

A single-molecule approach shows that tropomyosin isoforms can either allow or prohibit productive interactions of myosin Va with the actin track. These data support the idea that an “actin–Tpm code” is interpreted by myosin isoforms and their cargo adapter proteins to allow for spatial and temporal sorting of actomyosin function in the cell.
Abstract
Tropomyosin (Tpm) isoforms decorate actin with distinct spatial and temporal localization patterns in cells and thus may function to sort actomyosin processes by modifying the actin track affinity for specific myosin isoforms. We examined the effect of three Tpm isoforms on the ability of myosin Va (myoVa) to engage with actin in vitro in the absence or presence of the cargo adapter melanophilin (Mlph), which links myoVa to Rab27a-melanosomes for in vivo transport. We show that both the myosin motor domain and the cargo adapter Mlph, which has an actin-binding domain that acts as a tether, are sensitive to the Tpm isoform. Actin–Tpm3.1 and actin–Tpm1.8 were equal or better tracks compared to bare actin for myoVa-HMM based on event frequency, run length, and speed. The full-length myoVa-Mlph complex showed high-frequency engagement with actin-Tpm3.1 but not with actin-Tpm1.8. Actin–Tpm4.2 excluded both myoVa-HMM and full-length myoVa-Mlph from productive interactions. Of importance, Tpm3.1 is enriched in the dendritic protrusions and cortical actin of melanocytes, where myoVa-Mlph engages in melanosome transport. These results support the hypothesis that Tpm isoforms constitute an “actin–Tpm code” that allows for spatial and temporal sorting of actomyosin function in the cell.
INTRODUCTION
Actomyosin interactions drive many cellular processes, including organelle trafficking, cytokinesis, and maintenance of cell shape and polarity. A large diversity of myosin isoforms, grouped into >30 classes, allows for specialized motors to accomplish this wide variety of distinct cellular tasks (Hartman and Spudich, 2012). How are specific myosins targeted to their sites of action? The two cytoplasmic β- and γ-actin isoforms alone cannot provide the necessary track diversity. However, there are >40 tropomyosin (Tpm) isoforms in humans, generated from four genes by the use of multiple promoters and alternative splicing (Dufour et al., 1998; Cooley and Bergtrom, 2001). These isoforms differentially modify cellular actin structures in both space and time, generating filaments that are essential for polarity, embryonic development, and tissue function (for reviews, see Gunning et al., 2005, 2008; Vindin and Gunning, 2013). Tpm isoforms may provide a sorting mechanism by which a set of tracks works efficiently with only selected motor–cargo complexes.
Tpm forms an α-helical coiled-coil dimer that binds head to tail and stabilizes the actin filament (Cooper, 2002; Tojkander et al., 2011). By analogy to the well-studied striated muscle isoforms (reviewed in Lehman et al., 2013), Tpm in nonmuscle cells likely regulates actomyosin interactions, an idea supported by a number of biochemical and cellular studies. Tpm isoforms differentially affect the motility and actin-activated ATPase activity of nonmuscle myosin II in vitro (Barua et al., 2014). Myosin I is inhibited by Tpm in vitro (Collins et al., 1990; McIntosh et al., 2015), and its cellular localization corresponds to regions with dynamic actin and low Tpm concentration (Tang and Ostap, 2001). Budding and fission yeast class V myosins, in contrast, require Tpm for processive movement on actin (Hodges et al., 2012; Clayton et al., 2014). Thus there is strong evidence that the large variety of Tpm isoforms could be responsible for adapting the cytoskeletal actin filaments to their different purposes and relevant myosins.
Cargo transport processes require adapter proteins that link the cargo to the motor. Melanophilin (Mlph) links Rab27a melanosomes to full-length myosin Va (myoVa) to enable melanosome transport into the dendritic protrusions of melanocytes (Wu et al., 2002a, b). Full-length myoVa exists in a folded, autoinhibited conformation at physiologic ionic strength (reviewed in Trybus, 2008). Mlph activates full-length myoVa for transport by competing for the globular tail and disrupting the autoinhibited state (Sckolnick et al., 2013). Adapter proteins may also contribute to sorting by sensing different actin–Tpm topologies. A C-terminal positively charged region of Mlph binds to actin and is essential for melanosome localization in vivo (Kuroda et al., 2003). In vitro, this region of Mlph tethers myoVa to the actin track, resulting in enhanced track affinity (Sckolnick et al., 2013).
Most actin in vivo is bound to one of the many Tpm isoforms, suggesting that myoVa-mediated transport may be guided by the Tpm topology of the actin cytoskeleton. To test this hypothesis, we used single-molecule total internal reflection fluorescence (TIRF) microscopy to determine the effect of three Tpm isoforms found in melanocytes on the transport ability of myoVa. Our results show that Tpm isoforms differentially affect run frequency, run length, and speed of myoVa transport in vitro. Of importance, the isoform that supports efficient movement of myoVa-Mlph in vitro (Tpm3.1) is located in the dendritic processes of melanocytes, the site of myoVa-directed melanosome transport. This study supports the idea that the Tpm topology sorts myosins and cargoes to different actin structures inside the cell and ultimately allows for the correct spatial placement of biological processes.
RESULTS AND DISCUSSION
Tropomyosin isoforms bias actin track selection by myoVa
We examined the effect of Tpm isoform on myoVa motility at the single-molecule level by TIRF microscopy. We used full-length myoVa activated from its autoinhibited state by Mlph (myoVa-Mlph), which is the physiologically relevant transport complex. In addition, we used a truncated, constitutively active myoVa–heavy meromyosin (myoVa-HMM) to parse out the relative contributions of the motor domain versus Mlph to Tpm isoform–dependent changes in movement. We expressed in bacteria three Tpm isoforms (Tpm1.8, Tpm3.1, Tpm4.2) shown by reverse transcriptase PCR (RT-PCR) to be present in melanocytes (Figure 1). All experiments were carried out in 150 mM salt, conditions under which full-length myoVa is poised to be activated by Mlph and physiologic electrostatic protein–protein interactions are favored.
FIGURE 1:
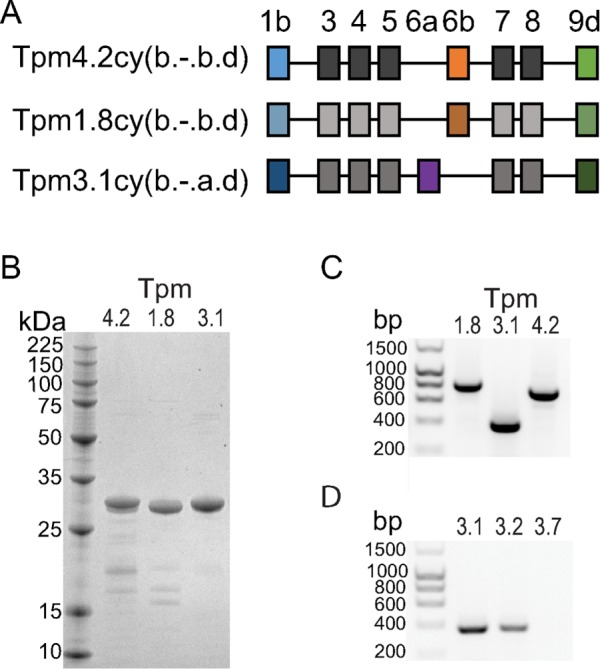
Low–molecular weight cytoplasmic Tpm isoforms used here. (A) Exon organization of Tpm4.2cy(b.-.b.d) (Tm4), Tpm1.8cy(b.-.b.d) (Tm5a), and Tpm3.1cy(b.-.a.d) (Tm5NM1). The new nomenclature for Tpm isoforms is described in Geeves et al. (2015). (B) 12.5% SDS–PAGE gel of purified Tpm isoforms. (C) RT-PCR confirmed the presence of Tpm-1.8, 3.1, and 4.2 in melanocytes. TPM-specific primer pairs were used (TPM1 exons 1b/9d for Tpm1.8, TPM3 exons 6a/9d for Tpm3.1, and TPM4 exons 1b/9d for Tpm4.2). (D) RT-PCR showed higher expression of Tpm3.1 than Tpm3.2 in melan-a melanocytes. Primer pairs from TPM isoform–specific exons were used (exons 6a/9d for Tpm3.1, exons 6b/9d for Tpm3.2, exons 6a/9c for Tpm3.7 negative control).
We used a single-molecule approach to compare processive movement of myoVa-HMM on bare actin and actin decorated with Tpm1.8, Tpm3.1, or Tpm4.2 (Figure 2, A–C). Run frequency, defined as the number of moving motors per micromolar motor per micrometer of actin per unit time, was used to assess the probability with which the motor domain engages with the various tracks. Strikingly, Tpm isoform had a noticeable effect on the number of motors that can engage with the track and undergo productive runs. The projection of all movement trajectories of myoVa-HMM in regions with comparable actin–Tpm density over 100 s illustrates this difference in run frequency (Figure 2B and Supplemental Figure S1A). The run frequency of myoVa-HMM on actin–Tpm3.1 and actin–Tpm1.8 increased 1.5- and 2.0-fold, respectively, relative to that on bare actin. In striking contrast, actin–Tpm4.2 mostly excluded myoVa-HMM from productive runs (Figure 2C).
FIGURE 2:

Effect of Tpm isoform on myoVa movement in single-molecule and ensemble assays. (A) Motion of single molecules of Qdot-labeled myoVa-HMM on actin tracks. (B) Projection of all movement tracks of myoVa-HMM over 100 s. Supplemental Figure S1A shows actin visualization. (C) Run frequency, the number of moving motors per actin length per micromolar motor per time, varies with Tpm isoform. Error bars, SEM. Data are from three independent experiments using two protein preparations. Fields analyzed were 30 × 30 μm2, had >100 μm of actin, and lasted 100 s. Values were normalized to bare actin. Supplemental Table S1 lists values. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with bare actin (ANOVA). (D) Movement of rhodamine-phalloidin–labeled actin over ensembles of myoVa-HMM attached via a biotin tag at the C-terminus to a NeutrAvidin-coated coverslip. (E) Filament speeds, determined by tracking individual filaments and fitting histograms to a Gaussian distribution. Error bars, SD. Actin-Tpm4.2 (blue) shows no interaction with myoVa-HMM. Speeds are listed in Supplemental Table S2. (F) Effect of Tpm isoform on median time of engagement of actin with myoVa ensembles. Representative data from one experiment show that the probability of filament attachment to the motor follows an exponential decay. Mean detachment time t1/2 was determined by fitting data from for independent experiments with a single exponential; inset shows mean values for t1/2. Error bars, SEM. *p < 0.05, compared with bare actin (ANOVA). (G) Motion of single molecules of the full-length myoVa-Mlph complex on actin tracks. The Mlph N-terminus is labeled with a streptavidin-Qdot. (H) Projection of all movement tracks of myoVa-Mlph over 100 s. Supplemental Figure S1B shows actin visualization. (I) Run frequency determined as in C. Right axis, frequency relative to myoVa-HMM on bare actin (scaled as in C). Supplemental Table S1 lists values. ***p < 0.001 compared with bare actin (ANOVA). Conditions: pH 7.4, 150 mM KCl, 23°C.
To complement the single-molecule experiments with an approach in which actin filaments were not adhered to the coverslip surface, we used an ensemble motility assay in which myoVa-HMM was attached to the coverslip, allowing actin filaments to be moved in solution (Figure 2D). Mean speeds of actin–Tpm3.1 and actin–Tpm1.8 were comparable to those of bare actin (Figure 2E and Supplemental Table S2). In contrast, actin-Tpm4.2 was such an unfavorable track that only transient interactions between myoVa-HMM and actin-Tpm4.2, with no directed movement, were observed (Supplemental Figure S1C and Supplemental Movie S1). We also quantified the length of time that actin filaments were engaged with myoVa-HMM before dissociating as a measure of affinity. Methylcellulose, a standard additive to prevent filament disengagement, was omitted. Actin filaments were sheared into short fragments to ensure comparable filament lengths for bare actin and actin–Tpm. Median times to filament dissociation increased for actin–Tpm1.8 compared with bare actin (analysis of variance [ANOVA] with Dunnett’s multiple comparisons test, p < 0.05; Figure 2F and Supplemental Table S2).
Both the single-molecule and ensemble motility approaches suggest that Tpm isoform regulates motor engagement. These data imply that the motor domain senses the Tpm isoform, consistent with high-resolution electron cryomicroscopy studies, which showed that the myosin motor domain directly interacts with Tpm (Behrmann et al., 2012). Specificity likely arises from the different sequences of the Tpm isoforms.
To assess the effect of Mlph on track selectivity, we compared run frequencies with full-length myoVa-Mlph on different actin tracks in the single-molecule assay (Figure 2G). Previously we showed that Mlph acts as a molecular tether, enhancing the run frequency of myoVa threefold on bare actin compared with constitutively active myoVa-HMM (Sckolnick et al., 2013). Here we show that Mlph tethering is Tpm-isoform dependent. A single-molecule binding assay demonstrates that Mlph by itself has twofold greater affinity for bare actin and actin-Tpm3.1 than with actin–Tpm1.8 and actin–Tpm4.2 (Supplemental Figure S1D). Consistent with this observation, the increase in run frequency due to tethering is greatest with bare actin and actin–Tpm3.1, whereas actin–Tpm1.8 and actin–Tpm4.2 are less favorable tracks for myoVa-Mlph (Figure 2H–I). These data strongly imply that Tpm isoforms modulate interactions of both the myoVa motor domain and cargo adapter proteins with actin.
MyoVa plus actin–Tpm3.1 is a favorable motor-track combination
In single-molecule assays, speeds and run lengths were additional parameters that we quantified to investigate the mechanism by which Tpm isoforms affect transport. The presence of Tpm3.1 reduced the speed of myoVa-HMM by 20%, indicating that the motor has a slower stepping rate on this track (ANOVA, Dunnett’s multiple comparisons test: p < 0.01; Figure 3A and Supplemental Table S1). The run length of myoVa-HMM on actin–Tpm3.1 was not affected (Figure 3B and Supplemental Table S1).
FIGURE 3:

Speeds and run lengths of myoVa-HMM and full-length myoVa-Mlph in a single-molecule assay. (A) Histogram and Gaussian fit of speeds for myoVa-HMM in the presence of no Tpm (black), Tpm3.1 (red; p < 0.01 compared with bare actin; ANOVA), Tpm1.8 (green), or Tpm4.2 (blue; p < 0.001 compared with bare actin; ANOVA). (B) Characteristic run lengths were determined from exponential fits to histograms. Error bars are 95% confidence intervals. ***p < 0.001 compared with bare actin (Kruskal–Wallis test). (C) Histogram and sum-of-two-Gaussians-fit of speeds for myoVa-Mlph in the presence of no Tpm (black), Tpm3.1 (red), Tpm1.8 (green), or Tpm4.2 (blue). Inset, relative percentage of slow vs. fast speeds; colors match those in the histogram. (D) Characteristic run lengths determined as in B. The first bar in each group (solid color) shows results from pooled data. The second bar (thin diagonal stripe) shows data from motors moving <500 nm/s, and the third bar (wide diagonal stripe) shows data from motors moving at >500 nm/s. Error bars are 95% confidence intervals. *p < 0.05 compared with bare actin (Kruskal–Wallis test). Run lengths of just the slow or the fast runs on actin-Tpm3.1 are not statistically different from those on bare actin. Values are listed in Supplemental Table S1. Conditions: pH 7.4, 150 mM KCl, 23°C.
The speed distribution for the full-length myoVa-Mlph complex is bimodal (Figure 3C). With the fluorescent quantum dot attached to Mlph, we ensure that the data reflect only motors with bound Mlph. The relative proportion of slow- and fast-moving motors was estimated from a combination of peak height and SD (see Materials and Methods). On bare actin, half of the myoVa-Mlph complexes moved at a speed of the same magnitude as that of myoVa-HMM, and the other half was slower. Our previous work showed that the slow speed (<500 nm/s) results from Mlph binding to the track and functioning as a tether to slow the movement of active full-length myoVa (Sckolnick et al., 2013). The faster speed (>500 nm/s) is likely due to Mlph that is bound to myoVa but not interacting with the actin track. Tpm3.1 increased the relative ratio of fast to slow runs compared with bare actin (Figure 3C, inset), implying that Mlph acts as a tether on actin–Tpm3.1 less frequently than on bare actin. A single-molecule binding assay showed that Mlph alone bound to bare actin with the same apparent affinity as to actin–Tpm3.1 (Supplemental Figure S1D), and thus it is possible that myoVa can alter the position of Tpm3.1 on actin so as to hinder Mlph binding. The ability of myosin to affect Tpm position on actin is well documented (Lehman et al., 2013).
The two distinct speed populations and the fact that we never observe a motor switching speeds within a run suggest that once Mlph is engaged as a tether, this behavior persists throughout the run. This may be related to how Mlph interacts with the track. We previously showed that Mlph bound to myoVa moves discontinuously in a steplike behavior with a distance of ∼100 nm between dwell points, the length of approximately three 36-nm steps of myoVa (Sckolnick et al., 2013).
Consistent with the interpretation that Mlph acts as a tether, the slow runs (<500 nm/s) had longer run lengths than the fast runs (>500 nm/s) for both bare actin and actin–Tpm3.1 (Figure 3D and Supplemental Table S1). Overall run lengths of myoVa-Mlph on actin-Tpm3.1 are shorter than on bare actin (Kruskal–Wallis test with Dunn’s multiple comparisons: p < 0.05), consistent with most runs on actin–Tpm3.1 being fast and untethered.
Actin–Tpm4.2 is an unfavorable track for myoVa transport
The presence of Tpm4.2 virtually excludes myoVa-HMM and myoVa-Mlph from interacting with the track (Figure 2, Supplemental Figure S1, A–C, and Supplemental Movie S1). Nonetheless, numerous fields can be scanned to collect sufficient data to determine speeds and run lengths. As with Tpm3.1, speed was ∼20% less than with bare actin (ANOVA, Dunnett’s multiple comparisons test: p < 0.001; Figure 3A and Supplemental Table S1). The run length of myoVa-HMM was also reduced on actin–Tpm4.2 compared with bare actin (Kruskal–Wallis test with Dunn’s multiple comparisons: p < 0.001; Figure 3B and Supplemental Table S1).
Full-length myoVa-Mlph on actin–Tpm4.2, shows the characteristic bimodal speed distribution, with both the fast and slow speeds similar to that for bare actin. Run length overall, as well as separated for slow and fast populations, was also comparable to that for bare actin. The proportion of slow motors was less than on bare actin, indicating poor tethering efficiency by Mlph. Consistent with these observations, Mlph had a lower apparent binding affinity for actin–Tpm4.2 than for bare actin or actin-Tpm3.1 (Supplemental Figure S1D). By all criteria, actin–Tpm4.2 is a poor track for myoVa transport.
Actin–Tpm1.8 is a good track for myoVa-HMM but a poor track for myoVa-Mlph
Actin–Tpm1.8 is a favorable track for myoVa-HMM, as evidenced by the approximately twofold increase in run frequency compared with bare actin in the single-molecule assay (Figure 2C). As with the other Tpm isoforms, speeds are ∼20% reduced (ANOVA, Dunnett’s multiple comparisons test: p < 0.001). Run lengths are not statistically significantly different from those for bare actin (Figure 3, A and B, and Supplemental Table S1).
Full-length myoVa-Mlph does not benefit from the tethering of the cargo adapter on actin–Tpm1.8. The run frequency is much lower than on bare actin (Figure 2I). Whereas the speed distribution is bimodal and two-thirds of the motors make up the slow peak, the magnitude of slowing is less on this track than on any of the other tracks. (Figure 3C and Supplemental Table S1). Not surprisingly, Mlph by itself has a lower affinity for actin-Tpm1.8 than for bare actin (Supplemental Figure S1D). Run lengths are comparable to those on bare actin.
Tpm localization in melanocytes reflects transport capabilities
Tpm3.1 distribution in melanocytes was imaged with an antibody specific for exon 9d of the TPM3 gene, which detects both Tpm3.1 and Tpm3.2. RT-PCR shows that melanocytes express less Tpm3.2 (Figure 1D), and thus this antibody predominantly reports Tpm3.1 localization. Tpm3.1 is present throughout all dendritic protrusions, as well as on cortical actin and long filamentous actin structures throughout the cell (Figure 4, A–C). Tpm3.1 allows myoVa-Mlph to move efficiently in vitro and is in the correct cellular location to decorate actin tracks for melanosome transport.
FIGURE 4:

Immunofluorescence images showing localization of Tpm isoforms in melan-a melanocytes. (A, D, G) Stained with antibodies against Tpm3.1, 4.2, or 1.8 (red); (B, E, H) phalloidin to visualize total actin (green); (C, F, I) merged images. The antibody against Tpm3.1 also reacts with Tpm3.2, which is present in melanocytes. The antibody against Tpm1.8 also reacts with other Tpm isoforms generated from the TPM1 gene, including Tpm1.6, 1.7, and 1.9, which are present in melanocytes. Scale bar, 20 μm.
Tpm4.2 distribution was visualized with an antibody specific for exon 9d of the TPM4 gene. It was not present in the dendritic protrusions and was mostly confined to the cell body and the area surrounding the nucleus (Figure 4, D–F). The in vitro movement of myoVa-Mlph on actin–Tpm4.2 is very poor, and this Tpm isoform does not extend into the dendritic protrusions, where actin-based melanosome transport is essential.
No antibody specifically reacts with Tpm1.8; the commercially available antibody against exon 9d of TPM1 also detects other isoforms derived from TPM1. Sequencing of RT-PCR products showed that in addition to Tpm1.8, Tpm1.6, 1.7, and 1.9 are expressed in melanocytes, whereas Tpm1.3 and 1.4 are not. Therefore it was not surprising that immunofluorescence showed staining throughout the cell (Figure 4, G–I). This observation suggests that, in addition to Tpm3.1, at least one more Tpm isoform is present in the dendritic processes.
Conclusion
This study provides a glimpse at how a cell might spatially regulate a myriad of different myosin functions that all happen simultaneously and require precise localization for correct cell and tissue function (Figure 5). Actomyosin biochemistry can be directly influenced by Tpm isoforms (Collins et al., 1990; Hodges et al., 2012; Barua et al., 2014; McIntosh et al., 2015). In addition, cargo adapter proteins can affect actomyosin function. The affinity of Mlph for actin, which provides an additional attachment point for myoVa and increases the affinity of myoVa-Mlph complexes significantly over myoVa alone, is Tpm-isoform dependent. Therefore we propose that track selection results from the myosin motor domain and the cargo adapter Mlph interacting independently and synergistically with actin–Tpm. Tpm isoforms designate cellular actin structures to function in specific processes (e.g., attachment, cell shape, or cargo transport), and myosin isoforms will preferentially interact with specific actin–Tpm structures (e.g., stress fibers, dynamic branched actin structures, or filamentous actin track), translating the Tpm landscape into a functional map of myosin activity. Analogous to the “tubulin code” for sorting microtubule motors (for reviews, see Verhey and Hammond, 2009; Kaul et al., 2014; Sirajuddin et al., 2014), myosin motors may respond to an “actin–Tpm code.” Understanding the role of Tpm isoforms in cytoskeletal actin topology will be essential in the study of all myosin-based cellular processes.
FIGURE 5:

Simplified schematic of a cell, illustrating the idea that different actomyosin processes in the cell may be spatially regulated by the “actin–Tpm code.” Actin structures with different functions and distinct Tpm isoforms bound will recruit only specific myosin motors. For example, the myosin-I isoform myo1b preferentially interacts with branched dynamic actin that is not decorated with Tpm, and there is evidence that other myosin-I isoforms show a similar preference (Ostap, 2008). Some nonmuscle (NM) myosin II isoforms interact with actin structures organized into stress fibers in a Tpm isoform–dependent manner (Ostap, 2008). We propose here that cortical actin and other filamentous actin structures decorated with different Tpm isoforms can act as selective transport tracks for motors such as myosin V and myosin VI.
MATERIALS AND METHODS
Expressed protein
Full-length mouse myoVa (melanocyte isoform) with a C-terminal FLAG tag (DYKDDDDK) and truncated myoVa-HMM with C-terminal biotin tag and C-terminal FLAG-tag were expressed and purified as previously described (Armstrong et al., 2012). Mouse melanophilin (accession number NP_443748) was cloned into baculovirus expression vector pAcSG2 with N-terminal SNAP tag and C-terminal Flag tag. Recombinant virus was produced from Sf9 cells using linearized Baculo Gold DNA (BD Biosciences, San Diego, CA) and GeneJuice (EMD Millipore Novagen, Billerica MA). Cells were infected with recombinant virus and incubated with shaking for 72 h at 27°C. Cells were pelleted and resuspended in 10 mM imidazole, pH 7.0, 0.3 M NaCl, 1 mM ethylene glycol tetraacetic acid (EGTA), 7% sucrose, and protease inhibitors (0.5 mM 4-(2-aminoethyl)benzenesulfonyl fluoride, 5 μg/ml leupeptin, and 0.78 mg/ml benzamidine). The cells were lysed by sonication, and the lysate was clarified for 40 min at 250,000 × g. The supernatant was incubated with Anti-Flag M2 affinity resin (Sigma-Aldrich, St. Louis, MO) for 1 h at 4°C with mixing. Protein was eluted with 0.1 mg/ml Flag peptide (Sigma-Aldrich) at 0.6 ml/min and then concentrated in a centrifugal filter (Amicon Ultra 15; EMD Millipore) and dialyzed against 50% glycerol, 10 mM imidazole, pH 7.0, 0.2 M NaCl, 1 mM EGTA, and 1 mM dithiothreitol (DTT). Protein was stored at −20°C.
For attachment of SNAP-Biotin (NEB, Ipswich, MA) or SNAP-Surface 488 Fluorophore (NEB), SNAP-tagged Mlph was dialyzed against 50 mM Tris-Cl, pH 7.5, 0.2 M NaCl, and 1 mM DTT and clarified (5 min, 10,000 × g). For the SNAP-tag reaction, 5 μM SNAP-tagged Mlph was incubated with 10 μM SNAP-tag substrate in 50 mM Tris-Cl, pH 7.5, 0.2 M NaCl, 0.1% Tween-20, and 1 mM DTT for 30 min at 37°C. Removal of excess SNAP substrate was done by dialysis against 10 mM imidazole, pH 7.4, 0.2 M NaCl, and 1 mM DTT. For storage, the protein was further dialyzed against 10 mM imidazole, pH 7.4, 0.2 M NaCl, 1 mM DTT, and 50% glycerol.
Human TPM4.2 and TPM3.1 cDNAs (gifts from Justine Stehn and Peter Gunning, University of New South Wales, Sydney, Australia) were modified by PCR to include an Ala-Ser dipeptide before the Met start codon to generate a mimic of N-acetylation and cloned into a pET3 vector. Rat TPM1.8 (without N-terminal Ala-Ser) in pET11d was a gift from Sarah Hitchcock (Robert Wood Johnson Medical School, Piscataway, NJ). All Tpm isoforms were expressed in BLR(DE3)-competent cells grown in lysogeny broth, Lennox formulation (Thermo Fisher Scientific, Waltham, MA). They were induced with 0.4 mM isopropyl-β-d-thiogalactoside and grown overnight at 27°C. Cell pellets were resuspended in lysis buffer (10 mM imidazole, pH 7.2, 100 mM NaCl, 2 mM EDTA, 1 mM DTT) supplemented with protease inhibitors (0.2 mM 4-(2-aminoethyl)benzenesulfonyl fluoride, 0.5 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, and 1 mg/ml benzamidine) and lysed by sonication. The lysate was clarified at 23,000 × g for 20 min. The supernatant was heated to near boiling in a water bath for 7 min with constant stirring. The heat-precipitated protein was pelleted at 23,000 × g for 20 min. The pH of the supernatant was adjusted to 0.2 below the theoretical pI of the specific Tpm using 0.2 M HCl and left at room temperature for 30 min. The precipitate was pelleted at 26,000 × g for 20 min and resuspended in 7 ml of dialysis buffer (10 mM imidazole, 50 mM NaCl, 1 mM EGTA, 1 mM DTT, 2 μg/ml leupeptin) and dialyzed against the same buffer overnight. The protein was further purified on a MonoQ ion exchange column (GE Healthcare Life Sciences, Uppsala, Sweden) with a 0.1–0.5 M NaCl salt gradient. For storage, protein was dialyzed against 10 mM imidazole, pH 7.4, 50 mM NaCl, 1 mM EGTA, 1 mM DTT, 2 μg/ml leupeptin, and 50 vol% glycerol. Chicken skeletal actin was prepared from acetone powder (Pardee and Spudich, 1982).
Single-molecule processivity assay
MyoVa or myoVa-HMM was diluted to 0.4 μM in buffer A (10 mM imidazole, pH 7.4, 300 mM KCl, 4 mM MgCl2, 1 mM EGTA, 10 mM DTT), mixed with a twofold molar excess of actin and 1 mM MgATP, and spun for 15 min at 400,000 × g to remove any myosin unable to dissociate from actin in the presence of ATP. Processivity assays were performed at room temperature in 25-μl flow chambers. Flow cells were first incubated with 0.1 mg/ml N-ethylmaleimide–modified myosin in buffer A for 4 min, blocked with 1 mg/ml bovine serum albumin (BSA) in buffer B (10 mM imidazole, pH 7.4, 25 mM KCl, 4 mM MgCl2, 1 mM EGTA, 10 mM DTT) for 2 min, rinsed with three volumes buffer B, incubated with 0.5 μM rhodamine-phalloidin–labeled actin filaments and 4 μM Tpm in buffer B for 4 min, and then equilibrated with three volumes of buffer C-150 (10 mM imidazole, pH 7.4, 150 mM KCl, 4 mM MgCl2, 1 mM EGTA, 50 mM DTT, 1 mg/ml BSA, 0.1 mg/ml CaMΔall [ Krementsov et al., 2004], 1 mM MgATP, an oxygen scavenger system [3 mg/ml glucose, 0.1 mg/ml glucose oxidase, 0.18 mg/ml catalase], and an ATP-regenerating system [0.5 mM phosphoenolpyruvate and 100 U/ml pyruvate kinase]) and 4 μM Tpm (unless stated otherwise). This Tpm concentration is sufficient to decorate actin fully (Barua et al., 2014). To ensure single-motor conditions, 655-nm streptavidin-coated quantum dots (Qdots; Invitrogen) were added to HMM in fivefold excess (Hodges et al., 2012). For experiments with Mlph, Mlph and myoVa were mixed to give a 10:1 M ratio and incubated for 10 min at room temperature (23°C). Qdots were added to the Mlph-motor complex as a 10-fold excess over the motor to ensure that most moving Qdots had single motors bound. Final dilution of motors gave a concentration of 5 nM in buffer C-150 supplemented with 1.25 μM Tpm (unless stated otherwise). The microscope setup has been described previously (Armstrong et al., 2012). Images were taken at 10-Hz frame rate using 2 × 2–pixel binning. Image stacks were analyzed using ImageJ v1.44k (National Institutes of Health, Bethesda, MD). We required motors to move continuously for five consecutive frames to be analyzed. Qdot position versus time was tracked with using the MtrackJ plug-in for ImageJ (Meijering et al., 2012). Speeds were calculated as distance traveled/time. Speeds of normally distributed myoVa-HMM were estimated by least-squares fitting to cumulative distribution functions. For statistical analysis, ANOVA with Dunnett’s multiple comparisons test was performed in GraphPad Prism (GraphPad Software). The bimodal speed distribution of myoVa-Mlph was binned such that at least three bins characterized each peak. Histograms were then fitted to the sum of two Gaussians by least squares fitting. For statistical analysis, a Kruskal–Wallis test with Dunn’s multiple comparisons test was performed in GraphPad Prism. The fractional area of a particular peak of the bimodal distributions was estimated from peak amplitude A and SD as follows:
Run length was calculated as total distance traveled between first and last tracked frame. Run lengths were not analyzed from any tracks starting outside the time and space coordinates of the movie and for tracks terminating at the end of a filament or at the edge of the field of view. The characteristic run lengths (distance after which one-third of motors had detached) were estimated by least-squares fitting to cumulative distribution functions. For statistical analysis, a Kruskal–Wallis test with Dunn’s multiple comparisons test was performed in GraphPad Prism.
Run frequency was calculated as number of processive runs/movie/actin length/myosin concentration. To normalize, the run frequency on bare actin was set to 1 for both conditions. For statistical analysis, ANOVA with Holm–Sidak multiple comparisons test was performed in GraphPad Prism. Comparison between myoVa-HMM and myoVa-Mlph was done for bare conditions only, and the average value of two experiments was used.
Unless noted otherwise, data were obtained from at least three independent sets of experiments using two different preparations of all proteins. At least 100 motors were tracked; exact numbers analyzed are listed in Supplemental Table S1.
Ensemble motility assay
The base motility buffer contained, 25 mM imidazole, pH 7.4, 4 mM MgCl2, 1 mM EGTA, 10 mM DTT, and 50 or 150 mM KCl. MyoVa-HMM was spun in the presence of F-actin as described to remove ATP-insensitive myosin heads. The protein concentration of the supernatant was determined using the Bio-Rad protein assay and then diluted to 10–20 μg/ml. Nitrocellulose-coated flow cells were first incubated with 0.2 mg/ml biotinylated BSA for 60 s, rinsed three times with 0.5 mg/ml BSA, incubated with 0.05 mg/ml NeutrAvidin for 60 s, and rinsed three times with 150 mM salt motility buffer before incubating with the diluted myoVa-HMM for 60 s. After three rinses with 50 mM salt motility buffer, 12 nM tetramethylrhodamine isothiocyanate-phalloidin–labeled F-actin was added to the flow cell twice for 30 s each and rinsed with 50 mM motility buffer. The assay was performed in 150 mM salt motility buffer with 2 mM MgATP, an oxygen-scavenging system (3 mg/ml glucose, 0.13 mg/ml glucose oxidase, 0.05 mg/ml catalase), and an ATP-regenerating system (0.5 mM phosphoenolpyruvate and 100 U/ml pyruvate kinase). Tpm, 4 μM, was added to all steps, including and after the incubation with actin filaments. Motility was performed at 30°C. Actin movement was observed using an inverted microscope (Zeiss Axiovert 10) equipped with epifluorescence, a Rolera MGi Plus Digital camera, and dedicated computer with the Nikon NIS Elements software package. Image stacks were analyzed using ImageJ v1.44k. Filaments were tracked by hand using the MtrackJ plug-in (Meijering et al., 2012) or the MultiTracker plug-in for ImageJ. The coordinates generated from the MultiTracker ImageJ plug-in were analyzed in Excel to calculate the speed and frequency of motile and nonmotile actin filaments. Motion >10 pixels/frame or <1 pixel/frame was considered noise, based on the resolution of our system. To determine whether filaments were undergoing directed motion, we used mean-squared displacement (MSD) analysis (Nelson et al., 2009):
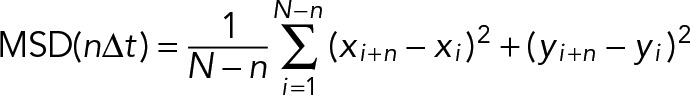 (1) (1)
|
where N is the total number of frames (up to 60), n is the number of frames for a given time interval, Δt is the time interval, and (xi, yi) are the coordinates at time i. The slope, or α-value, of the function of log(MSD) versus log(n Δt) was used to differentiate between motile (α > 1.5) and nonmotile (α ≤ 1.5) filaments. Motile filament speeds represent the means of the nonzero instantaneous displacements, whereas nonmotile filaments were assigned the speed of 0 μm/s. The mean speed was determined as the best-fit value of the mean of a Gaussian distribution fit to the frequency distribution of resultant speeds >0 μm/s in GraphPad Prism. To determine filament engagement times with the motor-coated surface, actin was vortexed for 1 min to ensure comparable lengths of bare and Tpm-decorated actin. Methylcellulose was omitted from all assays so as to allow for filament dissociation. The duration of filament movement was timed from initial until final detachment. Cumulative frequency distributions (n > 30) were fitted with a single exponential, and the characteristic time t1/2 after which one-half of all filaments had typically detached was used as a measure for filament affinity to myoVa-HMM. Results represent data from at least three independent experiments using two different myoVa-HMM preparations. For each experiment, filaments moving in at least four different 64-μm2 areas were analyzed. For statistical analysis, ANOVA with Dunnett’s multiple comparisons test was performed in GraphPad Prism.
Mlph binding to actin
Glycerol stock of Mlph-488 was diluted 1:4 in buffer A and clarified for 10 min at 10,000 × g. Flow cells were prepared in the same way as for the processivity assay. Serial dilutions (6 nM to 0.5 μM) of Mlph-488 in buffer D (10 mM imidazole, pH 7.4, 150 mM KCl, 4 mM MgCl2, 1 mM EGTA, 50 mM DTT, 1 mg/ml BSA, and an oxygen-scavenger system [3 mg/ml glucose, 0.1 mg/ml glucose oxidase, 0.18 mg/ml catalase]) supplemented with 5 μM Tpm were prepared. Images were recorded using a Nikon eclipse Ti-U inverted microscope (Nikon Instruments, Melville, NY) equipped with a PlanApo objective lens (100×, 1.49 numerical aperture [NA]) by through-the-objective total internal reflectance microscopy. A 532-nm DPSS, 50-mW laser (Lasever, Ningbo, China) was used for excitation. Images were obtained using an intensified high-resolution 10-bit digital camera (XR/Turbo-Z running Piper Control software; Stanford Photonics, Palo Alto, CA). Image analysis was performed using ImageJ 1.43u. Starting with the lowest concentration, Mlph was introduced into the flow cells and imaged for at least 25 frames at 2 Hz with 250-ms exposure. Higher concentrations were introduced thereafter without washing in between. Binding of Mlph-488 was determined by counting the number of fluorescent spots detected on an actin filament. Values from 15–25 images were averaged and normalized to actin length within the field of view. Apparent binding affinities were calculated from a fit of number of Mlph bound per actin length as a function of Mlph concentration to the one-site specific binding equation Mlph bound = Mlph bound at saturation × [Mlph]/(Kd,app + [Mlph]) using GraphPad Prism.
Cell culture
The melan-a mouse melanocyte cell line (Bennett et al., 1987) was a gift from Dorothy Bennett (St. George’s, University of London, London, United Kingdom). The cells were maintained in RPMI 1640, 10% fetal calf serum, and 200 nM 12-O-tetradecanoyl phorbol acetate at 37°C in 10% CO2 atmosphere.
Antibodies
Sheep polyclonal antibody α/9d (AB5441; EMD Millipore) was diluted 1:400. Sheep polyclonal antibody γ/9d (AB5447; EMD Millipore) was diluted 1:100, and rabbit polyclonal antibody WD4/9d (5449; EMD Millipore) was diluted 1:400. Alexa Fluor 546 donkey anti-sheep antibody (A21098; Thermo Fisher Scientific) was diluted 1:1000 in phosphate-buffered saline (PBS). Alexa Fluor 546 goat anti-rabbit antibody (A-11035; Thermo Fisher Scientific) was diluted 1:1000 in PBS.
Immunocytochemistry
Cells plated on glass coverslips were fixed for 15 min at room temperature in 4% formaldehyde in PBS, washed three times with PBS, permeabilized in 1% Triton X-100 for 10 min, washed three times with PBS, and blocked for 15 min in 2% BSA in PBS. Coverslips were incubated first with primary antibodies and after three washes with PBS with the secondary antibodies diluted in PBS for 1 h each. Coverslips were then washed three times with PBS and incubated with 250 nM Alexa Fluor 488–phalloidin in PBS for 1 h. Samples were mounted in ProLong Gold Antifade mountant (Molecular Probes, Eugene, OR) and stored in the dark. Samples were examined by confocal microscopy using a Zeiss LSM 510 META confocal scanning laser microscope (Zeiss MicroImaging, Thornwood, NY) and a 25× PlanNeofluar (1.3 NA) oil immersion objective lens.
RT-PCR
RNA was isolated from melan-a melanocytes using an RNeasy Mini Kit (Qiagen, Valencia, CA). First-strand cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Invitrogen, Carlsbad, CA). TPM-specific oligonucleotides were chosen from the mouse cDNA as follows: TPM1 exon 1b, TGCGGAGAAAGATCCGGAGCCT; TPM1 exon 9d, GGTTTCACGGTAAGCATGTG; TPM3 exon 6a, CGTTGCCGAGAGATGGATGAG; TPM3 exon 6b, GTGTTCTGAGCTGGAGGA; TPM3 exon 9c, CTTGCTTAGGGCGAACAGTGAC; TPM3 exon 9d, GCCCTCAGTTTCAAGGTCAG; TPM4 exon 1b, GCAGGCAGACGACGCA; and TPM4 exon 9d, GGTGTTAAAACAGTACTCTTCTGG. Reactions were performed for 30 cycles using PCR Master Mix (Promega). RT-PCR products were resolved in agarose gels and visualized by staining with SYBR Safe DNA gel stain (Invitrogen). Bands were cut out, gel purified, and sequenced using SimpleSeq (Eurofins Genomics, Louisville, KY).
Supplementary Material
Acknowledgments
We thank A. Heaslip for advice with cell culturing, N. Bishop and N. Bouffard (Microscopy Imaging Center, University of Vermont, Burlington, VT) for help with confocal microscopy, G. Kennedy (Instrumentation and Model Facility) for TIRF microscopy design and implementation, P. Gunning for helpful discussions, and M. Lord for input during the initial stages of the project. Confocal microscopy was performed on a Zeiss 510 META laser scanning confocal microscope supported by National Institutes of Health Award Number 1510RR019246 from the National Center for Research Resources. This work was supported by National Institutes of Health Grants GM078097 to K.M.T. and GM094229 to D.M.W.
Abbreviations used:
- DTT
dithiothreitol
- Mlph
melanophilin
- myoVa
full-length myosin Va
- myoVa-HMM
truncated, constitutively active myoVa-heavy meromyosin
- RT-PCR
reverse transcriptase PCR
- Tpm
tropomyosin.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-09-0641) on August 17, 2016.
REFERENCES
- Armstrong JM, Krementsova E, Michalek AJ, Heaslip AT, Nelson SR, Trybus KM, Warshaw DM. Full-length myosin Va exhibits altered gating during processive movement on actin. Proc Natl Acad Sci USA. 2012;109:E218–E224. doi: 10.1073/pnas.1109709109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barua B, Nagy A, Sellers JR, Hitchcock-DeGregori SE. Regulation of nonmuscle myosin II by tropomyosin. Biochemistry. 2014;53:4015–4024. doi: 10.1021/bi500162z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrmann E, Muller M, Penczek PA, Mannherz HG, Manstein DJ, Raunser S. Structure of the rigor actin-tropomyosin-myosin complex. Cell. 2012;150:327–338. doi: 10.1016/j.cell.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DC, Cooper PJ, Hart IR. A line of non-tumorigenic mouse melanocytes, syngeneic with the B16 melanoma and requiring a tumour promoter for growth. Int J Cancer. 1987;39:414–418. doi: 10.1002/ijc.2910390324. [DOI] [PubMed] [Google Scholar]
- Clayton JE, Pollard LW, Sckolnick M, Bookwalter CS, Hodges AR, Trybus KM, Lord M. Fission yeast tropomyosin specifies directed transport of myosin-V along actin cables. Mol Biol Cell. 2014;25:66–75. doi: 10.1091/mbc.E13-04-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins K, Sellers JR, Matsudaira P. Calmodulin dissociation regulates brush border myosin I (110-kD-calmodulin) mechanochemical activity in vitro. J Cell Biol. 1990;110:1137–1147. doi: 10.1083/jcb.110.4.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooley BC, Bergtrom G. Multiple combinations of alternatively spliced exons in rat tropomyosin-alpha gene mRNA: evidence for 20 new isoforms in adult tissues and cultured cells. Arch Biochem Biophys. 2001;390:71–77. doi: 10.1006/abbi.2001.2347. [DOI] [PubMed] [Google Scholar]
- Cooper JA. Actin dynamics: tropomyosin provides stability. Curr Biol. 2002;12:R523–R525. doi: 10.1016/s0960-9822(02)01028-x. [DOI] [PubMed] [Google Scholar]
- Dufour C, Weinberger RP, Gunning PW. Tropomyosin isoform diversity and neuronal morphogenesis. Immunol Cell Biol. 1998;76:424–429. doi: 10.1046/j.1440-1711.1998.00765.x. [DOI] [PubMed] [Google Scholar]
- Geeves MA, Hitchcock-DeGregori SE, Gunning PW. A systematic nomenclature for mammalian tropomyosin isoforms. J Muscle Res Cell Motil. 2015;36:147–153. doi: 10.1007/s10974-014-9389-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunning P, O’Neill G, Hardeman E. Tropomyosin-based regulation of the actin cytoskeleton in time and space. Physiol Rev. 2008;88:1–35. doi: 10.1152/physrev.00001.2007. [DOI] [PubMed] [Google Scholar]
- Gunning PW, Schevzov G, Kee AJ, Hardeman EC. Tropomyosin isoforms: divining rods for actin cytoskeleton function. Trends Cell Biol. 2005;15:333–341. doi: 10.1016/j.tcb.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Hartman MA, Spudich JA. The myosin superfamily at a glance. J Cell Sci. 2012;125:1627–1632. doi: 10.1242/jcs.094300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges AR, Krementsova EB, Bookwalter CS, Fagnant PM, Sladewski TE, Trybus KM. Tropomyosin is essential for processive movement of a class V myosin from budding yeast. Curr Biol. 2012;22:1410–1416. doi: 10.1016/j.cub.2012.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul N, Soppina V, Verhey KJ. Effects of alpha-tubulin K40 acetylation and detyrosination on kinesin-1 motility in a purified system. Biophys J. 2014;106:2636–2643. doi: 10.1016/j.bpj.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krementsov DN, Krementsova EB, Trybus KM. Myosin V: regulation by calcium, calmodulin, and the tail domain. J Cell Biol. 2004;164:877–886. doi: 10.1083/jcb.200310065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda TS, Ariga H, Fukuda M. The actin-binding domain of Slac2-a/melanophilin is required for melanosome distribution in melanocytes. Mol Cell Biol. 2003;23:5245–5255. doi: 10.1128/MCB.23.15.5245-5255.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman W, Orzechowski M, Li XE, Fischer S, Raunser S. Gestalt-binding of tropomyosin on actin during thin filament activation. J Muscle Res Cell Motil. 2013;34:155–163. doi: 10.1007/s10974-013-9342-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh BB, Holzbaur EL, Ostap EM. Control of the initiation and termination of kinesin-1-driven transport by myosin-Ic and nonmuscle tropomyosin. Curr Biol. 2015;25:523–529. doi: 10.1016/j.cub.2014.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijering E, Dzyubachyk O, Smal I. Methods for cell and particle tracking. Methods Enzymol. 2012;504:183–200. doi: 10.1016/B978-0-12-391857-4.00009-4. [DOI] [PubMed] [Google Scholar]
- Nelson SR, Ali MY, Trybus KM, Warshaw DM. Random walk of processive, quantum dot-labeled myosin Va molecules within the actin cortex of COS-7 cells. Biophys J. 2009;97:509–518. doi: 10.1016/j.bpj.2009.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostap EM. Tropomyosins as discriminators of myosin function. Adv Exp Med Biol. 2008;644:273–282. doi: 10.1007/978-0-387-85766-4_20. [DOI] [PubMed] [Google Scholar]
- Pardee JD, Spudich JA. Purification of muscle actin. Methods Enzymol. 1982;85B:164–181. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- Sckolnick M, Krementsova EB, Warshaw DM, Trybus KM. More than just a cargo adapter, melanophilin prolongs and slows processive runs of myosin Va. J Biol Chem. 2013;288:29313–29322. doi: 10.1074/jbc.M113.476929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirajuddin M, Rice LM, Vale RD. Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat Cell Biol. 2014;16:335–344. doi: 10.1038/ncb2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang N, Ostap EM. Motor domain-dependent localization of myo1b (myr-1) Curr Biol. 2001;11:1131–1135. doi: 10.1016/s0960-9822(01)00320-7. [DOI] [PubMed] [Google Scholar]
- Tojkander S, Gateva G, Schevzov G, Hotulainen P, Naumanen P, Martin C, Gunning PW, Lappalainen P. A molecular pathway for myosin II recruitment to stress fibers. Curr Biol. 2011;21:539–550. doi: 10.1016/j.cub.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Trybus KM. Myosin V from head to tail. Cell Mol Life Sci. 2008;65:1378–1389. doi: 10.1007/s00018-008-7507-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey KJ, Hammond JW. Traffic control: regulation of kinesin motors. Nat Rev Mol Cell Biol. 2009;10:765–777. doi: 10.1038/nrm2782. [DOI] [PubMed] [Google Scholar]
- Vindin H, Gunning P. Cytoskeletal tropomyosins: choreographers of actin filament functional diversity. J Muscle Res Cell Motil. 2013;34:261–274. doi: 10.1007/s10974-013-9355-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XFS, Rao K, Zhang H, Wang F, Sellers JR, Matesic LE, Copeland NG, Jenkins NA, Hammer JA. Identification of an organelle receptor for myosin-Va. Nat Cell Biol. 2002b;4:271–278. doi: 10.1038/ncb760. [DOI] [PubMed] [Google Scholar]
- Wu XF, Wang F, Rao K, Sellers JR, Hammer JA. Rab27a is an essential component of melanosome receptor for myosin Va. Mol Biol Cell. 2002a;13:1735–1749. doi: 10.1091/mbc.01-12-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.