

Abstract
Despite recent advances in therapeutic strategies for lung cancer, mortality is still increasing. Therefore, there is an urgent need to identify effective novel drugs. In the present study, we implement drug repositioning for lung adenocarcinoma (LUAD) by a bioinformatics method followed by experimental validation. We first identified differentially expressed genes between LUAD tissues and nontumor tissues from RNA sequencing data obtained from The Cancer Genome Atlas database. Then, candidate small molecular drugs were ranked according to the effect of their targets on differentially expressed genes of LUAD by a random walk with restart algorithm in protein–protein interaction networks. Our method identified some potentially novel agents for LUAD besides those that had been previously reported (eg, hesperidin). Finally, we experimentally verified that atracurium, one of the potential agents, could induce A549 cells death in non-small-cell lung cancer-derived A549 cells by an MTT assay, acridine orange and ethidium bromide staining, and electron microscopy. Furthermore, Western blot assays demonstrated that atracurium upregulated the proapoptotic Bad and Bax proteins, downregulated the antiapoptotic p-Bad and Bcl-2 proteins, and enhanced caspase-3 activity. It could also reduce the expression of p53 and p21Cip1/Waf1 in A549 cells. In brief, the candidate agents identified by our approach may provide greater insights into improving the therapeutic status of LUAD.
Keywords: lung adenocarcinoma, drug repositioning, bioinformatics, protein-protein interaction network, atracurium
Introduction
Lung cancer is estimated to have the second highest incidence of all cancers in the US with over 163,000 deaths in 2014.1,2 Non-small-cell lung cancer (NSCLC) may represent >80% of all lung cancer cases.3 The most common subtypes of NSCLC are adenocarcinoma and squamous cell carcinoma. More than half of the NSCLCs are constituted by lung adenocarcinoma (LUAD).4 A 5-year survival rate of only 17% reflects LUAD’s known heterogeneity; the complex cellular, molecular, and tumor microenvironmental factors presented in each individual; and poor therapy options.1,2,5 Furthermore, there is still a lack of effective treatment for LUAD.5,6 Thus, there is an urgent need for the identification of novel drugs that will provide clinicians with useful assistance in patient prognosis and potential therapeutic options.7
However, the identification of novel drugs is time consuming, costly, and risky. The average research and development cost, in the past 15 years, for developing a new drug is over one billion US dollars.8 Anticancer agents are especially costly.9 Therefore, drug repositioning, which discovers new applications for known drugs, offers a promising alternative to reduce the total time and cost because of existing safety, toleration, and efficacy data. Recently, with the development of bioinformatics and high-throughput genome-wide data, network-based drug repositioning has emerged.10 Drug repositioning emphasizes interactions among drugs, targets, and diseases and highlights the network concept.
Here, we identified potential novel drugs for LUAD by a network-based algorithm followed by experimental verification. Differentially expressed genes (DEGs) between LUAD tissues and nontumor tissues were identified. Then, known small molecular drugs were ranked according to the effect of their targets on DEGs of LUAD by a random walk with restart (RWR) algorithm. Finally, we experimentally verified that atracurium, one of the potential agents, could induce NSCLC-derived A549 cell death, and Western blot assay demonstrated that atracurium upregulated the proapoptotic Bad and Bax, and downregulated the antiapoptotic p-Bad and Bcl-2 proteins. Furthermore, atracurium also enhanced the caspase-3 activity and could also reduce the expression of p53 and p21Cip1/Waf1 in A549 cells. In brief, the candidate agents identified by our approach may provide great insights into improving the therapeutic status for LUAD.
Materials and methods
Protein–protein interaction networks
Protein–protein interactions (PPIs) were accessed from the HPRD (Human Protein Reference Database) and STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) databases. The HPRD contains manually entered information extracted from the literature by expert biologists who read, interpreted, and analyzed the published data.11 The HPRD release 9, which contains 37,070 PPIs among 9,465 proteins, was downloaded in this study. The STRING database contains comprehensive information from numerous sources, including experimental data, computational prediction methods, and public text collections.12 The PPIs and their confidence scores were downloaded from STRING 9. Only PPIs with confidence scores >900 were obtained.
Drug data
Drugs and their targets were downloaded from Drugbank (version 4.0, http://www.drugbank.ca/),13 which contained 7,759 drugs and 4,300 proteins. After converting protein identification names and mapping the targets to both the STRING and HPRD networks, 12,604 drug–target relations between 4,452 drugs and 1,617 proteins were retained for further study.
Differential gene expression analysis of LUAD
LUAD level 3 mRNA expression data derived from the IlluminaHiSeq RNASeqV2 platform were obtained from The Cancer Genome Atlas (TCGA) public data portal (https://tcga-data.nci.nih.gov/tcga/). This profile contained 445 LUAD samples and 19 normal samples. Fold change and edgeR methods were used to identify DEGs. edgeR is an R Bioconductor package for the analysis of gene expression data arising from RNA sequencing technologies.14 Genes with Benjamini–Hochberg adjusted false discovery rate (FDR) <0.01 in the edgeR method and fold change >2 or <0.5 were considered as DEGs. The DEGs were mapped to both the STRING and HPRD networks. There were 927 DEGs that existed in both the STRING and HPRD databases.
Random walk
To identify the potential drugs for LUAD, we developed a novel method to evaluate the effects of each candidate drug on LUAD by assessing the influence of corresponding drug targets on the DEGs of LUAD in the context of PPIs. To do this, we implemented an RWR algorithm to calculate the impact power score (IPS) for each candidate drug.15
RWR simulates a walker starting on given seed nodes, which, at each step, moves randomly from the current node to neighbors in the network based on the probabilities of the edges between the two nodes. In this study, let P0 be the initial probability vector and Ps be a vector in which the i-th element holds the probability of the random walker at node i at step s. Let γ be the restart probability of the random walker in each step at the source nodes and M be the normalized PPI network. Then, the probability at s+1 can be described as follows:
| (1) |
After several steps, the probability will achieve a stable state, and this can be defined as P∞ by performing the iteration until the difference between Ps and Ps+1 falls below a given cutoff (measured by L1 norm).
In this paper, target genes of each drug are considered as the seed nodes, while the DEGs of LUAD are considered as candidates in this analysis. The initial probability P0 is formed such that probabilities are assigned equally to the seed nodes, with a sum equal to 1, while the probabilities of nonseed nodes are 0. The restart probability is set to 0.7 as it was in the previous study.15 Then the final stable probability P∞ of each DEG can be achieved by an iterative process until the difference between Ps and Ps+1 falls below 10−10. The probability value of each DEG of LUAD presents the impact of drug targets on them in the PPI network. Then the IPS of each drug can be measured by , where n is the number of DEGs of LUAD. This process was performed in the HPRD and STRING networks, respectively, to obtain robust results.
Reagents
A549 cells were obtained from Harbin Medical University (Heilongjiang, People’s Republic of China). RPMI 1640 medium was purchased from Thermo (Beijing, People’s Republic of China). Fetal bovine serum was obtained from Gibco GRL (Grand Island, NY, USA). Penicillin–streptomycin solution, trypsin, phosphate-buffered saline (PBS), dimethyl sulfoxide (DMSO), MTT, normal melting point agarose, acridine orange and ethidium bromide (AO/EB), and cell lysis solution were purchased from Solarbio (Beijing, People’s Republic of China). This study was approved by the Ethics Committee of Harbin Medical University, and conformed with the tenets of the Declaration of Helsinki.
Cell viability assay
A549 cells were treated with atracurium at different concentrations (10, 50, 100, 150, and 200 μg/L). Cell viability was determined by an MTT assay16 to evaluate the possible cytotoxic effects of the test samples. A549 cells in the absence or presence of samples at different concentrations (4×103 cells/well) were cultured in 96-well plates for 48 hours. Aliquots (20 μL) of 5 mg/mL MTT in PBS were added to each well in the 96-well plate. The plates were incubated for another 4 hours. The culture medium was then discarded. The plates were carefully washed twice with PBS buffer. Aliquots of DMSO (150 μL) were added to each well and oscillated for 15 minutes to extract the insoluble formazan that had formed. A microplate reader (TECAN, Mannedorf, Switzerland) was used to measure the absorbance at a wavelength of 570 nm. A549 cell viability was calculated as: Survival (%) = A/B × 100%, where A is the average optical density (OD) of the atracurium-treated cells and B is the average OD of the control wells (culture medium with cells).
Electron microscopy
A549 cells were cultured in 60 mm plates, washed with PBS solution, and fixed with 2% (v/v) paraformaldehyde (PFA) containing 2.5% (w/v) glutaraldehyde (Paesel-Lorei, Duisburg, Germany) buffered in Hank’s-modified salt solution at 4°C for 4 hours. The cells were further fixed in 1% (w/v) OSO4 solution buffered by 0.1 M cacodylate (pH 7.2) at 4°C for 2 hours, and then scraped off and dehydrated in ethanol. Dehydration was completed in propylene oxide. The specimens were embedded in Araldite (Serva, Heidelberg, Germany). Ultrathin sections were produced on an FCR Reichert Ultracut ultramicrotome (Leica Microsystems, Wetzlar, Germany), mounted on pioloform-coated copper grids, and contrasted with lead citrate. Specimens were analyzed and documented with a 10A electron microscope (Zeiss, Oberkochen, Germany).
AO/EB fluorescence staining
The A549 cells were incubated with AO/EB mixing solution for 5 minutes (Solarbio, http://solarbio.en.alibaba.com). Cellular morphological changes were examined by fluorescence microscopy (3,200×). The percentage of apoptotic cells was calculated by the following formula: Apoptotic rate (%) = Number of apoptotic cells/Number of all cells counted.
Western blotting analysis
Total protein sample was extracted from A549 cells. Protein concentration was determined by BCA Protein Assay Kit (Beyotime Institute of Biotechnology, Jiangsu, People’s Republic of China). The protein samples (80 μg) were fractionated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE, 8%–10% polyacrylamide gels) and transferred to nitrocellulose membranes (EMD Millipore, Billerica, MA, USA). The membranes were then blocked with milk powder at room temperature for 2 hours and incubated overnight at 4°C with the primary antibody. The following day, the membranes were washed and incubated with a secondary rabbit or mouse polyclonal antibody for 1 hour at room temperature. Western blot bands were visualized by enhanced chemiluminesence reagent (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA) and quantified using Odyssey v1.2 software by measuring the band intensity (area × OD) for each group and normalizing to glyceraldehyde 3-phosphate dehydrogenase.
Caspase-3 activity assay
The caspase-3 activity was analyzed using a Caspase-3 Activity Assay Kit (Beyotime Institute of Biotechnology) according to the manufacturer’s instructions, using substrate peptides Ac-DEVD-pNA (p-nitroanilide), Ac-IETD-pNA, and Ac-LEHD-pNA, respectively. Briefly, the supernatant of cell lysate was mixed with buffer containing the substrate peptides for caspase attached to pNA. The release of pNA was quantified by determining the absorbance with an enzyme-linked immunosorbent assay (ELISA) reader at 405 nm. The caspase activities were expressed as percentage over control.
Data analysis
All experimental data were expressed as mean ± standard deviation. Analysis of variance or Student’s t-test was used to compare the mean values for multiple-group or two-group comparisons, using the SPSS 13.0 software. Values of P<0.05 were considered to be statistically significant.
Results
DEGs between LUAD and normal samples
In order to identify DEGs of LUAD, we obtained level 3 expression profiles of LUAD samples and normal samples from TCGA database (http://cancergenome.nih.gov/). After mapping the DEGs to the HPRD and STRING networks, a total of 927 genes were obtained (see the section “Materials and methods” for details).
We then identified deregulated pathways by using the DAVID v6.7 program (Database for Annotation, Visualization and Integrated Discovery).17 We identified 12 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways with a cutoff P-value <0.05 (Table 1). Some pathways were significantly related to the development of LUAD, such as pathways in cancer (hsa05200) and the cell cycle (hsa04110).
Table 1.
The significant enriched pathways related to LUAD and the corresponding annotated genes
| Term | P-value | Genes |
|---|---|---|
| hsa04080:Neuroactive ligand–receptor interaction | 2.75E–09 | CGA, GRIK2, ADCYAP1R1, LHCGR, PTH1R, GRIK5, PRSS1, FPR2, VIPR1, GCGR, EDNRB, KISS1R, AGTR2, NMUR1, GRIN2D, PRSS3, CALCRL, TUBB3, GHR, GABRG2, GABRG3, PTH2R, GABRA4, RXFP1, GABRA3, RXFP2, GRIN1, NTSR1, FSHR, GH2, SSTR4, GRM4, GABRR1, CHRM2, GRIA1, P2RX3, F2, MC4R, ADRA1A, TSHR, LHB, CTSG, MTNR1A, OPRD1 |
| hsa04610:Complement and coagulation cascades | 3.42E–08 | F11, KNG1, C7, F12, MASP1, C4BPB, F7, PROC, C8G, F13B, C8A, C8B, VWF, F5, FGA, FGB, F2, CFD, CPB2, PLAU |
| hsa04110:Cell cycle | 1.21E–04 | E2F2, CDC6, PKMYT1, TTK, CHEK1, CDC20, PTTG1, CDC25C, MCM4, CDC25A, CCNE2, CCNB1, CCNE1, CDKN2A, CCNB2, MAD2L1, PLK1, BUB1, BUB1B, CCNA2, SMC1B |
| hsa04114:Oocyte meiosis | 5.92E–04 | ADCY8, SGOL1, PKMYT1, AURKA, CDC20, PTTG1, CDC25C, CCNE2, CCNB1, CCNE1, CCNB2, MAD2L1, CALML3, PLK1, BUB1, CAMK2B, CALML5, SMC1B |
| hsa04950:Maturity onset diabetes of the young | 7.61E–04 | HNF1A, HNF4A, ONECUT1, FOXA3, SLC2A2, PKLR, PAX6, HNF4G |
| hsa04512:ECM–receptor interaction | 8.33E–04 | TNXB, COL3A1, ITGA11, COL2A1, CHAD, HMMR, VWF, CD36, ITGA8, TNR, COMP, COL1A1, THBS2, COL11A1, SPP1 |
| hsa04916:Melanogenesis | 0.010485 | ADCY8, WNT3A, EDN1, PRKCG, EDNRB, WNT1, WNT3, CALML3, CAMK2B, CREB3L3, CALML5, WNT6, WNT7A, TUBB3 |
| hsa04614:Renin-angiotensin system | 0.020692 | AGTR2, AGT, MME, CMA1, CTSG |
| hsa04510:Focal adhesion | 0.033515 | CAV2, CAV1, TNXB, COL3A1, ITGA11, PRKCG, ACTN2, COL2A1, CHAD, VWF, RAC3, PAK3, COMP, ITGA8, TNR, COL1A1, EGF, FIGF, THBS2, COL11A1, SPP1 |
| hsa04020:Calcium signaling pathway | 0.033556 | TNNC2, TNNC1, ADCY8, GRIN1, LHCGR, PRKCG, NTSR1, CACNA1S, ITPKA, EDNRB, CALML3, CHRM2, GRIN2D, P2RX3, ADRA1A, CAMK2B, CACNA1E, CALML5, CACNA1B |
| hsa04360:Axon guidance | 0.0372 | DCC, NGEF, SEMA6A, EPHA6, SEMA6D, EPHA8, RAC3, PAK3, EFNA2, PLXNB3, EFNA3, SEMA3A, SLIT2, EPHB2, SLIT3 |
| hsa05200:Pathways in cancer | 0.048267 | FGF19, DCC, E2F2, MMP9, WNT3A, EGLN3, FGF10, ZBTB16, MMP1, CCNE2, CCNE1, WNT1, CDKN2A, WNT3, RAC3, SLC2A1, HHIP, EGF, WNT6, FIGF, IL6, RET, EPAS1, KLK3, RXRG, PRKCG, BIRC5, RAD51, CBLC, WNT7A |
Abbreviation: LUAD, lung adenocarcinoma.
Potential drugs for LUAD
After walking the HPRD and STRING networks, the candidate drugs were ranked according to the IPS (see the section “Materials and methods” for details). Table 2 shows the top 50 drugs in the HPRD network. To obtain a robust result, we also implemented our method in the STRING network. In the top 5% of the ranked drugs (top 227 drugs), 145 were identified in two PPI networks (P<0.01; hypergeometric text, Figure 1A). Furthermore, the corresponding targets of the top 5% drugs significantly overlapped (P<0.01; hypergeometric text, Figure 1B). We selected one of the top ranked drugs, atracurium, for the following analysis.
Table 2.
The top 50 ranked drugs
| Drug ID | Drug name | Target | Score | Rank |
|---|---|---|---|---|
| DB00416 | Metocurine Iodide | CHRNA2 | 0.966581 | 1 |
| DB00565 | Cisatracurium besylate | CHRNA2 | 0.966581 | 1 |
| DB00732 | Atracurium | CHRNA2 | 0.966581 | 1 |
| DB00657 | Mecamylamine | CHRNA2 | 0.966581 | 1 |
| DB02457 | Undecyl-phosphinic acid butyl ester | LIPF | 0.953846 | 5 |
| DB04551 | Fructose-1,6-diphosphate | PKLR | 0.923036 | 6 |
| DB04869 | Olcegepant | CALCA | 0.906767 | 7 |
| DB05760 | MK-0974 | CALCA | 0.906767 | 7 |
| DB05333 | TC-2403-12 | CHRNB2 | 0.903221 | 9 |
| DB01336 | Metocurine | CHRM2;CHRNA2 | 0.856775 | 10 |
| DB05458 | ABT-089 | CHRNA4;CHRNB2 | 0.85602 | 11 |
| DB06097 | GSK-923295 | CENPE | 0.851175 | 12 |
| DB02071 | WAY-151693 | MMP13 | 0.83319 | 13 |
| DB07013 | Tert-butyl 4-([{4-(but-2-yn-1-ylamino)phenyl}sulfonyl]methyl)-4-(hydroxyamino) carbonyl]piperidine-1-carboxylate | MMP13 | 0.83319 | 13 |
| DB08561 | Benzyl 6-benzyl-5,7-dioxo-6,7-dihydro-5H-(1,3)thiazolo(3,2-c)pyrimidine-2-carboxylate | MMP13 | 0.83319 | 13 |
| DB04760 | Pyrimidine-4,6-dicarboxylic acid bis-(4-fluoro-3-methyl-benzylamide) | MMP13 | 0.83319 | 13 |
| DB08388 | 5-(2-ethoxyethyl)-5-(4-[4-fluorophenoxy]phenoxy)pyrimidine-2,4,6(1h,3h,5h)-trione | MMP13 | 0.83319 | 13 |
| DB02049 | 2-(4-[4-{4-chloro-phenoxy}-Benzenesulfonyl]-tetrahydro-pyran-4-Yl)-N-hydroxy-acetamide | MMP13 | 0.83319 | 13 |
| DB04759 | Pyrimidine-4,6-dicarboxylic acid bis-(3-methyl-benzylamide) | MMP13 | 0.83319 | 13 |
| DB04761 | Pyrimidine-4,6-dicarboxylic acid bis-([pyridin-3-ylmethyl]-amide) | MMP13 | 0.83319 | 13 |
| DB08490 | 4-(4-[4-chloro-phenoxy]-benzenesulfonylmethyl)-tetrahydro-pyran-4-carboxylic acid hydroxyamide | MMP13 | 0.83319 | 13 |
| DB07827 | 4-([1-methyl-2,4-dioxo-6-{3-phenylprop-1-yn-1-yl}-1,4-dihydroquinazolin-3{2h}-yl] methyl)benzoic acid | MMP13 | 0.83319 | 13 |
| DB03149 | Phenylalanylmethane | CMA1 | 0.83092 | 23 |
| DB07680 | ([1S]-1-[5-chloro-1-benzothien-3-yl]-2-[2-naphthylamino]-2-oxoethyl)phosphonic acid | CMA1 | 0.83092 | 23 |
| DB01135 | Doxacurium chloride | BCHE;CHRM2;CHRNA2 | 0.826912 | 25 |
| DB04027 | D-Arginine | CKM | 0.826264 | 26 |
| DB02490 | (Diaminomethyl-methyl-amino)-acetic acid | CKM | 0.826264 | 26 |
| DB01245 | Decamethonium | ACHE;BCHE;CHRNA2 | 0.821877 | 28 |
| DB07077 | (R)-1-(4-[4-{hydroxymethyl}-1,3,2-dioxaborolan-2-yl]phenyl)guanidine | F11 | 0.818087 | 29 |
| DB07299 | 4-methyl-pentanoic acid (1-[4-guanidino-1-{thiazole-2-carbonyl}-butylcarbamoyl]-2-methyl-propyl)-amide | F11 | 0.818087 | 29 |
| DB07887 | (R)-1-(4-[4-{hydroxymethyl}-1,3,2-dioxaborolan-2-yl]benzyl)guanidine | F11 | 0.818087 | 29 |
| DB07023 | (1R)-2-([amino{imino}methyl]amino)-1-(4-[{4r}-4-{hydroxymethyl}-1,3,2-dioxaborolan-2-yl]phenyl)ethyl nicotinate | F11 | 0.818087 | 29 |
| DB07022 | 3-Hydroxypropyl 3-([{7-(amino[imino]methyl)-1-naphthyl}amino]carbonyl) benzenesulfonate | F11 | 0.818087 | 29 |
| DB07074 | 6-Carbamimidoyl-4-(3-hydroxy-2-methyl-benzoylamino)-naphthalene-2-carboxylic acid methyl ester | F11 | 0.818087 | 29 |
| DB07212 | N-(7-carbamimidoyl-naphthalen-1-yl)-3-hydroxy-2-methyl-benzamide | F11 | 0.818087 | 29 |
| DB07071 | (R)-1-(4-[4-{hydroxymethyl}-1,3,2-dioxaborolan-2-yl]phenethyl)guanidine | F11 | 0.818087 | 29 |
| DB08486 | 2-(4-[{3,5-dimethylanilino}-carbonyl-methyl]-phenoxy)-2-methylpropionic acid | HBA1;HBB;HBA2 | 0.817855 | 37 |
| DB07645 | Sebacic acid | HBA1;HBB;HBA2 | 0.817855 | 37 |
| DB02126 | 4-Carboxycinnamic acid | HBA1;HBB;HBA2 | 0.817855 | 37 |
| DB08262 | 2,6-dicarboxynaphthalene | HBA1;HBB;HBA2 | 0.817855 | 37 |
| DB07428 | 4-([5-methoxy-2-methylphenoxy]methyl)pyridine | HBA1;HBB;HBA2 | 0.817855 | 37 |
| DB08077 | 2-(4-[{([3,5-dichlorophenyl]amino)carbonyl}amino]phenoxy)-2-methylpropanoic acid | HBA1;HBB;HBA2 | 0.817855 | 37 |
| DB07427 | 2-([2-methoxy-5-methylphenoxy]methyl)pyridine | HBA1;HBB;HBA2 | 0.817855 | 37 |
| DB08632 | 1,3,5-benzenetricarboxylic acid | HBA1;HBB;HBA2 | 0.817855 | 37 |
| DB00483 | Gallamine triethiodide | ACHE;CHRM2;CHRNA2 | 0.815138 | 45 |
| DB04703 | Hesperidin | AURKB | 0.812062 | 46 |
| DB01996 | 3-Methylpyridine | MMP3;MMP13 | 0.81043 | 47 |
| DB03033 | 1-Methyloxy-4-sulfone-benzene | MMP3;MMP13 | 0.81043 | 47 |
| DB02697 | Hydroxyaminovaline | MMP3;MMP13 | 0.81043 | 47 |
| DB03944 | 5-(1-[3,4-dimethoxy-benzoyl]-1,2,3,4-tetrahydro-quinolin-6-Yl)-6-methyl-3,6-dihydro-(1,3,4)thiadiazin-2-one | TNNC1 | 0.809827 | 50 |
Figure 1.

Venn diagram showing the overlap of the (A) top 5% of the ranked drugs and (B) targets of top 5% of the drugs between the HPRD and STRING networks.
Abbreviations: HPRD, Human Protein Reference Database; STRING, Search Tool for the Retrieval of Interacting Genes/Proteins.
We then mapped all the DEGs into a HPRD network and extracted the first two neighbor nodes. Figure 2 shows the relationship between DEGs and drug targets, which are colored red and yellow, respectively.
Figure 2.
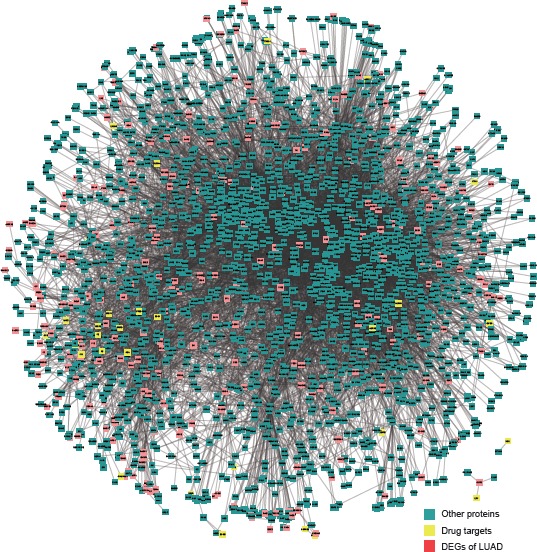
The relationship between DEGs and candidate drug targets in the HPRD network which are colored red and yellow, respectively.
Abbreviations: HPRD, Human Protein Reference Database; DEG, differentially expressed gene; LUAD, lung adenocarcinoma.
Atracurium suppresses the viability of A549 cells
The antiproliferative effect of atracurium on A549 cells was examined by exposing the cells to different concentrations (10, 50, 100, 150, or 200 μg/L) of atracurium for 24 hours. Cell growth was inhibited in a dose-dependent manner (Figure 3A). In the presence of 150 μg/L atracurium, A549 cells exhibited ~50% inhibition of proliferation after treatment for 24 hours. As such, this concentration and treatment time were used in subsequent experiments.
Figure 3.

Atracurium-induced apoptosis in A549 cells.
Notes: (A) Effects of atracurium on cell viability in A549 cells. After treatment of the cells with different concentrations of atracurium, cell viability was analyzed by an MTT assay. The data are expressed as mean ± SD, (n=6 batches of cells in each group), *P≤0.05 vs control group. (B) Representative image of acridine orange/ethidium bromide staining of A549 cells. (C) Micromorphological changes in cellular organelles examined by transmission electron microscopy. The data are expressed as mean ± SEM, n=3 for each group, *P≤0.05 vs control group.
Abbreviations: SD, standard deviation; SEM, standard error of the mean; Ctrl, control.
Atracurium induces apoptosis in A549 cells
To investigate whether atracurium regulates apoptosis, AO/EB staining and electron microscopy were used to detect apoptotic cells. The results from our fluorescence microscopic analysis are shown in Figure 3B. Atracurium induced a substantial number of apoptotic cells (P<0.05). Under electron microscope, cells with atracurium exhibited robust changes in microstructure, including cell surface microvillus reduction, nuclear chromatin condensation, imagination, and membrane blistering (Figure 3C).
FCePW activates proapoptotic signaling pathways
To explore the mechanisms by which atracurium induced apoptosis in A549 cells, we mapped the DEGs of LUAD and the target of atracurium (CHRNA2) into the PPI network. Figure 4A shows the subnetwork influenced by CHRNA2, in which CHRNA2 and DEGs are colored yellow and red, respectively. Also, besides DEGs of LUAD, there were many other cancer-related genes in this subnetwork (Figure 4B), suggesting that key apoptosis pathways were involved in this process. We then annotated these genes into a KEGG pathway (pathway in cancer) (Figure 4C).
Figure 4.


The top five neighbor nodes of CHRNA2 (the target of atracurium) are extracted from the HPRD network.
Notes: (A) After extraction, the DEGs are mapped to this subnetwork. CHRNA2, DEGs of LUAD, and cancer-related proteins are colored yellow, red, and blue, respectively. (B) The apoptosis-related genes (eg, P53 and BX) between CHRNA2 and DEGs and (C) results of annotating these proteins (with an asterisk [*]) into the pathway of cancer (hsa05200) (C) are presented.
Abbreviations: HPRD, Human Protein Reference Database; DEG, differentially expressed gene; LUAD, lung adenocarcinoma.
Next, we then measured some of the downstream proteins in the atracurium apoptotic pathway, including Bax, Bad, p-Bad, Bcl-2, p53, and p21Cip1/Waf1. Figure 5 demonstrates that atracurium upregulated Bad, p53, p21, and Bax (Figure 5A, B, D, and E) and downregulated p-Bad and Bcl-2 expression (Figure 5A and B). In addition, relative caspase-3 activity was significantly increased 2.5-fold by atracurium (Figure 5C).
Figure 5.

The Western blot analysis used to evaluate the protein expression in A549 cells after treatment with atracurium.
Notes: (A) Bax, Bcl-2, (B) p-Bad, Bad, (C) activation of caspase-3; (D) p53 protein level, and (E) p21Cip1/Waf1 protein level. Atracurium reduces the expression of p53 and p21Cip1/Waf1 in A549 cells. The data are expressed as mean ± SEM, n=3 for each group. *P≤0.05 vs control group.
Abbreviations: GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Ctrl, control; SEM, standard error of the mean.
Discussion
In this paper, we implemented drug repositioning for LUAD by using a network-based method. The integration of large-scale genomic, transcriptomic, and proteomic data in a network framework has provided new insights into a network-based view of drug discovery and development.18 The emergence of network medicine not only offers a better and more complete understanding of molecular complexities of diseases,19 but also serves as a promising tool for establishing new relationships among diseases that enable drug repositioning.20
In this research, by using the gene expression profile of LUAD, we first identified DEGs. Then, we identified pathways by the previously mentioned DEGs to explore the mechanism of development of LUAD. Table 1 shows the significant enriched pathways related to LUAD and the corresponding annotated genes. Some of these pathways have been reported to play important roles in LUAD, (eg, pathways in cancer [hsa05200; P=0.048267]) and cell cycle (hsa04110; P=1.21×10−4; Table 1).
Next, we prioritized the candidate drugs by accessing the effects of corresponding drugs on DEGs through an RWR algorithm in two PPI networks (HPRD and STRING). We found that the top 5% of the drugs or the corresponding targets of the top 5% of the drugs were significantly overlapped, suggesting that our methods were robust (Figure 1A and B). Table 2 shows the top 50 drugs in the HPRD network. Some drugs are existing anticancer drugs. For example, Birsu et al21 found that after hesperidin treatment, NSCLC-derived A549 cells exhibited decreasing cell proliferation and increasing caspase-3 and other apoptosis-related activities.
Interestingly, we found that atracurium, one of the top ranked drugs, had not been reported to have therapeutic effects on LUAD. Atracurium is a nondepolarizing skeletal muscle relaxant. Its cis-isomer cisatracurium besylate, which was also identified as a top ranked drug, is known to have a favorable safety profile with respect to the induction of histamine release.22 The effects of these drugs on cancer have not been widely studied. But Yabasin et al had indicated the anticancer effect of cisatracurium besylate on lung cancer cells (A549) in vitro.22 Xu et al23 reported that a mutation in CHRNA2, the target of atracurium and cisatracurium besylate, was related to salivary gland carcinomas. Furthermore, due its unique liver- and kidney-independent degradation, atracurium might be a potential chemotherapeutic drug in clinic due to its safety.24 However, to our knowledge, there were no direct reports about atracurium and LUAD. To investigate the effects of atracurium on LUAD, the A549 cells were treated with different concentrations (10, 50, 100, 150, or 200 μg/L) of atracurium, and we found that cell growth was inhibited in a dose-dependent manner (Figure 3A). Furthermore, the results of AO/EB staining and electron microscopy showed that atracurium induces a substantial number of apoptotic cells (Figure 3B and C), suggesting that atracurium might have a potential therapeutic effect on LUAD.
To further detect the mechanism of atracurium-induced A549 cell apoptosis, we extracted the top five neighbor nodes of CHRNA2, the target of atracurium, in the HPRD network, and then mapped the DEGs to this subnetwork (Figure 4A). We found that there were many apoptosis-related genes (eg, TP53 and BAX) between CHRNA2 and DEGs (Figure 4B), indicating that atracurium might directly/indirectly affect apoptosis signaling pathways (Figure 4C). To test this, Western blot was used to determine the level of some key proteins, including Bax, Bad, p-Bad, Bcl-2, p53, and p21Cip1/Waf1. Caspase-3 activity was also tested. It was reported that Bax, Bcl-2, and caspase-3 play key roles in apoptosis.25 The upregulation of Bax and downregulation of Bcl-2 and the increasing ratio of Bax/Bcl-2 and caspase3 activity were reported to trigger apoptosis.26 The results showed that the level of Bcl-2 protein after atracurium treatment was significantly lower than in the control group (P<0.05), whereas the level of Bax protein was significantly higher than in the control group (P<0.05). The ratio of Bax/Bcl-2 significantly increased; also, the caspase-3 activity was significantly increased (Figure 5A–C). These data indicate that the caspase-3-dependent apoptotic signaling plays an essential role in the apoptotic effects of atracurium.
We also examined the levels of p53 and p21Cip1/Waf1. Recent studies revealed that p53/p21 pathway mediates lung cancer A549 cells apoptosis.27,28 Our Western blot assay showed that the levels of p53 and p21Cip1/Waf1 were significantly augmented in the atracurium group compared with the control group (Figure 5D and E), suggesting that apoptotic effects on A549 cells are largely mediated through p53-dependent pathways.
Conclusion
We implemented drug repositioning for LUAD by random walking the PPI network. From the top ranked drugs, we demonstrated that atracurium could induce apoptosis in A549 human lung cancer cells and the apoptotic effect of atracurium on A549 cells might be mediated via the modulation of caspase-3, p21, and p53 activities. Though more in vitro and in vivo studies are needed to establish the antitumor activity and mechanisms of atracurium, our findings provide fundamental insight of the usefulness of atracurium in human lung cancer therapy.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Kilvaer TK, Paulsen EE, Hald SM, et al. Lymphangiogenic markers and their impact on nodal metastasis and survival in non-small cell lung cancer – a structured review with meta-analysis. PloS One. 2015;10(8):e0132481. doi: 10.1371/journal.pone.0132481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Youlden DR, Cramb SM, Baade PD. The international epidemiology of lung cancer: geographical distribution and secular trends. J Thorac Oncol. 2008;3(8):819–831. doi: 10.1097/JTO.0b013e31818020eb. [DOI] [PubMed] [Google Scholar]
- 4.Detterbeck FC, Postmus PE, Tanoue LT. The stage classification of lung cancer: diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013;143(Suppl 5):e191S–e210S. doi: 10.1378/chest.12-2354. [DOI] [PubMed] [Google Scholar]
- 5.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359(13):1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin J, Beer DG. Molecular predictors of prognosis in lung cancer. Ann Surg Oncol. 2012;19(2):669–676. doi: 10.1245/s10434-011-1967-x. [DOI] [PubMed] [Google Scholar]
- 7.Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kato S, Moulder SL, Ueno NT, et al. Challenges and perspective of drug repurposing strategies in early phase clinical trials. Oncoscience. 2015;2(6):576–580. doi: 10.18632/oncoscience.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamb A, Wee S, Lengauer C. Why is cancer drug discovery so difficult? Nat Rev Drug Discov. 2007;6(2):115–120. doi: 10.1038/nrd2155. [DOI] [PubMed] [Google Scholar]
- 10.Wu Z, Wang Y, Chen L. Network-based drug repositioning. Mol Biosyst. 2013;9(6):1268–1281. doi: 10.1039/c3mb25382a. [DOI] [PubMed] [Google Scholar]
- 11.Keshava Prasad TS, Goel R, Kandasamy K, et al. Human protein reference database – 2009 update. Nucleic Acids Res. 2009;37(Database issue):D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szklarczyk D, Franceschini A, Kuhn M, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Law V, Knox C, Djoumbou Y, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42(Database issue):D1091–D1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohler S, Bauer S, Horn D, Robinson PN. Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet. 2008;82(4):949–958. doi: 10.1016/j.ajhg.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caballero L, Climent V, Hernandez-Romero D, Quintanilla MA, de la Morena G, Marin F. Enzyme replacement therapy in Fabry disease: influence on cardiac manifestations. Curr Med Chem. 2010;17(16):1679–1689. doi: 10.2174/092986710791111297. [DOI] [PubMed] [Google Scholar]
- 17.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 18.Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4(11):682–690. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 19.Goh KI, Choi IG. Exploring the human diseasome: the human disease network. Brief Funct Genomics. 2012;11(6):533–542. doi: 10.1093/bfgp/els032. [DOI] [PubMed] [Google Scholar]
- 20.Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12(1):56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Birsu Cincin Z, Unlu M, Kiran B, Sinem Bireller E, Baran Y, Cakmakoglu B. Anti-proliferative, apoptotic and signal transduction effects of hesperidin in non-small cell lung cancer cells. Cell Oncol. 2015;38(3):195–204. doi: 10.1007/s13402-015-0222-z. [DOI] [PubMed] [Google Scholar]
- 22.Yabasin IB, Ibrahim MM, Adam A, et al. Anticancer effects of vecuronium bromide and cisatracurium besylate on lung cancer cells (A549), in vitro. Biomed Aging Pathol. 2014;4(4):49–353. [Google Scholar]
- 23.Xu L, Tang H, Chen DW, El-Naggar AK, Wei P, Sturgis EM. Genome-wide association study identifies common genetic variants associated with salivary gland carcinoma and its subtypes. Cancer. 2015;121(14):2367–2374. doi: 10.1002/cncr.29381. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Kisor DF, Schmith VD, Wargin WA, Lien CA, Ornstein E, Cook DR. Importance of the organ-independent elimination of cisatracurium. Anesth Analg. 1996;83(5):1065–1071. doi: 10.1097/00000539-199611000-00029. [DOI] [PubMed] [Google Scholar]
- 25.Singh J, Khan M, Pujol A, Baarine M, Singh I. Histone deacetylase inhibitor upregulates peroxisomal fatty acid oxidation and inhibits apoptotic cell death in abcd1-deficient glial cells. PloS One. 2013;8(7):e70712. doi: 10.1371/journal.pone.0070712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanif F, Perveen K, Jawed H, et al. N-(2-hydroxyphenyl)acetamide (NA-2) and temozolomide synergistically induce apoptosis in human glioblastoma cell line U87. Cancer Cell Int. 2014;14(1):133. doi: 10.1186/s12935-014-0133-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi EY, Shin KC, Lee J, Kwon TK, Kim S, Park JW. Treatment with a small synthetic compound, KMU-193, induces apoptosis in A549 human lung carcinoma cells through p53 up-regulation. Asian Pac J Cancer Prev. 2015;16(14):5883–5887. doi: 10.7314/apjcp.2015.16.14.5883. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Yang Y, Lei L, Tian M. Rhizoma paridis saponins induces cell cycle arrest and apoptosis in non-small cell lung carcinoma A549 cells. Med Sci Monit. 2015;21:2535–2541. doi: 10.12659/MSM.895084. [DOI] [PMC free article] [PubMed] [Google Scholar]