

Abstract
Background
T follicular helper (Tfh) cells underpin T-cell dependent humoral immunity and the success of most vaccines. Tfh cells also contribute to human immune disorders such as autoimmunity, immunodeficiency and malignancy. Understanding the molecular requirements for the generation and function of Tfh cells will provide strategies for targeting these cells to modulate their behavior in the setting of these immunological abnormalities.
Objective
To determine the signaling pathways and cellular interactions required for the development and function of Tfh cells in humans.
Methods
Human primary immunodeficiencies (PIDs) resulting from monogenic mutations provide a unique opportunity to assess the requirement for particular molecules in regulating human lymphocyte function. Circulating Tfh (cTfh) cell subsets, memory B cells and serum Ig levels were quantified and functionally assessed in healthy controls as well as patients with PIDs resulting from mutations in STAT3, STAT1, TYK2, IL21, IL21R, IL10R, IFNGR1/2, IL12RB1, CD40LG, NEMO, ICOS or BTK.
Results
Loss-of function (LOF) mutations in STAT3, IL10R, CD40LG, NEMO, ICOS or BTK reduced cTfh frequencies. STAT3, IL21/R LOF and STAT1 gain-of function mutations skewed cTfh differentiation towards a phenotype characterized by over-expression of IFNγ and programmed death -1 (PD-1). IFNγ inhibited cTfh function in vitro and in vivo, corroborated by hypergammaglobulinemia in patients with IFNGR1/2, STAT1 and IL12RB1 LOF mutations.
Conclusion
Specific mutations impact the quantity and quality of cTfh cells, highlighting the need to assess Tfh cells in patients by multiple criteria, including phenotype and function. Furthermore, IFNγ functions in vivo to restrain Tfh-induced B cell differentiation. These findings shed new light on Tfh biology and the integrated signaling pathways required for their generation, maintenance and effector function, and explain compromised humoral immunity in some PIDs.
Keywords: T follicular helper cells, humoral immunity, primary immunodeficiencies, cytokine signaling
INTRODUCTION
Naïve CD4+ T cells differentiate into distinct populations of effector cells with specialized functions. Such fine-tuning ensures the generation of appropriate immune responses that efficiently clear pathogens and generate long-term protective immunity following infection or vaccination1. The CD4+ T cells responsible for mediating the differentiation of naïve B cells into memory cells and plasma cells, thereby providing effective humoral immunity against T-dependent (TD) antigen (Ag), are T follicular helper (Tfh) cells2–5. Tfh cells express elevated levels of CXCR5, PD-1, Bcl-6, and several molecules involved in T-cell/B-cell interactions and localize to follicles of secondary lymphoid tissues2–4. Differentiation of naïve CD4+ T cells into Tfh cells is a complex process requiring integration of signals delivered by dendritic cells, B cells, cytokines, specific signaling pathways and transcription factors2–5. The critical role of Tfh cells in eliciting long-lived humoral immunity is evidenced by impaired generation of germinal centers (GCs), memory B cells and Abs to TD Ag in mice and humans who lack genes that promote Tfh formation2–6. Ab-mediated autoimmune conditions can also be caused by dysregulated Tfh function7–9. Thus, delineating molecular requirements underlying Tfh generation and function are important in understanding how these cells operate and in identifying pathways that could be targeted in the settings of vaccination, immunodeficiency, or autoimmunity.
Although studies of mice and some human immune disorders have taught us much about Tfh cells, our understanding of human Tfh biology remains incomplete, largely due to limited access to lymphoid tissues where Tfh cells are located. However, progress has been made by studying circulating CD4+CXCR5+ T cells as correlates of tissue Tfh cells. Subsets of circulating Tfh (cTfh) cells have been reported, with CCR6+, CCR6+PD-1+ or CCR7loPD-1hi subsets being superior to other subsets in providing B cell help10, 11. These subsets correlated with antibody (Ab) responses to influenza virus following vaccination in young adults, but not older adults12, are increased in autoimmune diseases8, 10, 13–16, and decreased in HIV-infection11. Similarly, CD4+CXCR5+PD1+ CXCR3− T cells were identified as the circulating counterpart of lymphoid Tfh cells and their frequencies positively correlated with neutralizing Abs in HIV infection17. Although these studies generally confirmed that PD-1+CXCR3−/CCR6+ cTfh cells are a reliable correlate of Tfh cells in human lymphoid tissue, the identity of the circulating B-helper human CD4+ T cells remains contentious, as other studies demonstrated that CXCR5+CXCR3+ or even CXCR5− CD4+ T cells exhibit detectable B-helper function11, 15, 19, 20 and correlate with influenza vaccine responsiveness18, 19
To assess the molecular requirements for the generation and function of human cTfh cells, we investigated >110 individuals with 14 different monogenic mutations that underlie primary immunodeficiencies (PIDs). Our findings identify mutations that have distinct quantitative and/or qualitative effects on human cTfh cells, providing an explanation for humoral immune defects in some PIDs as well as insights into mechanisms regulating human Tfh differentiation and function.
Methods
Human samples
Peripheral blood mononuclear cells (PBMCs) were isolated from healthy controls (Australian Red Cross) and PID patients. Human spleens were obtained from cadaveric organ donors (NSW Organ Transplant Registry). All studies were approved by Institutional Human Research Ethics Committees.
Antibodies and Reagents
eFluor660-anti-IL-21, PerCP-Cy5.5-anti-IFNγ, FITC-anti-CD45RA, biotin-PD-1 were from eBiosciences. Alexa647-anti-CXCR5 and anti-pSTAT1, APC-anti-CD10, APC-Cy7-anti-CD4, BV605-anti-IgG, PE-anti-pSTAT3 and anti-CCR6, Pe-Cy7-anti-CD25 and anti-CD27, PerCpCy5.5-anti-CD127, biotin-anti-IgA, SA-PerCpCy5.5, and recombinant IFNγ were from Becton Dickinson. BV421-anti-CXCR3, Pacific Blue-anti-CD20 and SA-BV605 were from Biolegend.
Lymphocyte phenotyping and isolation
T cells: PBMCs were incubated with mAbs to CD4, CD45RA, CD127, CD25, CXCR5, CXCR3, CCR6 and PD-1 and proportions of regulatory T cells (CD4+CD127loCD25hi), total memory (CD4+CD45RA−), cTfh (CD4+CD45RA−CXCR5+), as well as subsets of non-cTfh memory and cTfh cells defined according to CXCR3 and CCR6 expression were determined10, 20. To isolate these subsets, Tregs were excluded and the remaining population sorted into naïve (CD45RA+ CXCR5−CXCR3−CCR6−), non-Tfh memory (CD45RA−CXCR5−) and cTfh cells. Subsets of non-cTfh and cTfh cells were identified according to differential CXCR3 and CCR6 expression10. All populations were sorted on a FACS ARIA (Becton Dickinson) to > 98% purity.
B cells: PBMCs were incubated with mAbs to CD20, CD27, CD10, IgG and IgA, and the frequency of total memory (CD20+CD27+CD10−) and switched memory B cells determined21, 22.
Expression of phospho-STATs
Epstein Barr virus transformed lymphoblastoid cell lines (EBV-LCLs) established from healthy donors, IFNGR2LOF, or STAT1LOF were stimulated with IFNγ or IL-21 for 30 mins. Cells were fixed, permeabilised and stained for anti-pSTAT1 and anti-pSTAT321.
Analysis of CD4+ T cell function in vitro
Isolated CD4+ T cell populations were cultured with T cell activation and expansion (TAE) beads (anti-CD2/CD3/CD28; Miltenyi Biotech) in 96 well round bottomed well plates. After 5 days, supernatants were harvested and production of IL-4, IL-5, IL-10, IL-13, IL-17A, IL-17F, IFNγ and TNFα determined by cytometric bead arrays (Becton Dickinson); secretion of IL-22 (eBioscience) and CXCL13 (R&D systems) was determined by ELISA. For cytokine expression, activated CD4+ T cells were re-stimulated with PMA (100 ng/ml)/ionomycin (750 ng/ml) for 6 hours, with Brefeldin A (10 µg/ml) added after 2 hours. Cells were then fixed and expression of intracellular cytokines detected20, 22, 23. For gene expression, RNA was extracted, and transcribed into cDNA. Expression of TBX21, GATA3, RORC and BCL6 was determined by qPCR and standardized to GAPDH20, 24.
T-B cell co-culture assays and Ig determination
CD4+ T cell subsets were treated with mitomycin C (100 µg/ml, Sigma) and then co-cultured at a 1:1 ratio (50 × 103/200µl/well) with allogeneic total splenic B cells20, 22, 24. In some experiments, exogenous IFNγ was added to the cultures. After 7 days Ig secretion was determined by ELISA20, 21. Serum IgG and IgM levels were determined by nephelometry.
Statistical analysis
Significant differences were determined using a one-way ANOVA (Prism; GraphPad Software).
RESULTS
Cytokine and transcription factor expression by human naïve and memory CD4+ T cells
To determine the requirements for generating human Tfh cells in vivo, we first defined parameters that identify different CD4+ T cell subsets in peripheral blood. Memory cells were the predominant producers of all cytokines examined, producing ~5–100 fold higher levels of Th1 (IFNγ), Th2 (IL-4, IL-5, IL-13), and Th17 (IL-17A, IL-17F, IL-22) cytokines, as well as B-cell helper/Tfh cytokines (IL-10, IL-21), than naïve cells (Figure 1A–D). Memory CD4+ T cells also expressed significantly higher levels of TBX21 (T-bet), GATA3 and RORC (RORγt) than naïve cells (Figure 1E–G). There was no significant difference in BCL6 expression between naïve and memory cells (Figure 1H), consistent with other studies reporting Bcl-6 levels are similar in circulating human CD4+ T cell subsets8, 10, 12, 15, 17, 25.
Figure 1. Identification of effector subsets within populations of human memory CD4+ T cells.
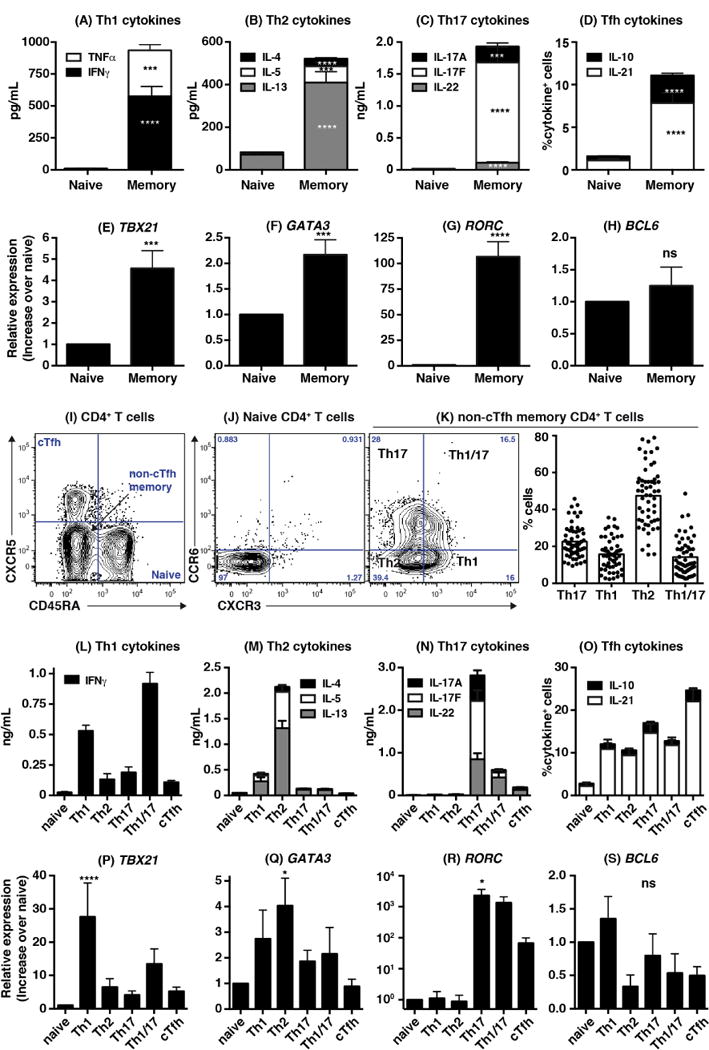
(A–H): Naïve and memory CD4+ T cells were sorted from healthy controls and stimulated with TAE (anti-CD2/CD3/CD28) beads. Secretion/expression of the indicated cytokines (A–D; mean ± SEM; n=25–27) or transcription factors (E–H; mean ± SEM; n=12–19) were determined after 5 days. (I) Resolving blood naïve (CD45RA+CXCR5−), non-Tfh memory (CD45RA−CXCR5−) and cTfh (CD45RA−CXCR5+) cells from healthy controls. (J, K) CXCR3 and CCR6 expression on naive and non-Tfh memory cells; (K) depicts % of Th1 (CXCR3+CCR6−), Th2 (CXCR3−CCR6−), Th17 (CXCR3-CCR6+) and Th1/17 (CXCR3+CCR6+) subsets amongst the non-Tfh memory population (n=55–58). (L–S) Secretion/expression of the indicated cytokines (L–O; mean ± SEM; n=10–15) or transcription factors (P–S; mean ± SEM; n=7–10) by naïve, Th1, Th2, Th17, Th1/Th17 and cTfh subsets after 5 days of culture with TAE beads. Significant differences (one-way ANOVA) between naïve and memory CD4+ T cells or subsets are indicated.
Delineation of memory CD4+ T cells into defined populations of Th1, Th2, Th17 and cTfh cells and subsets
Human memory Th1, Th2, Th17 and cTfh cells can be defined according to differential expression of CXCR3, CCR6 and CXCR526, 27, with Th1 cells being CD45RA−CXCR5−CXCR3+CCR6−, Th17 cells CD45RA−CXCR5−CXCR3−CCR6+, Th2 cells CD45RA−CXCR5−CXCR3−CCR6−, and cTfh cells CD45RA−CXCR5+ (Figure 1I–K). In contrast, CD45RA+ naïve cells lack these chemokine receptors (Figure 1I, J). We extended these findings by demonstrating Th1 cells were enriched for IFNγ secretion (Figure 1L), Th2 cells produced the greatest amounts of IL-4, IL-5 and IL-13 (Figure 1M), while Th17 cells secrete the most IL-17A, IL-17F and IL-22 (Figure 1N). Importantly, the highest proportion of IL-21-expressing cells was detected in the cTfh subset, which also contained a greater proportion of IL-10-expressing cells than Th1 and Th2 subsets (Figure 1O). Indeed, of the cytokines examined, IL-10 and IL-21 were the only ones produced by cTfh cells at levels significantly greater than naïve cells (Figure 1L–O; p<0.005), consistent with the B-cell helper function of these cytokines28. The proportions of cTfh cells expressing IL-10 and IL-21 were also significantly greater than naïve cells (p<0.005), Th1 cells (p<0.05), Th2 cells (p<0.05) and Th1/Th17 cells (p<0.05), but not Th17 cells. The memory CD4+ T cell population co-expressing CCR6 and CXCR3 (termed Th1/17 cells) produced both Th1 and Th17, but only low levels of Th2, cytokines (Figure 1L–O). Thus, these cells represent pro-inflammatory cells, consistent with their detection in inflamed tissues29. TBX21, GATA3 and RORC expression also significantly correlated with their respective production of Th1, Th2 and Th17 cytokines (Figure 1P–S).
Having successfully characterized Th1, Th2, Th17 and cTfh cells in peripheral blood, we next investigated cTfh cells using a similar approach: dividing them into subpopulations according to CCR6 and CXCR310 (Figure 2A, B) and assessing cytokine production and B-cell helper function. The different cTfh subsets produced lower levels of cytokines than memory (CD45RA−CXCR5−) cells, however IL-4 and IL-13 were enriched in the CXCR3−CCR6− (double negative; DN) subset, IL-17A/F and IL-22 in the CCR6+CXCR3− (hereafter referred to as CCR6+) subset, and IFNγ in the CXCR3+CCR6− (“CXCR3+”) and CXCR3+CCR6+ (double positive; DP) subsets (Figure 2C–F). In contrast, IL-10, IL-21 (Figure 2F) and CXCL13 (Figure 2G), which are produced by Tfh cells25, 30, were comparable in all cTfh subsets. Total cTfh cells induced greater B-cell differentiation than naïve and memory cells (Figure 2H) as well as Th1, Th2, Th17 and Th1/17 memory cell subsets (Figure 2I). Amongst cTfh subsets, the CCR6+ population consistently induced most Ig secretion by co-cultured B cells over the CXCR3+, DN and DP subsets (Figure 2J). Thus, consistent with recent studies, blood CD4+CXCR5+ T cells have Tfh function, with the CCR6+ subset being most effective10, 11, 17.
Figure 2. Delineating subsets of human circulating Tfh cells.
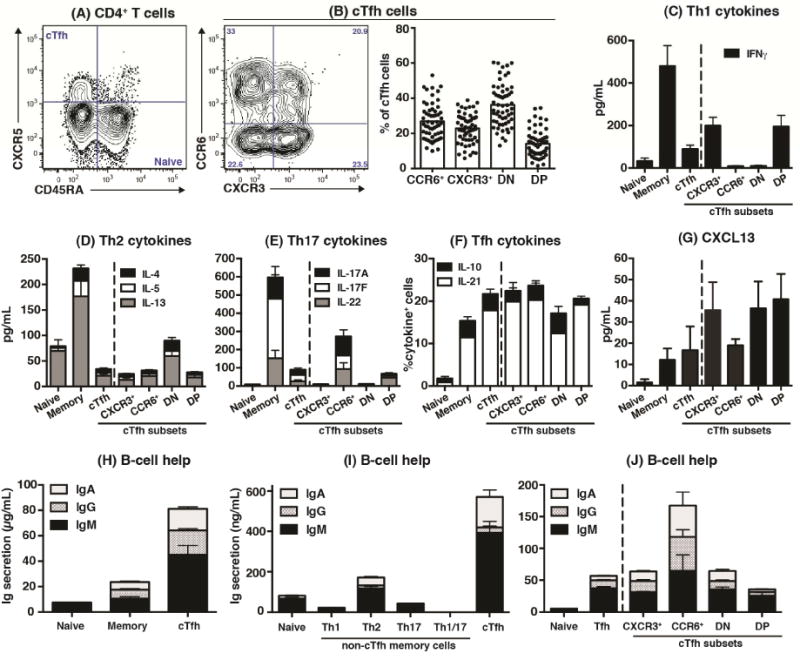
(A, B) cTfh cells were defined as CD45RA−CXCR5+ CD4+ T cells. CXCR3+CCR6−, CXCR3−CCR6−, CXCR3−CCR6+ and CXCR3+CCR6+ subsets were then detected (n=55–58). (C)–(G): Naïve, non-Tfh memory, total cTfh and CXCR3+CCR6−, CXCR3−CCR6+ CXCR3−CCR6− (DN), and CXCR3+CCR6+ (DP) cTfh subsets were sorted from peripheral blood and stimulated with TAE beads. After 5 days, secretion or expression of the indicated cytokines were determined (n=5–6). (H)–(J): purified subsets of blood CD4+ T cells were co-cultured with allogeneic B cells and TAE beads. IgM, IgG and IgA secretion was determined after 7 days.
Mutations causing PIDs impact CD4+ T cell function and B-cell memory formation
These data established the ability to identify effector functions of human CD4+ T cell subsets, thereby providing a framework to determine consequences of gene mutations on CD4+ T cell differentiation in vivo. We examined CD4+ T cell subsets and function in PID patients with mono- or bi-allelic mutations in surface receptors or cytokine signaling pathways that may impact humoral immunity in the context of Tfh formation and long-lived protective Ab responses4.
LOF mutations in CD40LG, NEMO, IL12RB1 or TYK2 compromised IFNγ production by memory CD4+ T cells (Figure 3A, see Table E1 in this articles Online Repository), consistent with known roles in eliciting Th1 responses31. Production of Th2 cytokines was enhanced by IL12RB1, IFNGR1/2 or TYK2 LOF mutations, confirming Th1 cells suppress Th2 cells32, as well as by LOF mutations in IL10R, IL21/R or STAT3 and GOF mutations in STAT1 (Figure 3B, Table E1). This revealed novel roles for IL-10, IL-21 and STAT3 as regulators of human Th2 immunity, which differ to studies in mice33, 34. B cells also clearly regulate CD4+ T cell differentiation, revealed by reduced IFNγ and increased Th2 cytokines in B-cell deficient individuals with BTK mutations (Figure 3A, B; Table E1). STAT3, NEMO, ICOS, IL12RB1 or TYK2 LOF mutations strongly diminished Th17 cytokines (Figure 3C) and RORC expression (not shown). STAT1 GOF and IL21/R LOF mutations also reduced Th17 cytokines but to a lesser extent than these other mutations (Figure 3C, Table E1). These findings are consistent with susceptibility of some individuals with these mutations to Candida infection (STAT3LOF, STAT1GOF, IL21R, IL12RB1, NEMO)31, 35, and the requirement for ICOS in human Th17 cell generation36. In contrast, STAT1LOF mutations had no or partial effect on IL-17A/F or IL-22 (Figure 3C, Table E1), mirroring intact immunity against Candida spp in these patients31, 35, while IL10R mutations resulted in excessive production of IL-17A/F and IL-22 (Table E1). IL-10 production was compromised by STAT3, TYK2, IL10R and IL21R LOF and STAT1 GOF mutations, but unaffected by other mutations, while IL-21 was reduced by STAT1 GOF, IL21R, IFNGR1/2 and NEMO LOF mutations (Figure 3D, Table E1).
Figure 3. Defects in cytokine production due to monogenic mutations causing different PIDs.
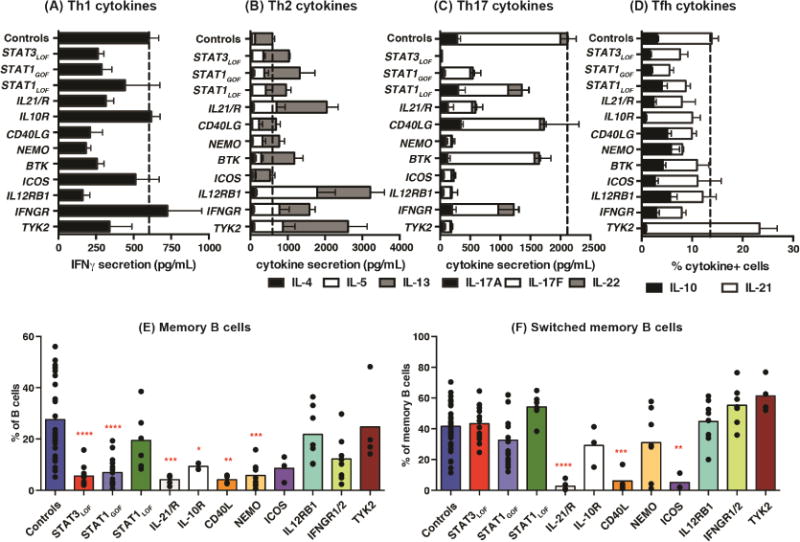
(A–D) memory CD4+ T cells were sorted from healthy controls or patients with the indicated gene mutations then stimulated with TAE beads. Production of (A) IFNγ, (B) IL-4, IL-5, IL-13 (Th2 cytokines), (C) IL-17A, IL-17F, IL-22 (Th17 cytokines), (D) IL-10, IL-21 (Tfh cytokines) was determined after 5 days (mean ± SEM). Controls: n=26–35; STAT3LOF: n=6; STAT1GOF: n=7–9; STAT1LOF: n=5–7; IL21/R: n=5; IL10R: n=2; CD40LG: n=4; NEMO: n=5–6; BTK: n=6; ICOS: n=4; IL12R: n=5; IFNGR1/2: n=6; TYK2: n=3. See Table E1 for more detailed results. (E–F) proportions of total (E) and class switched (F) memory B cells in healthy controls (n=37) and patients [STAT3LOF: n=11–12; STAT1GOF: n=16; STAT1LOF: n=7; IL21/R: n=5; IL10R: n=3–4; CD40LG: n=4; NEMO: n=7–10; ICOS: n=3; IL12R: n=8; IFNGR1/2: n=7–8; TYK2: n=4] were determined. Significant differences (one-way ANOVA) between controls and patients are indicated.
To establish regulators of human lymphocyte differentiation in the context of TD B-cell function, we assessed PID patients for memory B cells. There were marked reductions in memory B cells in patients with STAT3, IL21/R, IL10R, ICOS, CD40LG and NEMO LOF, and STAT1 GOF, mutations (Figure 3E). It is thus likely that signaling via IL-10R and IL-21R underlie the paucity of memory B cells in STAT3-deficient patients. Although memory B cells were reduced in patients with STAT3 LOF, STAT1 GOF, IL10R and NEMO mutations, the residual memory cells still underwent class switching. In contrast, a lack of CD40L, ICOS or IL-21/IL-21R signaling impaired isotype switching. The frequencies of memory B cells were unaffected by LOF mutations in STAT1, TYK2, IL12RB1 or IFNGR1/2, however there was a trend for more switched memory B cells in most of these patients (Figure 3F). Together, these data demonstrated the validity of using PID patients as models for defects in B and CD4+ T cell differentiation and cytokine production.
Specific gene mutations compromise the generation of human circulating Tfh cells
We next used these patients to examine the relationship between PID-associated gene mutations and phenotypically-defined subsets of CD4+ T cells. Th1 cells were significantly increased in STAT3LOF and STAT1GOF patients, while Th17 cells were significantly reduced in these as well as in NEMO, ICOS or BTK-deficient patients (Table 1), consistent with poor production of IL-17A/IL-17F, and IL-22 by memory cells from most of these patient groups (Figure 3C, Table E1). The reduction in CCR6+ CD4+ T cells due to STAT3 mutations likely reflects the inability of mutant STAT3 to induce CCR6, which has been shown to be a direct target of STAT3 in mice37. Th2- and Th1/17-phenotype cells were unaffected by most of the mutations examined (Table 1). When cTfh cells were measured, significant reductions were observed in patients with mutations in STAT3, CD40LG, BTK and ICOS - consistent with previous studies20, 38, 39 - as well as in IL10R or NEMO (Figure 4A, B). In our cohort of CD40LG-deficient patients, three had clinically milder disease than those with classic HIGM (ie detectable levels of IgG/A, less severe infections, later age of diagnosis). These patients had normal cTfh frequencies, thus correlating with disease severity (Figure 4A, B). While there were trends for fewer cTfh when signaling through IL-21R, IL-12R or TYK2 was compromised, these differences were not significant (Figure 4A, B). None of the other mutations examined impacted Tfh formation, as determined by quantifying circulating CD4+CXCR5+ T cells. In addition to Tfh cells that promote B-cell differentiation, there is also a population of cells termed T follicular regulatory (Tfr) cells that restrain Tfh function to regulate production of specific Abs40. These cells can be defined as CXCR5+ cells within the population of Tregs. Although specific gene mutations impacted the formation of cTfh cells (Figure 4A, B), the proportions of CXCR5+ Tregs (putative Tfr cells) in all groups of PID patients were similar to those in healthy controls. Thus, the effects of the gene mutations studied here are specific to cTfh cells, suggesting distinct origins of cTfh and cTfr cells. This is consistent with Tfr cells arising from Tregs, which are largely unaffected by the mutations studied here (Table 1), rather than Tfh cells40.
Table 1.
Tregs and CD4+CXCR5− Memory subsets in human PIDs
| Genetic diseases | Tregs | CXCR5+ Tregs (%Tregs) | Total memory | CD4+CXCR5− memory cells | |||
|---|---|---|---|---|---|---|---|
| Th1 (CXCR3+ CCR6−) | Th2 (CXCR3−CCR6−) | Th17 (CXCR3−CCR6+) | Th1/17 (CXCR3+ CCR6+) | ||||
| Controls (n=47–71) |
6.1 ± 0.3 | 8.2 ± 0.9 | 52.2 ± 1.8 | 15.5 ± 1.0 | 47.3 ± 1.8 | 23.0 ± 1.1 | 14.1 ± 1.1 |
|
STAT3LOF (n=12–22) |
7.4 ± 0.7 | 8.7 ± 1.7 | 29.6 ± 3.7*** | 34.4 ± 4.3**** | 46.3 ± 5.1 | 5.3 ± 0.7**** | 10.0 ± 1.5 |
|
STA T1GOF (n=16–23) |
5.6 ± 0.7 | 12.0 ± 1.6 | 41.0 ± 3.7 | 27.4 ± 3.4* | 56.9 ± 3.7 | 6.9 ± 1.05**** | 9.5 ± 1.6 |
|
STA T1LOF =5–6) |
7.5 ± 1.1 | 15.9 ± 5.5 | 38.0 ± 6.1 | 22.4 ± 6.2 | 37.9 ± 7.6 | 20.5 ± 4.4 | 19.2 ± 5.0 |
|
IL-21/R (n=5) |
5.8 ± 1.1 | 6.6 ± 2.4 | 32.2 ± 7.1 | 25.1 ± 11.2 | 57.6 ± 10.5 | 12.0 ± 3.3 | 5.3 ± 1.1 |
|
IL10R (n=4) |
6.4 ± 1.2 | 9.8 ± 5.3 | 31.5 ± 9.4 | 20.1 ± 17.0 | 52.8 ± 12.3 | 23.0 ± 10.9 | 4.1 ± 1.8 |
|
CD40LG (n=8) |
3.6 ± 0.8 | 6.0 ± 1.4 | 25.9 ± 3.8** | 6.9 ± 2.0 | 70.7 ± 6.0* | 17.9 ± 2.8 | 4.6 ± 2.0 |
|
NEMO (n=8–12) |
9.0 ± 1.7 | 8.3 ± 1.7 | 40.2 ± 7.6 | 2.6 ± 1.6 | 85.2 ± 4.0**** | 10.7 ± 2.3** | 1.4 ± 0.7** |
|
BTK (n=4–11) |
4.9 ± 0.7 | 5.5 ± 1.1 | 30.0 ± 5.2** | 12.3 ± 3.6 | 71.4 ± 6.5 | 11.2 ± 1.8* | 5.2 ± 1.6 |
|
ICOS (n=6) |
8.8 ± 0.9 | 10.1 ± 2.8 | 53.1 ± 7.1 | 30.5 ± 6.3 | 50.1 ± 4.5 | 10.4 ± 1.4* | 8.9 ± 1.7 |
|
IL12RB1 (n=3) |
4.1 ± 1.8 | 10.8 ± 2.2 | 16.4 ± 3.0* | 12.8 ± 2.1 | 67.8 ± 7.6 | 16.3 ± 8.4 | 3.0 ± 1.2 |
|
IFNGR1/2 (n=4–7) |
15.8 ±5.0**** | 10.5 ± 1.5 | 49.1 ± 7.0 | 15.7 ± 3.6 | 55.9 ± 5.8 | 16.8 ± 2.6 | 11.5 ± 2.9 |
|
TYK2 (n=4) |
5.4 ± 0.6 | 3.3 ± 1.2 | 56.2 ±10.5 | 18.3 ± 5.6 | 43.4 ± 5.3 | 18.8 ± 4.6 | 19.5 ± 5.1 |
PBMCs from healthy controls or patients with specific gene mutations were labeled with mAb against CD4, CD127, CD25, CD45RA, CXCR5, CXCR3 and CCR6. The proportions of Tregs amongst all CD4+ T cells, the proportions Tregs expressing CXCR5 (putative Tfr cells), proportions of memory cells within the non-Treg cell population, and the proportions of CD4+CD45RA−CXCR5− memory cells with a Th1 (CXCR3+CCR6−), Th2 (CXCR3−CCR6−), Th17 (CXCR3−CCR6+) or Th1/17 (CXCR3+CCR6+) phenotype were then determined by flow cytometric analysis. The values represent mean ± SEM for the indicated number of healthy controls or patients. Significant differences (one-way ANOVA) between healthy controls and patients are indicated.
Figure 4. Effect of monogenic mutations on the generation of human cTfh cells.
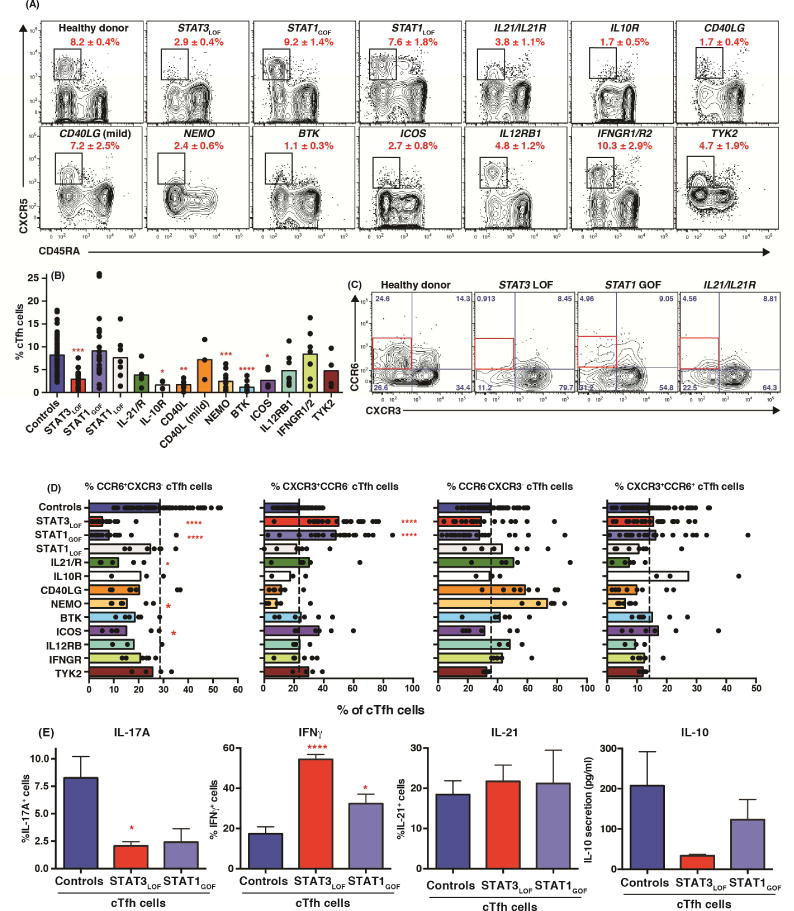
(A, B) cTfh cells were identified amongst the population of non-Tregs as CD45RA−CXCR5+ CD4+ T cells. cTfh frequencies in healthy donors (n=72) and patients with mutations in STAT3 (n=24), STAT1 (GOF [n=22]; LOF [n=7]), IL-21R/IL21 (n=5), IL10R (n=4), CD40LG (n=10), NEMO (n=11), BTK (n=11), ICOS (n=6), IL12RB1 (n=8), IFNGR1/2 (n=7) or TYK2 (n=4) were determined. (C, D) proportions of CXCR3+CCR6−, CXCR3−CCR6+, CXCR3−CCR6− or CXCR3+CCR6+ subsets amongst total cTfh cells [healthy donors: n=71; STAT3: n=21, STAT1: GOF n=20, LOF n=7; IL-21R/IL21: n=5; IL10R: n=3; CD40LG: n=8; NEMO: n=7; BTK: n=5; ICOS: n=6; IL12RB1: n=3; IFNGR1/2: n=6; TYK2: n=4]. (E) Production of IL-17A, IFNγ, IL-21 and IL-10 by sorted cTfh cells from healthy donors, STAT3LOF or STAT1GOF patients (mean ± SEM; n=3–4). Significant differences (one-way ANOVA) between controls and patients are indicated.
STAT3LOF or STAT1GOF mutations skew cTfh differentiation to a non-helper phenotype
When we analyzed cTfh subsets defined by CXCR3 and CCR6 expression10 (Figure 2D), we found significant reductions in CCR6+ cTfh cells – the most proficient B-helper cTfh population (Figure 2H–J)10 - in patients with STAT3LOF, STAT1GOF and IL21/IL21R mutations, and corresponding increases in CXCR3+ cTfh cells in STAT3LOF and STAT1GOF individuals (Figure 4C, D). The loss of CCR6+ cTfh cells in these patients was even more striking when ratios of CCR6+ to CXCR3+ cTfh cells were calculated (ie controls: 1.6; STAT3LOF: 0.18 [p<0.001]; STAT1GOF: 0.6 [p<0.05]; IL21/IL21R: 0.6). NEMO mutations resulted in more DN cTfh cells, while the DP subset was not affected by any mutations examined (Figure 4D). Thus, not only did STAT3LOF mutations compromise cTfh generation, they also prevented formation of the cTfh subset most capable of inducing B-cell help. This perturbation to cTfh differentiation was mirrored by STAT1GOF and to a lesser extent IL21/IL21RLOF mutations (Figure 4A–D).
These data suggest that cTfh cells from patients with STAT3LOF and STAT1GOF mutations would exhibit skewed cytokine production. Indeed, cTfh cells from STAT3LOF individuals exhibited 4-fold fewer IL-17A-expressing and 4-fold more IFNγ-expressing cells than healthy controls (Figure 4E). Although IL-21 was expressed by comparable frequencies of control and STAT3LOF cTfh cells, IL-10 secretion by STAT3-deficient cTfh cells was markedly reduced (6-fold) (Figure 4E). STAT1GOF mutant cTfh cells exhibited similar, but less extreme, perturbations to cytokine production (Figure 4E). Thus, altered phenotypes of cTfh cells in some PIDs correlated with altered function with respect to cytokine production, demonstrating these mutations not only impact cTfh generation but also their quality, predominantly yielding a population with limited B-cell helper capacity, which is consistent with impaired humoral immunity in STAT3LOF and STAT1GOF individuals21, 41.
IFNγ restrains Tfh-induced B cell differentiation
IFNγ can impede Ig secretion by human PBMCs42–46. Taken together with our findings of skewed differentiation of STAT3LOF and STAT1GOF cTfh cells to an IFNγ-secreting subset, we hypothesized that IFNγ production by CXCR3+ cTfh cells would compromise their B-helper function (Figure 2J)10. To investigate this, it was important to demonstrate that IFNγ directly signals in human B cells. IFNγ induced STAT1 phosphorylation in EBV-LCLs from healthy controls, but not STAT1 or IFNGR-deficient individuals, whilst IL-21-induced STAT3 activation was unaffected by such mutations (Figure 5A). Next, we co-cultured cTfh and allogeneic B cells with or without exogenous IFNγ. Exogenous IFNγ suppressed TD differentiation of normal B cells by 40–50%, but had no effect on Ig secretion by IFNGR1- or STAT1-deficient B cells (Figure 5B), demonstrating B-cell intrinsic signaling via IFNγR1/STAT1 mediates the repressive effect of IFNγ on Tfh-induced B-cell differentiation.
Figure 5. IFNγ suppresses B-cell differentiation in vitro and in vivo.
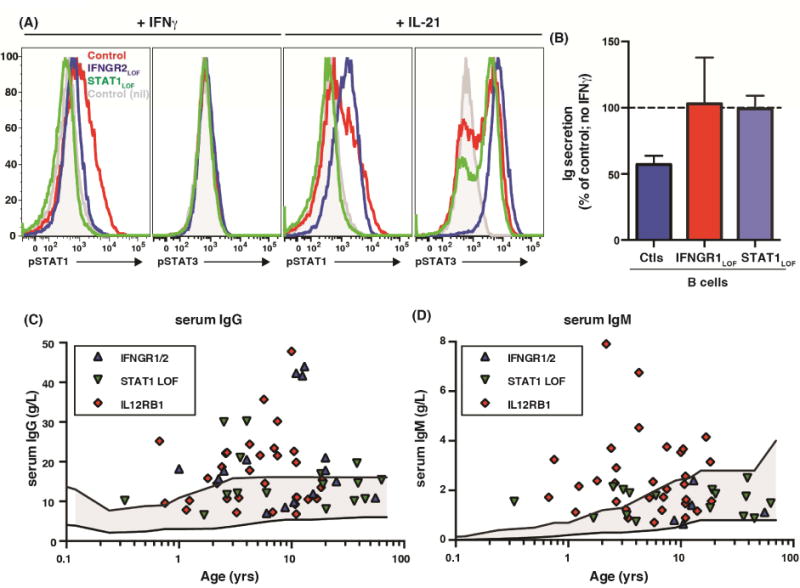
(A) Phosphorylation of STAT1 or STAT3 in EBV-LCLs from healthy controls (red histograms) or individuals with IFNGR2 (blue) or STAT1 (green) mutations in response to IFNγ or IL-21. Grey histogram: response of unstimulated cells from healthy controls. (B) cTfh cells from healthy controls were co-cultured with allogeneic normal, IFNγR1- or STAT1-deficient B cells in the absence or presence of exogenous IFNγ. Ig secretion was determined after 7 days. Values represent the mean % Ig secretion (± SEM) in the presence of IFNγ relative to it absence (defined as 100%). (C, D) serum levels of IgG and IgM in individuals with IFNGR1/IFNGR2, IL12RB1 or STAT1LOF mutations. The solid upper and lower lines correspond to normal serum Ig levels in age-matched controls
If IFNγ plays a substantial role in regulating Ig production in vivo, mutations in molecules regulating its production (ie IL12RB1) or signaling (ie IFNGR1 or STAT1) should cause hypergammaglobulinemia. Indeed, 60% (9/15), 38% (6/16) and 53% (17/32) of patients with IFNGR1/2, STAT1 and IL12RB1 LOF mutations, respectively, had serum IgG levels greater than age-matched controls (Figure 5C), while 60% and 25% of IL12RB1 or STAT1-deficient patients also had elevated serum IgM (Figure 5D). Notably, several patients whose Ig levels fell within the normal range were actually at the upper end of normal (Figure 5C, D). Thus, a key function of IFNγ in vivo is to suppress Ig production.
Mutations in STAT3, STAT1 and IL21R cause aberrant expression of PD-1 on CD4+ T cells
Although PD-1 is highly expressed on Tfh cells24, 47, only a subset of cTfh cells exhibit elevated PD-19, 11, 12, 15, 17. PD-1hi cTfh cells have been used as a biomarker of humoral immunity in health and disease8, 11, 12, 14, 15, 17. As cTfh development was perturbed by different PID-causing mutations, we examined PD-1 in these patients. PD-1 was expressed at low levels on naïve CD4+ T cells irrespective of genotype, and increased on non-cTfh memory cells from healthy controls and patients (Figure 6A, B). Non-Tfh memory cells from STAT1GOF and IL21/RLOF, and cTfh cells from STAT3LOF and STAT1GOF, individuals expressed significantly more PD-1 than controls (Figure 6A, B). None of the other mutations affected PD-1 expression, suggesting signaling via STAT3/STAT1, possibly downstream of IL-21, regulates PD-1 on memory CD4+ T cells.
Figure 6. Aberrant expression of PD-1 on cTfh cells from patients with STAT3LOF, STAT1GOF and IL21/RLOF mutations.
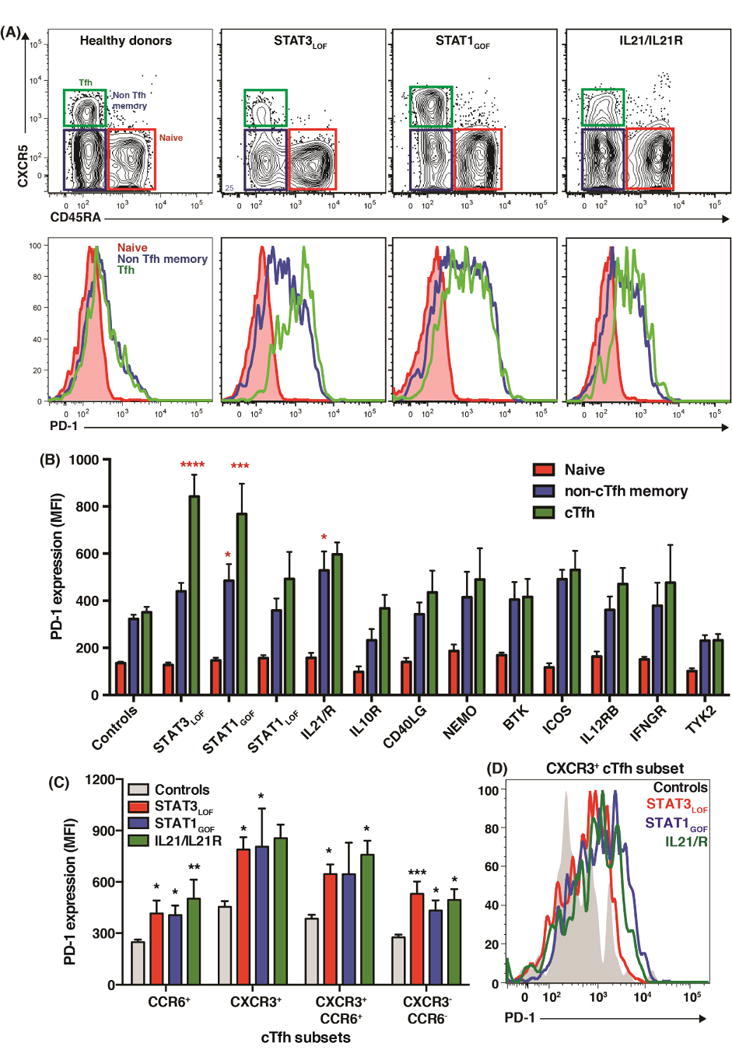
(A, B) PD-1 expression on naïve, non-Tfh memory and cTfh cells. Contour and histogram plots in (A) represent individual healthy controls or patients. The graph (B) depicts the average PD-1 expression (MFI ± SEM) for all individuals tested [healthy donors: n=45; STAT3: n=15; STAT1: GOF n=18, LOF n=7; IL-21R/IL21: n=5; IL10R: n=4; CD40LG: n=3; NEMO: n=5–6; BTK: n=5–6; ICOS: n=5; IL12RB1: n=4; IFNGR1/2: n=4; TYK2: n=4]. (C, D) PD-1 expression on CXCR3+CCR6−, CXCR3-CCR6+, CXCR3−CCR6− and CXCR3+CCR6+ cTfh subsets cells from healthy controls (n=25) or patients with STAT3LOF (n=10), STAT1GOF (n=10) or IL21/RLOF mutations (n=5). Significant differences (one-way ANOVA) between controls and patients are indicated.
As STAT3LOF, STAT1GOF and to a lesser extent IL21R mutations skewed cTfh differentiation towards a CXCR3+ phenotype and upregulated PD-1, we tested whether these two observations were related. PD-1 was expressed by all cTfh subsets from healthy controls, with significantly greater expression on CXCR3+ and CXCR3+CCR6+ subsets compared to CCR6+ and CCR6−CXCR3− subsets (Figure 6C). Despite this, PD-1 was significantly higher on all cTfh subsets from STAT3LOF patients, and for most cTfh subsets from STAT1GOF and IL21R-deficient patients compared to controls (Figure 6C, D). Thus, STAT3LOF, STAT1GOF or IL21/IL21R mutations dysregulate PD-1 expression.
Discussion
It was first reported in 1965 that thymus-derived cells are required for Ag-specific Ab responses48. While it was initially proposed that Th2 cells mediated B-cell differentiation32, it is now clear that this is mediated by Tfh cells. For this reason, Tfh cells are being used as a biomarker for humoral immunity8−15, 17, 18, 49. Furthermore, targeting Tfh cells may constitute a viable approach of enhancing the efficacy of some vaccines and treating diseases caused by autoantibodies2, 4, 5, 7. Tracking Tfh cells in humans requires clear understanding of their circulating counterparts. Recent studies revealed heterogeneity within circulating CD4+CXCR5+ T cells, with CCR7loPD1+ or CXCR3−/CCR6+PD1+ subsets emerging as the most efficient helpers for B-cell differentiation8, 11, 17. Notably, frequencies of these subsets correlated with in vivo Tfh behavior, such as generating neutralizing Abs following infection/vaccination12, 17, excessive activity in autoimmunity8, 9, 13–15, or impaired function in HIV infection11, 49. However, other studies found that CXCR3+ cTfh18 or CD4+CXCR5− T cells19 predicted Ab responses following vaccination. Thus, despite substantial advances in our understanding of human Tfh biology, the exact correlates between successful Ab responses and CD4+ T cells subsets remain controversial and present a roadblock to translating these findings to the clinic.
Here we examined a wide spectrum of PIDs to delineate quantitative and qualitative consequences of gene mutations on cTfh cells. Consistent with previous studies, cTfh cells were reduced by mutations in CD40LG, ICOS, BTK or STAT320, 38, 39. Our finding of reduced cTfh cells in NEMO-deficiency revealed another similarity between disease due to CD40LG and NEMO mutations, consistent with NEMO being required for CD40 signalling35. The significant reduction in cTfh cells in IL-10R-deficiency was unexpected, as IL-10/IL-10R-signalling in mice reduced Tfh formation4. This points to possible species-specific differences in the role of IL-10 in regulating human and murine Tfh cells, which parallels the distinct effects of IL-10 on B-cell differentiation in these species28. The deficits in cTfh cells in these groups of PID patients are likely a direct effect of the mutation, rather than being secondary to infection, as cTfh cells were detected at normal frequencies in STAT1 and IFNGR1/2-deficient individuals. Our findings highlight that interactions between CD4+ T cells, DCs and B cells are required for Tfh formation2–5. It is likely that CD40L/CD40/NEMO signaling in B cells contributes to the maintenance of cTfh cells in humans. However, a T-cell intrinsic role for NEMO cannot be discounted. The importance of B cells is also evident from studies reporting diminished cTfh frequencies in patients with severe B-cell deficiency due to E4750 or NFKB251 mutations. Our data also shed light on the division of labor between STAT3-activating cytokines in generating cTfh cells, as well as on the functionality and requirements for cTfh subset formation. Mutations in STAT3 or IL10R, but not IL21/IL21R, had comparable deleterious effects on cTfh frequencies, implying IL-10/STAT3 signaling is important in establishing the pool of cTfh cells. However, STAT3LOF, STAT1GOF and IL21/RLOF mutations, but not IL10R-deficiency, reduced CCR6+ cTfh cells most likely due to STAT3 directly regulating CCR6 expression37. The resultant population of cTfh cells in these individuals was skewed to a CXCR3+IFNγ++IL-10low phenotype resembling STAT3-deficient murine Tfh cells, which also adopt a Th1 fate52. This revealed a dominant role for IL21/STAT3 signaling in specifying cTfh subsets. These data demonstrate that specific mutations cause qualitative changes in cTfh cells that would not be apparent by quantifying CD4+CXCR5+ T cells.
The mechanism underlying greater helper function of CCR6+, and corresponding poor function of CXCR3+, cTfh cells is unknown. Although it was reported that CCR6+ cTfh cells produce more IL-21 than other subsets10, this was not confirmed by us or others11, 13, 17 and is unlikely to explain their ability to promote B-cell differentiation. Rather, based on several lines of evidence including our data, we propose this results from reduced IFNγ production. First, exogenous IFNγ reduced Ig production by human PBMCs43, 44 or in B/Tfh cell co-cultures45. This was not observed for IFNGR1 or STAT1-deficient B cells, demonstrating a B-cell intrinsic inhibitory effect of IFNγ. Second, Mycobacterium leprae-specific IgG levels negatively correlated with IFNγ production by PBMCs in response to M leprae Ags42. Third, serum Ig was elevated in many patients with mutations in the IFNγ signaling pathway. Fourth, there were trends for increased class switched memory B cells in individuals with IFNGR1/2, TYK2 or STAT1LOF mutations. Overall, IFNγ appears to attenuate Ag-specific Ab responses in humans in vivo, thereby explaining the poor helper function of CXCR3+ cTfh cells. As IFNγ is induced by IL-12/IL-12R/Tyk2 signalling and itself signals through STAT1, it is likely that the similar phenotypes detected in patients with IL12RB1, TYK2, IFNGR1/2 and STAT1 LOF mutations reflect the contribution of a common pathway to the regulation of B-cell differentiation and Ig secretion.
The abundance of IFNγ-producing cTfh cells in STAT3LOF and STAT1GOF patients, together with reduced CCR6+ cTfh cells, would thus contribute to impaired humoral immune responses21, 41. This would be further compounded by reduced IL-10 production, an important cytokine for B cell differentiation in the context of Tfh/B cell interactions22, 28, 45. CCR6+ cTfh cells are also reduced in HIV-infection11, where Tfh cells exhibit impaired B-helper function11, 49, 53. The similarities in cTfh dysregulation in HIV infection and PIDs identifies intrinsic (specific gene mutations) and extrinsic/environmental (chronic viral infection/pathogen exposure) factors as modifiers of Tfh differentiation and function. Thus, it would be interesting to determine whether Tfh cells in HIV+ patients also have skewed cytokine profile with increased IFNγ and reduced IL-10. Our findings regarding IFNγ and impaired humoral immunity also explain the paradoxical data of Ueno and colleagues that cTfh cells are reduced in IL-12Rβ1-deficiency, yet these patients have intact if not heightened Ab responses to vaccination or infection54. Here, while impaired IL-12R signaling may reduce cTfh cells54, there will also be less IL-12-induced IFNγ production, resulting in a qualitative change in the cTfh cytokine repertoire, with reduced IFNγ-mediated suppression of B cell responses.
A defining feature of Tfh cells is elevated PD-1 expression47. Although most cTfh cells express low levels of PD-1, a subset with elevated PD-1 corresponds to the most efficient population of B-helper cells8, 11, 12, 17. Given our finding of fewer cTfh cells in STAT3LOF patients, and skewing of cTfh subsets in STAT1GOF or STAT3LOF individuals to a phenotype with reduced B-helper function, it may be predicted that their cTfh cells would have reduced PD-1. However, this was not the case, as PD-1 was significantly increased on cTfh cells in these individuals, raising several important possibilities. First, as PD-1 engagement impedes Tfh formation in vivo55–57, exaggerated signaling through over-expressed PD-1 on STAT1GOF and STAT3LOF cTfh cells may further impair function. Interestingly, the PD-1 ligand PD-L1 is aberrantly expressed on CD4+ T cells from STAT1GOF patients41, revealing a possible autocrine mechanism of restrained cTfh function. Similarly, while PD-1 is expressed comparably on Tfh cells from healthy donors and HIV-infected individuals, more GC B cells from HIV-infected individuals expressed PD-L1 than healthy donors53. Importantly, this heightened expression impaired Tfh cell IL-21 production, suppressing effector function53, 58. PD-1hi-expressing Tfh cells also undergo accelerated death compared to cells expressing lower levels of PD-159. Thus, dysregulated expression of PD-1 or its ligand(s) could compromise Tfh function or survival in these PIDs41, 53, 58, 59. Interestingly, cTfh cells in STAT3-deficient individuals also expressed CD57 (MC Cook et al; submitted). While CD57 is expressed by a large proportion of Tfh cells in lymphoid tissues, it is usually absent from cTfh cells in healthy controls9, 17, 24. Expression of CD57 by Tfh cells has been associated with an exhausted/senescent phenotype, and a susceptibility to rapidly undergo death relative to CD57− CD4+ T cells60 (MC Cook et al; submitted). Thus, it is possible that cTfh cells in some PIDs correspond to “exhausted” cells, and this would further impact their function in the setting of humoral responses in these individuals. These possibilities are currently being investigated. Second, our data caution against solely quantifying cTfh cells with respect to PD-1 as a biomarker for humoral immunity. While this correlation has been established in vaccination12, HIV infection11, 17 and autoimmunity8, 9, 15, 16, it may be misleading in some monogenic PIDs. Thus, cTfh cells in these and other immunopathologies should be assessed for phenotype and function. The importance of doing so is highlighted by a recent study reporting impaired cTfh function in elderly individuals even though their cTfh cells expressed sufficient levels of IL-21 and ICOS12. Third, STAT3 signaling during Tfh differentiation is required to not only generate the CCR6+ subset, but also regulate PD-1 and minimize suppression through PD-1/PD-L1 interactions. The mechanism underlying STAT3-mediated PD-1 repression is unknown but may involve IL-21 as IL21R deficiency partially recapitulated this defect. Interestingly, as STAT3 can protect Tfh cells from the inhibitory effects of IFNα52, and IFNα induces PD-1 on murine CD4+ T cells61, elevated PD-1 on STAT3LOF cTfh cells may result from heightened IFNα signaling in the absence of STAT3.
Intriguingly, our findings also revealed overlapping cellular phenotypes due to STAT3LOF and STAT1GOF mutations including reduced production of Th17 cytokines by memory CD4+ T cells, skewed differentiation of and PD-1 expression by cTfh cells, and fewer memory B cells. These shared functional defects are consistent with similar clinical features of these distinct molecular entities, such as mucocutaneous candidiasis and impaired Ab responses21, 35, 41. While the mechanism underlying these common features is unknown, the data suggest that hypermorphic STAT1 suppresses STAT3, creating a situation mimicking STAT3-deficiency in STAT3-sufficient cells. Notably, there was greater variability in these defects in STAT1GOF patients, suggesting some mutations are more deleterious than others, consistent with the broader clinical phenotypes of affected individuals. Further studies will be required to elucidate exactly how STAT1GOF intersects with STAT3 to regulate function. Collectively, our study provides important insights into the molecular requirements and signaling pathways regulating human cTfh function and mechanisms for poor humoral immunity in some PIDs, such as elevated IFNγ but reduced IL-10 production and aberrant PD-1 expression. These defects would co-operate to limit cTfh function by suppressing B-cell help via IFNγ42–46 and PD-1 engagement impairing survival or production of IL-10 and IL-2153, 58, 59. Identifying these pathways potentially provides opportunities to manipulate Tfh function not only in immunodeficiency, but also autoimmunity.
Supplementary Material
Key Messages.
Loss-of function (LOF) mutations in STAT3, IL10R, CD40LG, NEMO, ICOS or BTK reduce cTfh frequencies.
STAT3, IL21/R LOF and STAT1 gain-of function mutations skew cTfh differentiation towards a phenotype typified by over-expression of IFN-γ and PD-1.
IFN-γ negatively regulates Ig production in vivo
Acknowledgments
This work was funded by project and program grants from the National Health and Medical Research Council (NHMRC) of Australia (to EKD, SGT, CSM, DAF, MCC; 596813, 1016953, 1066694, 1027400), the German Federal Ministry of Education and Research (BMBF 01EO1303, to BG and KW), and Rockefeller University Center for 541 Clinical and Translational science (5UL1RR024143, to JLC). CSM is a recipient of a Career Development Fellowship (1008820) and SGT a Principal Research Fellowship (1042925) from the NHMRC of Australia.
Abbreviations
- Tfh
T follicular helper cell
- cTfh
circulating Tfh
- TD
T-dependent
- SLE
systemic lupus erythematosus
- RA
rheumatoid arthritis
- GCs
germinal centres’
- LOF
loss-of function
- GOF
gain-of function
- PIDs
primary immunodeficiencies
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflicts of interest
References
- 1.O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. 2010;327:1098–102. doi: 10.1126/science.1178334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crotty S. Follicular helper CD4 T cells (TFH) Annu Rev Immunol. 2011;29:621–63. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 3.Ma CS, Deenick EK, Batten M, Tangye SG. The origins, function, and regulation of T follicular helper cells. J Exp Med. 2012;209:1241–53. doi: 10.1084/jem.20120994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tangye SG, Ma CS, Brink R, Deenick EK. The good, the bad and the ugly - TFH cells in human health and disease. Nat Rev Immunol. 2013;13:412–26. doi: 10.1038/nri3447. [DOI] [PubMed] [Google Scholar]
- 5.Crotty S. T Follicular Helper Cell Differentiation, Function, and Roles in Disease. Immunity. 2014;41:529–42. doi: 10.1016/j.immuni.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma CS, Uzel G, Tangye SG. Human T follicular helper cells in primary immunodeficiencies. Curr Opin Pediatr. 2014;26:720–6. doi: 10.1097/MOP.0000000000000157. [DOI] [PubMed] [Google Scholar]
- 7.Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol. 2012;8:337–47. doi: 10.1038/nrrheum.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity. 2013;39:770–81. doi: 10.1016/j.immuni.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 9.Simpson N, Gatenby PA, Wilson A, Malik S, Fulcher DA, Tangye SG, et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum. 2010;62:234–44. doi: 10.1002/art.25032. [DOI] [PubMed] [Google Scholar]
- 10.Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. 2011;34:108–21. doi: 10.1016/j.immuni.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boswell KL, Paris R, Boritz E, Ambrozak D, Yamamoto T, Darko S, et al. Loss of circulating CD4 T cells with B cell helper function during chronic HIV infection. PLoS Pathog. 2014;10:e1003853. doi: 10.1371/journal.ppat.1003853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herati RS, Reuter MA, Dolfi DV, Mansfield KD, Aung H, Badwan OZ, et al. Circulating CXCR5+PD-1+ response predicts influenza vaccine antibody responses in young adults but not elderly adults. J Immunol. 2014;193:3528–37. doi: 10.4049/jimmunol.1302503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Coz C, Joublin A, Pasquali JL, Korganow AS, Dumortier H, Monneaux F. Circulating TFH subset distribution is strongly affected in lupus patients with an active disease. PLoS One. 2013;8:e75319. doi: 10.1371/journal.pone.0075319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arroyo-Villa I, Bautista-Caro MB, Balsa A, Aguado-Acin P, Bonilla-Hernan MG, Plasencia C, et al. Constitutively altered frequencies of circulating follicullar helper T cell counterparts and their subsets in Rheumatoid Arthritis. Arthritis Res Ther. 2014;16:500. doi: 10.1186/s13075-014-0500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi JY, Hsi-enHo J, Pasoto SG, Bunin V, Kim S, Carrasco S, et al. Circulating follicular helper-like T cells in systemic lupus erythematosus: Association with disease activity. Arthritis Rheumatol. 2015 doi: 10.1002/art.39020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J, Shan Y, Jiang Z, Feng J, Li C, Ma L, et al. High frequencies of activated B cells and T follicular helper cells are correlated with disease activity in patients with new-onset rheumatoid arthritis. Clin Exp Immunol. 2013;174:212–20. doi: 10.1111/cei.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al. Human circulating PD-(+)1CXCR3(−)CXCR5(+) memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity. 2013;39:758–69. doi: 10.1016/j.immuni.2013.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bentebibel SE, Lopez S, Obermoser G, Schmitt N, Mueller C, Harrod C, et al. Induction of ICOS+CXCR3+CXCR5+ TH Cells Correlates with Antibody Responses to Influenza Vaccination. Sci Transl Med. 2013;5:176ra32. doi: 10.1126/scitranslmed.3005191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spensieri F, Borgogni E, Zedda L, Bardelli M, Buricchi F, Volpini G, et al. Human circulating influenza-CD4+ ICOS1+IL-21+ T cells expand after vaccination, exert helper function, and predict antibody responses. Proc Natl Acad Sci U S A. 2013;110:14330–5. doi: 10.1073/pnas.1311998110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma CS, Avery DT, Chan A, Batten M, Bustamante J, Boisson-Dupuis S, et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood. 2012;119:3997–4008. doi: 10.1182/blood-2011-11-392985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Avery DT, Deenick EK, Ma CS, Suryani S, Simpson N, Chew GY, et al. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med. 2010;207:155–71. doi: 10.1084/jem.20091706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma CS, Hare NJ, Nichols KE, Dupre L, Andolfi G, Roncarolo MG, et al. Impaired humoral immunity in X-linked lymphoproliferative disease is associated with defective IL-10 production by CD4+ T cells. J Clin Invest. 2005;115:1049–59. doi: 10.1172/JCI23139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. 2008;205:1551–7. doi: 10.1084/jem.20080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma CS, Suryani S, Avery DT, Chan A, Nanan R, Santner-Nanan B, et al. Early commitment of naive human CD4(+) T cells to the T follicular helper (T(FH)) cell lineage is induced by IL-12. Immunol Cell Biol. 2009;87:590–600. doi: 10.1038/icb.2009.64. [DOI] [PubMed] [Google Scholar]
- 25.Chevalier N, Jarrossay D, Ho E, Avery DT, Ma CS, Yu D, et al. CXCR5 expressing human central memory CD4 T cells and their relevance for humoral immune responses. J Immunol. 2011;186:5556–68. doi: 10.4049/jimmunol.1002828. [DOI] [PubMed] [Google Scholar]
- 26.Appay V, van Lier RA, Sallusto F, Roederer M. Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A. 2008;73:975–83. doi: 10.1002/cyto.a.20643. [DOI] [PubMed] [Google Scholar]
- 27.Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The who’s who of T-cell differentiation: human memory T-cell subsets. Eur J Immunol. 2013;43:2797–809. doi: 10.1002/eji.201343751. [DOI] [PubMed] [Google Scholar]
- 28.Moens L, Tangye SG. Cytokine-Mediated Regulation of Plasma Cell Generation: IL-21 Takes Center Stage. Front Immunol. 2014;5:65. doi: 10.3389/fimmu.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–61. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim CH, Lim HW, Kim JR, Rott L, Hillsamer P, Butcher EC. Unique gene expression program of human germinal center T helper cells. Blood. 2004;104:1952–60. doi: 10.1182/blood-2004-03-1206. [DOI] [PubMed] [Google Scholar]
- 31.Zhang SY, Boisson-Dupuis S, Chapgier A, Yang K, Bustamante J, Puel A, et al. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol Rev. 2008;226:29–40. doi: 10.1111/j.1600-065X.2008.00698.x. [DOI] [PubMed] [Google Scholar]
- 32.Coffman RL. Origins of the T(H)1–T(H)2 model: a personal perspective. Nat Immunol. 2006;7:539–41. doi: 10.1038/ni0606-539. [DOI] [PubMed] [Google Scholar]
- 33.Frohlich A, Marsland BJ, Sonderegger I, Kurrer M, Hodge MR, Harris NL, et al. IL-21 receptor signaling is integral to the development of Th2 effector responses in vivo. Blood. 2007;109:2023–31. doi: 10.1182/blood-2006-05-021600. [DOI] [PubMed] [Google Scholar]
- 34.Stritesky GL, Muthukrishnan R, Sehra S, Goswami R, Pham D, Travers J, et al. The transcription factor STAT3 is required for T helper 2 cell development. Immunity. 2011;34:39–49. doi: 10.1016/j.immuni.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2014;5:162. doi: 10.3389/fimmu.2014.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paulos CM, Carpenito C, Plesa G, Suhoski MM, Varela-Rohena A, Golovina TN, et al. The inducible costimulator (ICOS) is critical for the development of human T(H)17 cells. Sci Transl Med. 2010;2:55ra78. doi: 10.1126/scitranslmed.3000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32:605–15. doi: 10.1016/j.immuni.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bossaller L, Burger J, Draeger R, Grimbacher B, Knoth R, Plebani A, et al. ICOS deficiency is associated with a severe reduction of CXCR5+CD4 germinal center Th cells. J Immunol. 2006;177:4927–32. doi: 10.4049/jimmunol.177.7.4927. [DOI] [PubMed] [Google Scholar]
- 39.Martini H, Enright V, Perro M, Workman S, Birmelin J, Giorda E, et al. Importance of B cell co-stimulation in CD4(+) T cell differentiation: X-linked agammaglobulinaemia, a human model. Clin Exp Immunol. 2011;164:381–7. doi: 10.1111/j.1365-2249.2011.04377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Linterman MA, Liston A, Vinuesa CG. T-follicular helper cell differentiation and the co-option of this pathway by non-helper cells. Immunol Rev. 2012;247:143–59. doi: 10.1111/j.1600-065X.2012.01121.x. [DOI] [PubMed] [Google Scholar]
- 41.Romberg N, Morbach H, Lawrence MG, Kim S, Kang I, Holland SM, et al. Gain-of-function STAT1 mutations are associated with PD-L1 overexpression and a defect in B-cell survival. J Allergy Clin Immunol. 2013;131:1691–3. doi: 10.1016/j.jaci.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hussain R, Kifayet A, Dojki M, Dockrell HM. Selective correlation of interferon-gamma, tumour necrosis factor-alpha and granulocyte-macrophage colony-stimulating factor with immunoglobulin G1 and immunoglobulin G3 subclass antibody in leprosy. Immunology. 1999;98:238–43. doi: 10.1046/j.1365-2567.1999.00876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.King CL, Gallin JI, Malech HL, Abramson SL, Nutman TB. Regulation of immunoglobulin production in hyperimmunoglobulin E recurrent-infection syndrome by interferon gamma. Proc Natl Acad Sci U S A. 1989;86:10085–9. doi: 10.1073/pnas.86.24.10085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.King CL, Nutman TB. IgE and IgG subclass regulation by IL-4 and IFN-gamma in human helminth infections. Assessment by B cell precursor frequencies. J Immunol. 1993;151:458–65. [PubMed] [Google Scholar]
- 45.Kim JR, Lim HW, Kang SG, Hillsamer P, Kim CH. Human CD57+ germinal center-T cells are the major helpers for GC-B cells and induce class switch recombination. BMC Immunol. 2005;6:3. doi: 10.1186/1471-2172-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Geha RS, Jabara HH, Brodeur SR. The regulation of immunoglobulin E class-switch recombination. Nat Rev Immunol. 2003;3:721–32. doi: 10.1038/nri1181. [DOI] [PubMed] [Google Scholar]
- 47.Haynes NM, Allen CD, Lesley R, Ansel KM, Killeen N, Cyster JG. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol. 2007;179:5099–108. doi: 10.4049/jimmunol.179.8.5099. [DOI] [PubMed] [Google Scholar]
- 48.Miller JF, De Burgh PM, Grant GA. Thymus and the production of antibody-plaque-forming cells. Nature. 1965;208:1332–4. doi: 10.1038/2081332a0. [DOI] [PubMed] [Google Scholar]
- 49.Pallikkuth S, Parmigiani A, Silva SY, George VK, Fischl M, Pahwa R, et al. Impaired peripheral blood T-follicular helper cell function in HIV-infected nonresponders to the 2009 H1N1/09 vaccine. Blood. 2012;120:985–93. doi: 10.1182/blood-2011-12-396648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boisson B, Wang YD, Bosompem A, Ma CS, Lim A, Kochetkov T, et al. A recurrent dominant negative E47 mutation causes agammaglobulinemia and BCR(−) B cells. J Clin Invest. 2013;123:4781–5. doi: 10.1172/JCI71927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee CE, Fulcher DA, Whittle B, Chand R, Fewings N, Field M, et al. Autosomal-dominant B-cell deficiency with alopecia due to a mutation in NFKB2 that results in nonprocessable p100. Blood. 2014;124:2964–72. doi: 10.1182/blood-2014-06-578542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ray JP, Marshall HD, Laidlaw BJ, Staron MM, Kaech SM, Craft J. Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity. 2014;40:367–77. doi: 10.1016/j.immuni.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cubas RA, Mudd JC, Savoye AL, Perreau M, van Grevenynghe J, Metcalf T, et al. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med. 2013;19:494–9. doi: 10.1038/nm.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmitt N, Bustamante J, Bourdery L, Bentebibel SE, Boisson-Dupuis S, Hamlin F, et al. IL-12 receptor beta1 deficiency alters in vivo T follicular helper cell response in humans. Blood. 2013;121:3375–85. doi: 10.1182/blood-2012-08-448902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Butler NS, Moebius J, Pewe LL, Traore B, Doumbo OK, Tygrett LT, et al. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat Immunol. 2012;13:188–95. doi: 10.1038/ni.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hams E, McCarron MJ, Amu S, Yagita H, Azuma M, Chen L, et al. Blockade of B7-H1 (programmed death ligand 1) enhances humoral immunity by positively regulating the generation of T follicular helper cells. J Immunol. 2011;186:5648–55. doi: 10.4049/jimmunol.1003161. [DOI] [PubMed] [Google Scholar]
- 57.Kawamoto S, Tran TH, Maruya M, Suzuki K, Doi Y, Tsutsui Y, et al. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science. 2012;336:485–9. doi: 10.1126/science.1217718. [DOI] [PubMed] [Google Scholar]
- 58.Khan AR, Hams E, Floudas A, Sparwasser T, Weaver CT, Fallon PG. PD-L1(hi) B cells are critical regulators of humoral immunity. Nat Commun. 2015;6:5997. doi: 10.1038/ncomms6997. [DOI] [PubMed] [Google Scholar]
- 59.Wang C, Hillsamer P, Kim CH. Phenotype, effector function, and tissue localization of PD-1-expressing human follicular helper T cell subsets. BMC Immunol. 2011;12:53. doi: 10.1186/1471-2172-12-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marinova E, Han S, Zheng B. Human germinal center T cells are unique Th cells with high propensity for apoptosis induction. Int Immunol. 2006;18:1337–45. doi: 10.1093/intimm/dxl066. [DOI] [PubMed] [Google Scholar]
- 61.Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, et al. IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol. 2011;186:2772–9. doi: 10.4049/jimmunol.1003208. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.