

Abstract
DNA ligases are attractive therapeutics because of their involvement in completing the repair of almost all types of DNA damage. A series of DNA ligase inhibitors with differing selectivity for the three human DNA ligases were identified using a structure-based approach with one of these inhibitors being used to inhibit abnormal DNA ligase IIIα-dependent repair of DNA double-strand breaks (DSB)s in breast cancer, neuroblastoma and leukemia cell lines. Raghavan and colleagues reported the characterization of a derivative of one of the previously identified DNA ligase inhibitors, which they called SCR7 (designated SCR7-R in our experiments using SCR7). SCR7 appeared to show increased selectivity for DNA ligase IV, inhibit the repair of DSBs by the DNA ligase IV-dependent non-homologous end-joining (NHEJ) pathway, reduce tumor growth, and increase the efficacy of DSB-inducing therapeutic modalities in mouse xenografts. In attempting to synthesize SCR7, we encountered problems with the synthesis procedures and discovered discrepancies in its reported structure. We determined the structure of a sample of SCR7 and a related compound, SCR7-G, that is the major product generated by the published synthesis procedure for SCR7. We also found that SCR7-G has the same structure as the compound (SCR7-X) available from a commercial vendor (XcessBio). The various SCR7 preparations had similar activity in DNA ligation assay assays, exhibiting greater activity against DNA ligases I and III than DNA ligase IV. Furthermore, SCR7-R failed to inhibit DNA ligase IV-dependent V(D)J recombination in a cell-based assay. Based on our results, we conclude that SCR7 and the SCR7 derivatives are neither selective nor potent inhibitors of DNA ligase IV.
Keywords: Human DNA ligases, DNA ligase inhibitors, DNA double strand break repair, Non-homologous end-joining
1. Introduction
DNA ligation is required during DNA replication and to complete almost all DNA repair events. In human cells, the DNA ligases encoded by three LIG genes are responsible for joining interruptions in the phosphodiester backbone [1]. These enzymes have distinct but overlapping functions in cellular DNA metabolism. Interestingly, DNA ligase expression levels are frequently dysregulated in cancer. For example, the steady state levels of DNA ligase I (LigI) are usually elevated in cancer cell lines and tumor specimens [2,3]. This is presumed to reflect the increased proliferation that is a characteristic of cancer cells. In addition, a significant fraction of cancer cell lines have elevated levels of DNA ligase IIIα (LigIIIα) and reduced levels of DNA ligase IV (LigIV) [2]. Notably, this reciprocal change in DNA ligase levels has been shown to result in abnormal repair of DNA double-strand breaks in leukemia, breast cancer and neuroblastoma, with increased levels of LigIIIα correlating with reduced survival [4–6].
Given their dysregulation in cancer and almost ubiquitous involvement in DNA transactions, DNA ligases are potential therapeutic targets for the development of novel anti-cancer agents. There have been several attempts to identify DNA ligase inhibitors by screening of synthetic chemical and natural product libraries that have met with limited success. These have mainly involved radioactive-based assays and the screening of a relatively small number of compounds [7–9]. A series of small molecule inhibitors with differing specificities for the three human DNA ligases were identified by a structure-based approach using the atomic resolution structure of the DNA binding domain of human DNA ligase I complexed with nicked DNA [2,10]. As expected, several of these inhibitors were cytotoxic and, at subtoxic concentrations, they potentiated cell killing by DNA damaging agents [2]. Unexpectedly, this enhancement of cytotoxicity occurred in malignant cells, but not their non-neoplastic counterparts [2]. In further studies, a LigI/III inhibitor L67 was found to synergistically increase the cytotoxicity of a PARP inhibitor by inhibiting LigIIIα in therapy-resistant chronic myeloid leukemia and breast cancer cells lines with abnormal DNA repair characterized by elevated levels of LigIIIα and PARP-1 [5,6].
Using molecular modeling to predict the structure of the DNA ligase IV DNA binding domain with L189, the inhibitor of all three human DNA ligases identified in the previous structure-based approach [2], Raghavan and colleagues reported the identification of a derivative of L189, which they called SCR7 [11]. SCR7 appeared to selectively inhibit the repair of DSBs by the non-homologous end-joining (NHEJ) pathway in a DNA ligase IV-dependent manner as well as to both reduce tumor growth and increase the efficacy of DSB-inducing therapeutic modalities [11]. In attempting to synthesize SCR7 by the published procedure [11], we encountered problems with the synthesis procedures and discovered discrepancies in the reported structure of SCR7. Using three different preparations of SCR7, we found that it is a DNA ligase inhibitor with greater activity against DNA ligases I and III than DNA ligase IV and that it fails to inhibit DNA ligase IV-dependent V(D)J recombination in a cell-based assay.
2. Materials and methods
2.1. Purification of human DNA ligases
Human LigI and LigIIIβ were purified after expression in Escherichia coli as described [12,13]. Human LigIIIα/XRCC1 and LigIV/XRCC4 complexes were purified from insect cells infected with a single baculovirus expressing both subunits of the DNA ligase complex as described [12,14].
2.2. Preparation and purification of SCR7-G
A solution of benzaldehyde (466 mg, 4.4 mmol) in DMF (1.5 mL) and acetic acid (0.5 mL) was added to solid 4,5-diamino-6-hydroxy-2-mercaptopyrimidine (316 mg, 2.0 mmol). The reaction mixture was heated under reflux for 3 h, then cooled to room temperature, and added slowly to 10 mL of ice water. A yellow solid precipitated out which was collected by vacuum filtration and air dried. The solid was dissolved in 60 mL of chloroform, filtering the chloroform solution to remove insoluble material. The entire chloroform solution was loaded onto a silica gel column packed in dichloromethane. The column was eluted with 2:1 dichloromethane: ethyl acetate, and the first yellow band was collected. The solvent was removed to yield 257 mg (0.77 mmol, 39%).1H NMR (DMSO) δ 13.42 (br. s,) 12.82 (br. s, 1H), 7.43–7.34 (m, 10H).13C NMR (DMSO) δ 175.7,158.5, 155.9, 149.1, 147.0, 137.7, 137.1,129.8 (C-H)129.7 (C-H), 129.5 (C-H), 128.8 (C-H), 128.3 (C-H), 128.2 (C-H), 127.2. HRMS Calcd. for C18H13N4OS (M + H+) 333.0810. Found: 333.0802. IR (Nujol): 3407 (br), 1698, 1633, 1552, 1126, 722, 695, 665. M.P. 200–203 (d.).
2.3. Preparation and purification of SCR7-R
A solution of benzaldehyde (466 mg, 4.4 mmol) in DMF (2 mL) was added to solid 4,5-diamino-6-hydroxy-2-mercaptopyrimidine (316 mg, 2.0 mmol). The reaction mixture was heated under reflux under nitrogen and in the dark for 3 h, then cooled to room temperature and diluted with 3 mL of ethanol. The reaction mixture was added dropwise with stirring to 40 mL of diethyl ether to produce a yellow precipitate, which was collected. The solid was re-suspended in 20 mL of ethanol with heating and stirring to dissolve any oily residue that co-precipitated with the product. The ethanol-insoluble material was collected by vacuum filtration and dried under vacuum to yield pure product. Additional pure product crystallized out of the mother liquor over the course of 3 days, yielding a total of 150 mg (0.45 mmol, 22%) of free-flowing light yellow powder. 1H NMR (DMSO)
δ 12.07 (br. s, 1H), 12.00 (s, 1H), 7.83 (dd, J = 7.6, 1.4 Hz, 2H), 7.52 (d, J = 3.3 Hz, 1H, N-H), 7.39–7.26 (m, 8H), 6.02 (d, J = 3.3 Hz, 1H, C-H). 13C NMR (DMSO)d 173.1, 157.6, 146.7, 143.2, 140.1, 136.1, 129.7 (C-H), 129.2 (C-H), 128.63 (C-H), 128.56 (C-H), 127.0 (C-H), 126.4 (C-H), 104.3, 52.3 (C-H). HRMS Calcd. for C18H15N4OS (M+H+) 335.0967. Found: 333.0959. IR (Nujol): 3381 (br), 1652, 1565, 1085, 693, 665, 612. M.P. 273–280 (d.).
2.4. DNA ligase assays
DNA nick ligation was measured using a fluorescence-based ligation assay described previously [15]. Briefly, 100 fmol of purified LigI, LigIIIα/XRCC1, or LigIV/XRCC4, SCR7, were incubated in the presence or absence of an SCR7 derivative with fluorescent nicked DNA annealed to an upstream quencher (200 fmol) in ligation buffer (60 mM Tris–HCl [pH 7.4], 10 mM MgCl2, 5 mM DTT, 1 mM ATP, 50 μg/ml BSA, 4% DMSO, and 50 mM NaCl for LigI or 150 mM NaCl for LigIII and LigIV) at 25 °C in a total volume of 20 μL. Following incubation, reactions were further diluted to 200 μL with a 30-fold molar excess of the unlabeled competing oligonucleotide in annealing buffer (10 mM Tris–HCl [pH 7.4], 50 mM KCl, 1 mM EDTA and 5 mM MgCl2) and heated to 95 °C for 5 min. After cooling to 4 °C at a rate of 2 °C/min, fluorescence read at 519 nm (excitation at 495 nm) was measured immediately using the Synergy H4 microplate reader (BioTek). Relative ligation efficiency was calculated as a percentage of the amount of product formed by each uninhibited ligase. This corresponds to 106 fmol (53% ligation), 104 fmol (52% ligation), and 9.4 fmol (4.7% ligation) of product formed for LigI, LigIII, and LigIV respectively. Additionally, the ligation assay was repeated with concentration of ligases adjusted to give 50% ligation efficiency (94 fmol LigI, 96 fmol LigIII, and 1.1 pmol LigIV); however, this data was not significantly different. Data was analyzed by GraphPad Prism software.
DNA nick ligation and intermolecular joining of linear duplex DNAs were measured using 32P-labeled DNA substrates. The nick ligation substrate was prepared by annealing of three oligonucleotides: upstream-strand (5′-GGTAATTGTGAGCGCTCACAAGCGGTACGTCAACGGAACGCAAATCTCGAGATCACAGCAACTAGACCT), downstream strand (5′-32P-phosphorylated GAAGATGACGTGAGGAATTCTACC) and bottom strand (5′-biotinylated GGTAGAATTCCTCACGTCATCTTCAGGTCTAGTTGCTGTGATCTCGAGATTTGCGTTCCGTTGACGTACCTGCTTGTGAGCGCTCACAATTAC).
DNA ligase (0.05 pmol LigI, 0.05 pmol LigIIIα/XRCC1 or 0.2 pmol LigIV/XRCC4) in 5 μL of the dilution buffer (100 mM Tris-Cl pH7.5, 50 mM NaCl, 2 mM DTT, 1.6 mM ATP, 10% glycerol) was preincubated at room temperature with 4 μL of an SCR7 derivative diluted in 10% DMSO/25 mM Tris-Cl pH7.5. Subsequently the ligase/SCR7 mixture was supplemented with 11 μL of the reaction buffer (45.5 mM Tris-Cl pH7.5, 10.91 mM MgCl2, 0.455 mg/ml BSA, 0.91 mM DTT, 0.7% glycerol and 0.2 pmol DNA substrate) and incubated at 25 °C for 20 min. The concentration of SCR7 is indicated as its final concentration in each ligation reaction. After the reaction was terminated by transfer to ice and the addition of SDS to a final concentration of 0.5%, the DNA substrate was recovered with Streptavidin Magnesphere Paramagnetic Particles (Promega) and then suspended in formamide-dye solution prior to incubation at 100 °C for 5 min, and electrophoresis through a 7.5 M urea-containing 15% polyacrylamide gel. Ligation substrate and products were visualized using a Typhoon 7000 phosphorimager (GE Healthcare).
The double-strand ligation substrate, which has a 52 bp stem and a 5 nucleotide loop with a 4 nucleotide cohesive end at the 5′-terminus (Figs. 1–4 A), was prepared by ligation of three deoxyoligonucleotides: top-oligo (5′-GATCGAAGATGACGTGAGGAATTCTACCGCAGGGTAAG), hairpin-oligo (5′-phosphorylated- CGACGCATGACTCTAAAGCCC[T-biotinylated]CCTTTAGAGTC), and bottom-oligo (5′-phosphorylated ATGCGTCGCTTACCCTGCGGTAGAATTCCTCACGTCATCTTC), and purified by denaturing polyacrylamide gel electrophoresis. Before ligation assay, the Hairpin-52 was labeled at the 5′-terminus using T4 polynucleotide kinase and [γ-32P]ATP, followed by incubation with cold ATP.
Fig. 1.
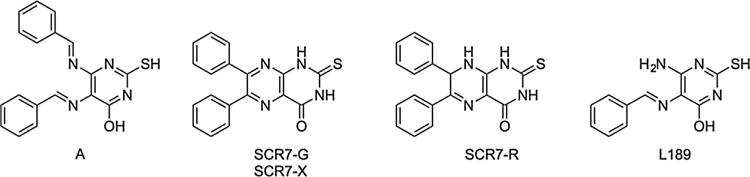
Structures of SCR7 derivatives. In (A) is shown the structure of SCR7 as reported by Srivastava et al. [11]. The structure of the major product generated by the synthesis protocol described by Srivastava et al. [11] and by a different synthesis method [20] (SCR7-G) and the compound sold as SCR7 by XcessBio (SCR7-X) are shown. Our structure determination of the SCR7 provided by Dr. Sathees Raghavan (SCR7-R) and the pan human DNA ligase inhibitor (L189) described by Chen et al. [2] are also shown.
Fig. 4.
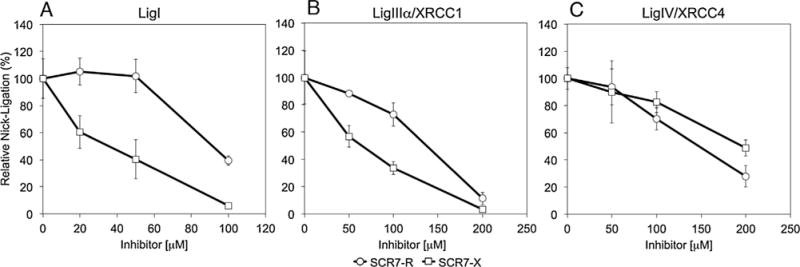
Effects of SCR7 derivatives on nick ligation by LigI, LigIIIα/XRCC1 and LigIV/XRCC4 using radioactively-labeled nicked DNA substrate. DNA ligation reactions with a nicked, fluorescent DNA substrate (0.2 pmol) were carried as described in Materials and Methods. LigI (0.1 pmol, Panel A), LigIIIα/XRCC1 (0.1 pmol, Panel B) and LigIV/XRCC4 (1.1 pmol, Panel C) were incubated with SCR7 derivatives; SCR7-G (triangle); SCR7-R (circle); SCR7-X (square). Data are the mean ± standard deviation of three independent experiments.
The double-stranded DNA ligation assay was carried out as described for the nick ligation assay with the following modifications: 0.2 pmol LigIIIα/XRCC1 or 1 pmol LigIV/XRCC4 was used; the reaction buffer contained 21.8% polyethylene glycol (average molecular weight 6000); electrophoresis was carried out in 7.5 M urea-containing 12.5% polyacrylamide gels.
2.5. V(D)J recombination assay
Transfections into the human pre-B cell line, Reh, were performed as described using pGG49 [16–18]. Aliquots of recovered plasmid were digested with DpnI and electrotransformed into DH10B Escherichia coli [19]. Bacterial transformants were plated on media containing ampicillin alone (100 mg/ml), and on ampicillin and chloramphenicol (100 mg/ml and 33 mg/ml, respectively). The frequency of recombination was quantitated as described [19]. For the V(D)J recombination assay, paired two-tailed Student’s t-tests were performed to compare V(D)J recombination efficiency of cells treated with SCR7 at 200 μM or only with the control amount of DMSO solvent to test the null hypothesis that the V(D)J recombination efficiency of cells treated with SCR7 at 200 μM or only with the control amount of DMSO solvent are the same.
3. Results
3.1. Synthesis and structure of SCR7
After carefully following the published procedure for SCR7 [11], we were unable to isolate a compound with the reported structure of SCR7 (Fig. 1). Instead, the synthesis protocol generated a mixture of L189, a previously described inhibitor of all three DNA ligases [2] plus a compound that we call SCR7-G with the structure shown in Fig. 1. We were able to separate and purify SCR7-G, which is a known compound that has been previously synthesized by a different method [20]. Using this method [20], we were able to synthesize and purify a compound that was physically and spectroscopically identical to SCR7-G. Next we purchased SCR7 from XcessBio (SCR7-X) and analyzed it by 1H and 13C spectroscopy. The spectra of SCR7-X matched that of SCR7-G rather than that predicted for the reported SCR7 structure [11]. In an attempt to determine the structure of the compound used in the published paper [11], we obtained a sample of SCR7 from Dr. Sathees C. Raghavan (designated SCR7-R in our experiments). We analyzed this material by 1H, 13C, DEPT and COSY NMR and found that the experimental data were not consistent with the structure determined for SCR7-G or the published structures of either SCR7 [11] or L189 [2]. Instead, we conclude that SCR7-R is closely related to SCR7-G and has the structure shown in Fig. 1. Since SCR7-R is a minor product of our synthesis protocol for SCR7-G, we developed a purification procedure for SCR7-R. Further details about the synthesis, structure determination, and chemical properties of these compounds will be reported in the chemical literature elsewhere.
3.2. Effect of SCR7 derivatives on DNA joining by LigI, LigIIIα/XRCC1 and LigIV/XRCC4
Given the discrepancies in the SCR7 structures, we purified the three human DNA ligases (Fig. 2) and determined the activities of SCR7-R, SCR7-G and SCR7-X. In initial studies, we employed a fluorescence-based nick ligation assay that we developed for high throughput screening of candidate DNA ligase inhibitors and subsequently adapted for kinetic studies [2,12,15]. Using this assay, we found that activities of the SCR7 derivatives were indistinguishable (Fig. 3). Notably, each of the SCR7 derivatives were more potent inhibitors of LigI than the other DNA ligases, with 300 μM causing about 50% inhibition. In contrast, both LigIIIα/XRCC1 and LigIV/XRCC4 were only inhibited about 25% by the SCR7 derivatives at 300 μM. Similar results were obtained with LigIIIβ purified from Escherichia coli (Fig. 2). These assays contained higher amounts of DNA ligase IV/XRCC4 (1.1 pmol) than the other DNA ligases (100 fmol) because of its lower catalytic activity. Similar results were obtained in assays with 100 fmol DNA ligase IV/XRCC4. Since these results contradicted the published study, which reported that SCR7 does not inhibit LigI and has a greater inhibitory effect on LigIV/XRCC4 than LigIIIα/XRCC1 [11], we did additional ligation assay studies with a radioactively–labeled oligonucleotide duplex substrate with a single central nick [2]. Although there was more variability in the assays with the radioactive substrate and the degree of inhibition was greater because of the lower amounts of substrate used in these assays, the SCR7 derivatives were again more potent inhibitors of LigI compared with LigIIIα/XRCC1 and LigIV/XRCC4 (Fig. 4).
Fig. 2.
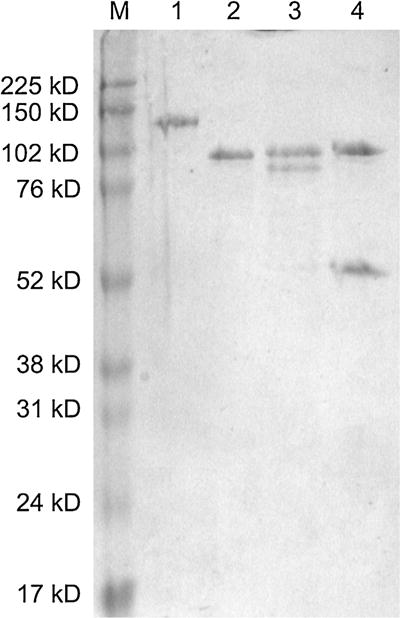
Purified human DNA ligases. Five pmoles of each DNA ligase were electrophoresed in an SDS-containing 10% polyacrylamide gel and then stained with Coomassie Brilliant Blue. Lane 1, LigI; lane 2, LigIIIβ; lane 3, LigIIIα/XRCC1; lane 4, LigIV/XRCC4.
Fig. 3.
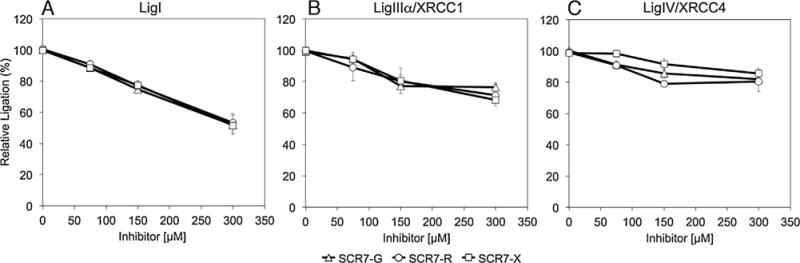
Effects of SCR7 derivatives on nick ligation by LigI, LigIIIα/XRCC1 and LigIV/XRCC4 using nicked, fluorescent DNA substrate. DNA ligation reactions with a labeled, nicked DNA substrate (0.2 pmol) were carried as described in Materials and Methods. LigI (0.05 pmol, Panel A), LigIIIα/XRCC1 (0.05 pmol, Panel B) and LigIV/XRCC4 (0.2 pmol, Panel C) were incubated with SCR7 derivatives; SCR7-G (triangle); SCR7-R (circle); SCR7-X (square). Data are the mean ± standard deviation of three independent experiments.
Since the in vivo substrate for LigIV is a duplex DNA end, we designed a novel assay to detect ligation of either one or both strands during inter-molecular ligation. The DNA substrate consists of an oligonucleotide that forms a partial duplex with a hairpin at one end and a linear duplex with a single-stranded end that is complementary to the single-stranded end of the hairpin duplex (Fig. 5A). Using this substrate, we examined the effects of the various SCR7 derivatives on the two human DNA ligases, LigIIIα/XRCC1and LigIV/XRCC4, with robust intermolecular joining activity [21]. Notably, each of the SCR7 derivatives were more potent inhibitors of LigIIIα/XRCC1 than LigIV/XRCC4 (Fig. 5B and C).
Fig. 5.
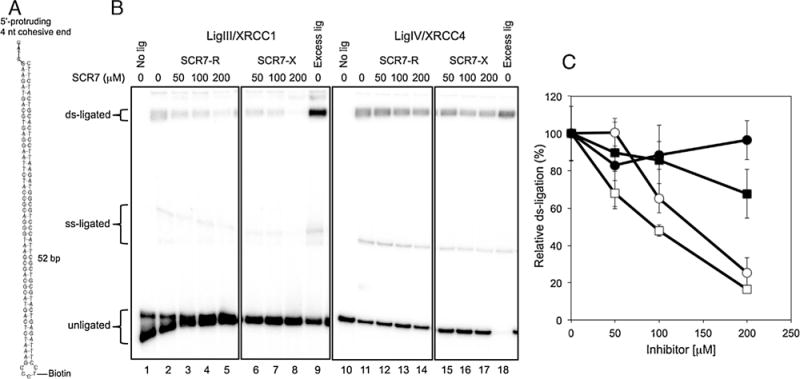
Effects of SCR7 derivatives on double-strand ligation by LigIIIα/XRCC1 and LigIV/XRCC4. (A) Schematic structure of the DNA substrate for double-strand ligation. The 5′ terminus is 32P-phosphorylated. (B) The DNA substrate (0.2 pmol) was incubated with either LigIIIα/XRCC1 (0.2 pmol) or LigIV/XRCC4 (1 pmol) in the absence or presence of the indicated concentrations of SCR7 derivatives prior to electrophoresis in 7 M urea-containing 12.5% polyacrylamide gels. The products resulting from ligation of both strands (ds-ligated) and only one strand (ss-ligated) as well as the original substrate (unligated) are indicated. (C) The results of the inter-molecular ligation assays are shown graphically. LigIIIα/XRCC1 (open symbols) and LigIV/XRCC4 (closed symbols) incubated with either SCR7-R (circles) or SCR7-X (squares). Ligation efficiency was calculated by setting the reactions with no inhibitor as 100%. Data are the mean ± standard deviation of three independent experiments.
3.3. Effect of SCR7 derivatives on V(D)J recombination
The repair of DNA DSBs generated by the RAG1/RAG2 endonuclease at specific sites flanking immunoglolublin gene segments in human cells is dependent on NHEJ [22,23]. This assay has been used extensively to identify the core NHEJ factors, including DNA ligase IV, and to provide insights into the mechanism of NHEJ [22–24]. We transfected the V(D)J recombination substrate, pGG49, into the human pre-B cell line, Reh. After cleavage of the substrate by the RAG enzyme complex, the two signal ends are joined by NHEJ in a manner that is entirely dependent on DNA ligase IV [23]. The effect of SCR7-R on V(D)J recombination signal joint formation was determined in plasmid substrates recovered 48 h after transfection. In assays with concentrations of SCR7-R up to 200 μM, there was no significant effect on the frequency of V(D)J recombination signal joint formation (p = 0.81, paired t-test) between treated and untreated cells (Table 1). It was not possible to test higher concentrations of SCR7-R because of cellular toxicity caused by SCR7-R. Thus, in accord with the results of our in vitro intermolecular ligation assays, we conclude that SCR7-R does not inhibit DNA ligase IV activity at concentrations up to 200 μM in an established cell-based assay for DNA ligase IV-dependent NHEJ.
Table 1.
Effects of SCR7-R on LigIV-dependent V(D)J recombination.
| pGG49 V(D)J Reconbination Efficiency in Reh Cells
| ||||
|---|---|---|---|---|
| SCR7 (200 μm) | DAC | DA | Rec% | |
| Experiment 1 | − | 57 | 4060 | 1.40% |
| + | 22 | 7350 | 0.30% | |
| Experiment 2 | − | 131 | 3550 | 3.60% |
| + | 6 | 140 | 3.90% | |
| Experiment 3 | − | 28 | 2210 | 1.30% |
| + | 4 | 260 | 1.70% | |
4. Discussion
Raghavan and colleagues described the synthesis and characterization of a compound, SCR7, that inhibited DNA ligase IV and blocked DNA ligase IV-dependent NHEJ in extracts and cell-based assays [11]. Furthermore, they reported that this compound reduced the growth of tumor xenografts, both in combination with genotoxic agents used clinically and as a single agent [11]. In this study, we have found that the published synthesis protocol does not generate the compound with the structure described by Raghavan and colleagues and that SCR7 provided by Dr. Raghavan (SCR7-R) also does not have the structure described [11]. We have determined the structure of SCR7-R and SCR7-G, the compounds generated by the synthesis protocol described by Raghavan and colleagues [11]. Furthermore, our analysis of commercially available SCR7 (from XcessBio and designated SCR7-X here) revealed that it has the same structure as SCR7-G rather than the structure listed by XcessBio.
Although high concentrations of SCR7-G and SCR7-R inhibited LigIV as reported [11], the SCR7 derivatives were more effective inhibitors of LigIIIα and, in particular, LigI. Although it is possible that differences in the DNA substrate underlie this discrepancy, it appears more likely that this reflects actual differences in how the compounds affect the different ligases. In the published study [11], multiple other bands were present in the DNA ligase preparations purified from Escherichia coli in addition to the DNA ligase polypeptides and protein partners identified by immunoblotting [11]. In contrast, the fractions of DNA LigIIIα/XRCC1 and LigIV/XRCC4 used in our study were purified after expression in insect cells, which permits higher and more reliable enzyme activity.
While it was reported that SCR7-R inhibited the joining of linear DNA molecules by extracts from rat testes [11], it has been established that extracts from mammalian cell lines have robust end joining activity that, in most cases, is not dependent on the core factors involved in the major NHEJ pathway [25–28]. Unlike the extract assay developed by Baumann and West [29], there is no evidence that the end joining carried out by the rat testes extract is dependent upon LigIV and the other core NHEJ factors. It was also reported that SCR7 reduced the frequency of NHEJ in cell-based assays with plasmid substrates. Notably, these assays used concentration of SCR7-R that were 10-fold lower than the concentration of SCR7-R used to inhibit purified DNA ligase IV by 50% in vitro. In our study, we have used a cellular plasmid-based quantitative assay for V(D)J recombination that has been shown by many laboratories to be dependent upon LigIV [23,24]. Concentrations of up to 200 μM SCR7-R failed to reduce LigIV-dependent V(D)J recombination. This is in contrast to the less direct assay for V(D)J recombination in lymphoid cellsused by Raghavan et al., which also reduced the numbers of B and T cells in mice treated with SCR7-R, suggesting toxicity [11]. It should be noted that there are many possible steps at which a compound might affect a PCR readout for V(D)J recombination in an animal model. In contrast, the cellular plasmid-based assay provides a direct and quantitative measure of LigIV-dependent V(D)J recombination.
In summary, both SCR7-R and its closely related derivative SCR7-G are weak inhibitors of LigIV and exhibit higher activity against LigI and LigIIIα/XRCC1. Furthermore, SCR7-R did not inhibit a V(D)J recombination assay that is dependent upon LigIV. Recent studies have shown that either knockdown of a core NHEJ factor [30] or incubation with SCR7-X [30,31] increases the efficiency of CRISPR-Cas9-induced precise genome editing. Since SCR7-X/SCR7-G enhanced CRISPR-CAS9 [30,31] at a concentration of SCR7-X/SCR7-G (1 μM) that was significantly lower than the concentration required to inhibit LigIV in vitro (> 200 μM), it appears likely that SCR7-X/SCR7-G is causing this effect by mechanisms other than inhibition of LigIV.
Acknowledgments
We thank Dr. Sathees Raghavan for the sample of SCR7-R. This work was supported by US National Institute of Health Grants, R01 GM47251, GM57479, ES01252 and P01CA92584, to AET) and by US National Institute of Health funding to MRL. Flow cytometry and microscopy were carried out in University of New Mexico Cancer Center Shared Resources supported by NCI Cancer Center Support Grant P30CA11800.
References
- 1.Ellenberger T, Tomkinson AE. Eukaryotic DNA ligases: structural and functional insights. Annu Rev Biochem. 2008;77:313–338. doi: 10.1146/annurev.biochem.77.061306.123941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen X, Zhong S, Zhu X, Dziegielewska B, Ellenberger T, Wilson GM, MacKerell AD, Jr, Tomkinson AE. Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res. 2008;68:3169–3177. doi: 10.1158/0008-5472.CAN-07-6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun D, Urrabaz R, Nguyen M, Marty J, Stringer S, Cruz E, Medina-Gundrum L, Weitman S. Elevated expression of DNA ligase I in human cancers. Clin Cancer Res. 2001;7:4143–4148. [PubMed] [Google Scholar]
- 4.Newman EA, Lu F, Bashllari D, Wang L, Opipari AW, Castle VP. Alternative NHEJ pathway components are therapeutic targets in high-Risk neuroblastoma. Mol Cancer Res MCR. 2015;13:470–482. doi: 10.1158/1541-7786.MCR-14-0337. [DOI] [PubMed] [Google Scholar]
- 5.Tobin LA, Robert C, Nagaria P, Chumsri S, Twaddell W, Ioffe OB, Greco GE, Brodie AH, Tomkinson AE, Rassool FV. Targeting abnormal DNA repair in therapy-resistant breast cancers. Mol Cancer Res MCR. 2012;10:96–107. doi: 10.1158/1541-7786.MCR-11-0255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tobin LA, Robert C, Rapoport AP, Gojo I, Baer MR, Tomkinson AE, Rassool FV. Targeting abnormal DNA double strand break repair in tyrosine kinase inhibitor-resistant chronic meyloid leukemias. Oncogene. 2013;32:1784–1793. doi: 10.1038/onc.2012.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan GT, Lee S, Lee IS, Chen J, Leitner P, Besterman JM, Kinghorn AD, Pezzuto JM. Natural-product inhibitors of human DNA ligase I. Biochem J. 1996;314(Pt 3):993–1000. doi: 10.1042/bj3140993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun D, Urrabaz R. Development of non-electrophoretic assay method for DNA ligases and its application to screening of chemical inhibitors of DNA ligase I. J Biochem Biophys Methods. 2004;59:49–59. doi: 10.1016/S0165-022X(02)00071-4. [DOI] [PubMed] [Google Scholar]
- 9.Tseng HM, Shum D, Bhinder B, Escobar S, Veomett NJ, Tomkinson AE, Gin DY, Djaballah H, Scheinberg DA. A high-Throughput scintillation proximity-Based assay for human DNA ligase IV Assay. Drug Dev Technol. 2011 doi: 10.1089/adt.2011.0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong S, Chen X, Zhu X, Dziegielewska B, Bachman KE, Ellenberger T, Ballin JD, Wilson GM, Tomkinson AE, MacKerell AD., Jr Identification and validation of human DNA ligase inhibitors using computer-aided drug design. J Med Chem. 2008;51:4553–4562. doi: 10.1021/jm8001668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srivastava M, Nambiar M, Sharma S, Karki SS, Goldsmith G, Hegde M, Kumar S, Pandey M, Singh RK, Ray P, et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell. 2012;151:1474–1487. doi: 10.1016/j.cell.2012.11.054. [DOI] [PubMed] [Google Scholar]
- 12.Chen X, Pascal J, Vijayakumar S, Wilson GM, Ellenberger T, Tomkinson AE. Human DNA ligases I, III, and IV-purification and new specific assays for these enzymes. Methods Enzymol. 2006;409:39–52. doi: 10.1016/S0076-6879(05)09003-8. [DOI] [PubMed] [Google Scholar]
- 13.Mackey ZB, Niedergang C, Murcia JM, Leppard J, Au K, Chen J, de Murcia G, Tomkinson AE. DNA ligase III is recruited to DNA strand breaks by a zinc finger motif homologous to that of poly(ADP-ribose) polymerase. Identification of two functionally distinct DNA binding regions within DNA ligase III. J Biol Chem. 1999;274:21679–21687. doi: 10.1074/jbc.274.31.21679. [DOI] [PubMed] [Google Scholar]
- 14.Della-Maria J, Zhou Y, Tsai MS, Kuhnlein J, Carney JP, Paull TT, Tomkinson AE. Human Mre11/Human Rad50/Nbs1 and DNA ligase III{alpha}/XRCC1 protein complexes act together in an alternative nonhomologous end joining pathway. J Biol Chem. 2011;286:33845–33853. doi: 10.1074/jbc.M111.274159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen X, Ballin JD, Della-Maria J, Tsai MS, White EJ, Tomkinson AE, Wilson GM. Distinct kinetics of human DNA ligases I, IIIalpha, IIIbeta, and IV reveal direct DNA sensing ability and differential physiological functions in DNA repair. DNA Repair. 2009;8:961–968. doi: 10.1016/j.dnarep.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gauss GH, Lieber MR. Mechanistic constraints on diversity in human V(D)J recombination. Mol Cell Biol. 1996;16:258–269. doi: 10.1128/mcb.16.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gauss GH, Lieber MR. Unequal signal and coding joint formation in human V(D)J recombination. Mol Cell Biol. 1993;13:3900–3906. doi: 10.1128/mcb.13.7.3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gauss GH, Lieber MR. DEAE-dextran enhances electroporation of mammalian cells. Nucleic Acids Res. 1992;20:6739–6740. doi: 10.1093/nar/20.24.6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lieber MR, Hesse JE, Mizuuchi K, Gellert M. Developmental stage specificity of the lymphoid V(D)J recombination activity. Genes Dev. 1987;1:751–761. doi: 10.1101/gad.1.8.751. [DOI] [PubMed] [Google Scholar]
- 20.Schneider HJ, Pfleiderer W. Pteridines: LXI. Synthesis and properties of thiolumazines. Chem Ber. 1974;107:3377–3394. [Google Scholar]
- 21.Chen L, Trujillo K, Sung P, Tomkinson AE. Interactions of the DNA ligase IV-XRCC4 complex with DNA ends and the DNA-dependent protein kinase. J Biol Chem. 2000;275:26196–26205. doi: 10.1074/jbc.M000491200. [DOI] [PubMed] [Google Scholar]
- 22.Alt FW, Zhang Y, Meng FL, Guo C, Schwer B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell. 2013;152:417–429. doi: 10.1016/j.cell.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grawunder U, Zimmer D, Fugmann S, Schwarz K, Lieber MR. DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol Cell. 1998;2:477–484. doi: 10.1016/s1097-2765(00)80147-1. [DOI] [PubMed] [Google Scholar]
- 24.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boe SO, Sodroski J, Helland DE, Farnet CM. DNA end-joining in extracts from human cells, Biochem. Biophys. Res Commun. 1995;215:987–993. doi: 10.1006/bbrc.1995.2561. [DOI] [PubMed] [Google Scholar]
- 26.Fairman MP, Johnson AP, Thacker J. Multiple components are involved in the efficient joining of double stranded DNA breaks in human cell extracts. Nucleic Acids Res. 1992;20:4145–4152. doi: 10.1093/nar/20.16.4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson AP, Fairman MP. The identification and characterization of mammalian proteins involved in the rejoining of DNA double-strand breaks in vitro. Mutat Res. 1996;364:103–116. doi: 10.1016/0921-8777(96)00028-6. [DOI] [PubMed] [Google Scholar]
- 28.Mason RM, Thacker J, Fairman MP. The joining of non-complementary DNA double-strand breaks by mammalian extracts. Nucleic Acids Res. 1996;24:4946–4953. doi: 10.1093/nar/24.24.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baumann P, West SC. DNA end-joining catalyzed by human cell-free extracts. Proc Natl Acad Sci U S A. 1998;95:14066–14070. doi: 10.1073/pnas.95.24.14066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kuhn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol. 2015;33:543–548. doi: 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- 31.Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33:538–542. doi: 10.1038/nbt.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]