10.1073/pnas.1115761109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellamy R, Ruwende C, Corrah T, et al. Variations in the NRAMP1 gene and susceptibility to tuberculosis in West Africans. N Engl J Med. 1998;338:640–644. doi: 10.1056/NEJM199803053381002. [DOI] [PubMed] [Google Scholar]
- Berry MPR, Graham CM, McNab FW, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisson-Dupuis S, El Baghdadi J, Parvaneh N, et al. IL-12Rβ1 deficiency in two of fifty children with severe tuberculosis from Iran, Morocco, and Turkey. PLoS One. 2011;6:e18524. doi: 10.1371/journal.pone.0018524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisson-Dupuis S, Bustamante J, El-Baghdadi J, et al. Inherited and acquired immunodeficiencies underlying tuberculosis in childhood. Immunol Rev. 2015;264:103–120. doi: 10.1111/imr.12272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova J-L, Abel L. Genetic dissection of immunity to mycobacteria: the human model. Annu Rev Immunol. 2002;20:581–620. doi: 10.1146/annurev.immunol.20.081501.125851. [DOI] [PubMed] [Google Scholar]
- Chimusa ER, Zaitlen N, Daya M, et al. Genome-wide association study of ancestry-specific TB risk in the South African Coloured population. Hum Mol Genet. 2014;23:796–809. doi: 10.1093/hmg/ddt462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobat A, Gallant CJ, Simkin L, et al. Two loci control tuberculin skin test reactivity in an area hyperendemic for tuberculosis. J Exp Med. 2009;206:2583–2591. doi: 10.1084/jem.20090892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobat A, Gallant CJ, Simkin L, et al. High heritability of anti-mycobacterial immunity in an area of hyperendemicity for tuberculosis disease. J Infect Dis. 2010;201:15–19. doi: 10.1086/648611. [DOI] [PubMed] [Google Scholar]
- Cobat A, Barrera LF, Henao H, et al. Tuberculin skin test reactivity is dependent on host genetic background in Colombian tuberculosis household contacts. Clin Infect Dis Off Publ Infect Dis Soc Am. 2012;54:968–971. doi: 10.1093/cid/cir972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobat A, Abel L, Alcaïs A, Schurr E. A general efficient and fexible approach for genome-wide association analyses of imputed genotypes in family-based designs. Genet Epidemiol. 2014a;38:560–571. doi: 10.1002/gepi.21842. [DOI] [PubMed] [Google Scholar]
- Cobat A, Poirier C, Hoal E, et al. Tuberculin skin test negativity is under tight genetic control of the chromosomal region 11p14–15 in settings of different TB endemicity. J Infect Dis. 2014b doi: 10.1093/infdis/jiu446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran WG. The combination of estimates from different experiments. Biometrics. 1954;10:101–129. [Google Scholar]
- Curtis J, Luo Y, Zenner HL, et al. Susceptibility to tuberculosis is associated with variants in the ASAP1 gene encoding a regulator of dendritic cell migration. Nat Genet. 2015 doi: 10.1038/ng.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Zhang X, Pan H, et al. Fine mapping of genetic polymorphisms of pulmonary tuberculosis within chromosome 18q11.2 in the Chinese population: a case-control study. BMC Infect Dis. 2011;11:282. doi: 10.1186/1471-2334-11-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst JD. The immunological life cycle of tuberculosis. Nat Rev Immunol. 2012;12:581–591. doi: 10.1038/nri3259. [DOI] [PubMed] [Google Scholar]
- Grant AV, El Baghdadi J, Sabri A, et al. Age-dependent association between pulmonary tuberculosis and common TOX variants in the 8q12–13 linkage region. Am J Hum Genet. 2013;92:407–414. doi: 10.1016/j.ajhg.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Daoud H, Rochefort D, et al. De novo mutations in FOXP1 in cases with intellectual disability, autism, and language impairment. Am J Hum Genet. 2010;87:671–678. doi: 10.1016/j.ajhg.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Xu X, Laird NM. The family based association test method: strategies for studying general genotype–phenotype associations. Eur J Hum Genet EJHG. 2001;9:301–306. doi: 10.1038/sj.ejhg.5200625. [DOI] [PubMed] [Google Scholar]
- Jepson A, Fowler A, Banya W, et al. Genetic regulation of acquired immune responses to antigens of Mycobacterium tuberculosis: a study of twins in West Africa. Infect Immun. 2001;69:3989–3994. doi: 10.1128/IAI.69.6.3989-3994.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann SHE, Lange C, Rao M, et al. Progress in tuberculosis vaccine development and host-directed therapies—a state of the art review. Lancet Respir Med. 2014;2:301–320. doi: 10.1016/S2213-2600(14)70033-5. [DOI] [PubMed] [Google Scholar]
- Mägi R, Morris AP. GWAMA: software for genome-wide association meta-analysis. BMC Bioinformatics. 2010;11:288. doi: 10.1186/1471-2105-11-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahasirimongkol S, Yanai H, Mushiroda T, et al. Genome-wide association studies of tuberculosis in Asians identify distinct at-risk locus for young tuberculosis. J Hum Genet. 2012;57:363–367. doi: 10.1038/jhg.2012.35. [DOI] [PubMed] [Google Scholar]
- Malik S, Abel L, Tooker H, et al. Alleles of the NRAMP1 gene are risk factors for pediatric tuberculosis disease. Proc Natl Acad Sci USA. 2005;102:12183–12188. doi: 10.1073/pnas.0503368102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer CG, Thye T. Host genetic studies in adult pulmonary tuberculosis. Semin Immunol. 2014;26:445–453. doi: 10.1016/j.smim.2014.09.005. [DOI] [PubMed] [Google Scholar]
- Möller M, Hoal EG. Current findings, challenges and novel approaches in human genetic susceptibility to tuberculosis. Tuberc Edinb Scotl. 2010;90:71–83. doi: 10.1016/j.tube.2010.02.002. [DOI] [PubMed] [Google Scholar]
- O’Garra A, Redford PS, McNab FW, et al. The immune response in tuberculosis. Annu Rev Immunol. 2013;31:475–527. doi: 10.1146/annurev-immunol-032712-095939. [DOI] [PubMed] [Google Scholar]
- Png E, Alisjahbana B, Sahiratmadja E, et al. A genome wide association study of pulmonary tuberculosis susceptibility in Indonesians. BMC Med Genet. 2012;13:5. doi: 10.1186/1471-2350-13-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puffer RR. Familial susceptibility to tuberculosis: its importance as a public health problem. Harvard University Press; 1944. [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabri A, Grant AV, Cosker K, et al. Association study of genes controlling IL-12-dependent IFN-γ immunity: STAT4 alleles increase risk of pulmonary tuberculosis in Morocco. J Infect Dis. 2014;210:611–618. doi: 10.1093/infdis/jiu140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulveda RL, Heiba IM, Navarrete C, et al. Tuberculin reactivity after newborn BCG immunization in mono- and dizygotic twins. Tuber Lung Dis Off J Int Union Tuberc Lung Dis. 1994;75:138–143. doi: 10.1016/0962-8479(94)90043-4. [DOI] [PubMed] [Google Scholar]
- Shi C, Sakuma M, Mooroka T, et al. Down-regulation of the forkhead transcription factor Foxp1 is required for monocyte differentiation and macrophage function. Blood. 2008;112:4699–4711. doi: 10.1182/blood-2008-01-137018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thye T, Vannberg FO, Wong SH, et al. Genome-wide association analyses identifies a susceptibility locus for tuberculosis on chromosome 18q11.2. Nat Genet. 2010;42:739–741. doi: 10.1038/ng.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thye T, Owusu-Dabo E, Vannberg FO, et al. Common variants at 11p13 are associated with susceptibility to tuberculosis. Nat Genet. 2012;44:257–259. doi: 10.1038/ng.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuoka SM, Kita Y, Shindou H, Shimizu T. Alkylglycerol monooxygenase as a potential modulator for PAF synthesis in macrophages. Biochem Biophys Res Commun. 2013;436:306–312. doi: 10.1016/j.bbrc.2013.05.099. [DOI] [PubMed] [Google Scholar]
- Vidal SM, Malo D, Vogan K, et al. Natural resistance to infection with intracellular parasites: isolation of a candidate for Bcg. Cell. 1993;73:469–485. doi: 10.1016/0092-8674(93)90135-d. [DOI] [PubMed] [Google Scholar]
- Wang X, Tang NLS, Leung CC, et al. Association of polymorphisms in the Chr18q11.2 locus with tuberculosis in Chinese population. Hum Genet. 2013;132:691–695. doi: 10.1007/s00439-013-1282-7. [DOI] [PubMed] [Google Scholar]
- World Health Organization (WHO) Global tuberculosis control 2011. WHO; 2011. http://www.who.int/tb/publications/global_report/en/. Accessed 23 Apr 2012. [Google Scholar]
- World Health Organization (WHO) Global tuberculosis report 2013. WHO; 2013. http://www.who.int/tb/publications/global_report/en/. Accessed 7 Aug 2014. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.