

In a Drosophila model of ALS, neuronal defects are associated with altered endosomal traffic of growth factor receptors and loss of growth-promoting signals. Manipulation of an endosomal recycling pathway suppresses these neuronal defects. The findings suggest that rerouting membrane traffic could be therapeutic in ALS.
Abstract
TAR DNA-binding protein 43 (TDP-43) is genetically and functionally linked to amyotrophic lateral sclerosis (ALS) and regulates transcription, splicing, and transport of thousands of RNA targets that function in diverse cellular pathways. In ALS, pathologically altered TDP-43 is believed to lead to disease by toxic gain-of-function effects on RNA metabolism, as well as by sequestering endogenous TDP-43 and causing its loss of function. However, it is unclear which of the numerous cellular processes disrupted downstream of TDP-43 dysfunction lead to neurodegeneration. Here we found that both loss and gain of function of TDP-43 in Drosophila cause a reduction of synaptic growth–promoting bone morphogenic protein (BMP) signaling at the neuromuscular junction (NMJ). Further, we observed a shift of BMP receptors from early to recycling endosomes and increased mobility of BMP receptor–containing compartments at the NMJ. Inhibition of the recycling endosome GTPase Rab11 partially rescued TDP-43–induced defects in BMP receptor dynamics and distribution and suppressed BMP signaling, synaptic growth, and larval crawling defects. Our results indicate that defects in receptor traffic lead to neuronal dysfunction downstream of TDP-43 misregulation and that rerouting receptor traffic may be a viable strategy for rescuing neurological impairment.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease arising from the death of upper and lower motor neurons and can be sporadic or inherited (Robberecht and Philips, 2013). One dominantly inheritable form of ALS is coupled with frontotemporal dementia (FTD) and is caused by aggregation-prone mutations in the RNA-associated protein TAR DNA-binding protein 43 (TDP-43; Kabashi et al., 2008; Rutherford et al., 2008; Sreedharan et al., 2008). Aggregates of wild-type TDP-43 are also found in sporadic ALS, suggesting a common underlying biology (Neumann et al., 2006). Mutant or pathologically altered wild-type TDP-43 may cause neurodegeneration both by sequestering endogenous TDP-43 away from its normal site of action in the nucleus and causing gain-of-function misregulation of cellular RNAs (Robberecht and Philips, 2013).
TDP-43 regulates a vast number of downstream RNA targets in both healthy and diseased neurons, making it difficult to identify the critical perturbation(s) that lead to disease (Polymenidou et al., 2011; Sephton et al., 2011; Tollervey et al., 2011). An alternative strategy is to take a “bottom-up” approach by defining disease-causing cellular defects downstream of TDP-43 and devising strategies to redirect these processes to a healthy state. Two primary lines of evidence suggest that altered endocytic traffic of neuronal growth factor receptors, leading to changes in their signaling capacity, could be a critical defect in ALS. First, growth factor signaling (in neurotrophin, insulin-like growth factor 1 [IGF-1], and vascular endothelial growth factor [VEGF] pathways) is required for motor neuron survival and is misregulated in ALS, leading to a switch from prosurvival to proapoptotic signaling (Gould and Oppenheim, 2010; Tovar et al., 2014). Second, mutations in components of the membrane traffic machinery are linked to multiple human neurodegenerative diseases, including ALS (Schreij et al., 2016), suggesting that intracellular transport is an important point of cellular dysfunction and a potential target for therapeutic intervention.
The Drosophila larval neuromuscular junction (NMJ) provides a powerful biological system in which to probe how neurons use membrane trafficking to manipulate trophic signals (Deshpande and Rodal, 2015). During larval development, a bone morphogenetic protein (BMP) pathway controls synaptic function and the morphological expansion of NMJ arbors (Marques and Zhang, 2006). In this pathway, the muscle-derived BMP ligand glass bottom boat (Gbb) binds to its coreceptors Wishful Thinking (Wit), Thickveins (Tkv), and Saxophone (Sax) on the motor neuron, and this complex is internalized into signaling endosomes. These activated BMP receptors control local cytoskeletal remodeling via LIM kinase (Eaton and Davis, 2005; Piccioli and Littleton, 2014) and phosphorylate the R-SMAD Mothers Against Decapentaplegic (Mad), which enters the motor neuron nucleus to activate the transcription of genes that promote synaptic growth, transmission, and homeostasis (Ball et al., 2010; Kim and Marques, 2012; Deshpande and Rodal, 2015). Neuronal Wit, Tkv, and Sax also regulate a separate, Gbb-independent pool of phosphorylated Mad (pMad), which remains localized to synapses and stabilizes postsynaptic type A glutamate receptors (Sulkowski et al., 2016). Previous work showed that BMP signaling from early endosomes can be attenuated by either recycling BMP receptors to the cell surface or targeting them for endolysosomal degradation (Deshpande and Rodal, 2015). Indeed, modest shifts in endosomal distribution of BMP receptors at the NMJ can dramatically alter both synaptic and nuclear pMad accumulation and synaptic growth (Rodal et al., 2011; Liu et al., 2014; West et al., 2015), similar to endocytic regulation of mammalian neuronal trophic factor signaling (Cosker and Segal, 2014).
Drosophila is also particularly well suited for studying TDP-43–induced neurodegeneration (for review, see Casci and Pandey, 2015). Overexpression or loss of TDP-43 leads to motor neuron–autonomous defects in larval NMJ growth, motor function, and adult lifespan (Feiguin et al., 2009; Li et al., 2010; Estes et al., 2011; Godena et al., 2011; Lin et al., 2011; Wang et al., 2011; Diaper et al., 2013; Miskiewicz et al., 2014). Here we study the relationship between synaptic growth signaling and membrane traffic in both gain-of-function and loss-of-function Drosophila TDP-43 models.
RESULTS
Misexpression of TDP-43 in Drosophila motor neurons impairs synaptic growth
To examine the relationship between membrane traffic and synaptic growth signaling upon TDP-43 misregulation, we first selected gain-of-function and loss-of-function Drosophila models of TDP-43. We used the binary GAL4/UAS system to compare expression levels and lifespan phenotypes for previously generated human TDP-43 (hTDP-43) overexpression transgenes (Figure 1, A and B; Lu et al., 2009; Hanson et al., 2010). Strongly overexpressing lines and either ubiquitous or motor neuron–specific loss of the Drosophila TDP-43 homologue tbph both caused lethality within a few days of eclosion (Feiguin et al., 2009). We selected one of these strongly overexpressing lines as a gain-of-function model to compare with tbph loss-of-function mutants.
FIGURE 1:

Misregulation of TDP-43 leads to defects in synaptic growth. (A) Comparison of hTDP-43–overexpression models. Immunoblot from adult heads showing panneuronal (Appl-GAL4)-driven expression levels of previously reported UAS-hTDP-43 lines. Expression levels were not altered by inclusion of an additional UAS transgene (UAS-lacZ). (B) Lifespan assays for Appl-GAL4–driven UAS-hTDP-43 lines. Only strongly overexpressing lines produced significant defects in lifespan similar to the tbph null (Feiguin et al., 2009). Control is w1118. (C) NMJ morphology in TDP-43–misexpressing larvae. Representative maximum intensity projections of muscle 4 NMJs. Arrows indicate representative small boutons; scale bar, 10 μm. (D) Quantification of mean type Ib bouton number and main axis (excluding satellite) bouton number ± SEM (E) Quantification of mean fraction of boutons <2 μm in diameter/total main axis boutons ± SEM (F) Quantification of mean α-HRP–positive type 1b NMJ area ± SEM. (G) Quantification of mean length of longest NMJ branch. Statistical significance was calculated using Kruskal–Wallis tests (D) or by one-way ANOVA (E–G). n is the number of NMJs.
To test the effects of gain and loss of TDP-43 on synaptic growth in Drosophila, we examined NMJ morphology in third-instar larvae. As previously reported for both human and Drosophila TDP-43 (Li et al., 2010; Lin et al., 2011), motor neuron overexpression of hTDP-43 caused a reduction in bouton number, an increase in the proportion of small (<2 μm) boutons along the main NMJ axis, a decrease in overall NMJ area, and a decrease in the length of the longest NMJ branch (Figure 1, C–G). Although we found no significant change in total bouton number or branch length on muscle 4 in tbph-null larvae (Feiguin et al., 2009), we observed a significant decrease in NMJ area and an increase in the proportion of small (<2 μm) boutons (Figure 1, C–G). Larvae heterozygous for tbph∆23 and a chromosomal deficiency deleting the tbph locus exhibited a similar increase in the proportion of small boutons to tbph∆23 homozygotes, indicating that this phenotype is not due to second-site mutations on the tbph∆23 chromosome (Figure 1, C–G). Taken together, our results indicate that both gain and loss of TDP-43 lead to synaptic growth defects and TDP-43 overexpression may cause more deleterious effects at the NMJ than with tbph loss of function, perhaps due to dose effects of overexpression (Figure 1, A and B) or toxic gain-of-function disruption of TDP-43 targets.
Reductions in bouton number and size are characteristics of reduced BMP signaling at the NMJ (Aberle et al., 2002; Marques et al., 2002; McCabe et al., 2003; Rawson et al., 2003; Choi et al., 2014). Indeed, we found that pMad was dramatically reduced at both hTDP-43–overexpressing and tbph loss-of-function NMJs (Figure 2A), consistent with reduced synaptic BMP signaling. To test whether TDP-43 misexpression affects nuclear BMP signaling, we examined pMad intensity in a subset of motor neuron cell bodies defined by the transcription factor even-skipped (eve; Landgraf et al., 1999) and found no significant difference between controls and TDP-43–misexpressing larvae (Figure 2B). We next examined a BMP transcriptional reporter consisting of a green fluorescent protein (GFP) insertion into an intron of target of wit (twit), a BMP transcriptional target (Kim and Marques, 2012; Sulkowski et al., 2016). Mean Twit::GFP levels were not changed in eve-positive motor neuron nuclei, and the distribution of Twit::GFP intensities in these nuclei was not significantly different between control and TDP-43–overexpressing larvae (Figure 2C). Finally, endogenous wit, tkv, and twit RNA levels and Wit protein levels were not significantly reduced in larval brains upon either overexpression or loss of function of TDP-43 (Figure 2D). By contrast, an early nonsense mutation in the BMP receptor wit (Marques et al., 2002) caused a significant increase in Tkv RNA and a decrease in both wit and twit RNAs, as well as loss of full-length Wit protein (Figure 2D), suggesting that TDP-43 misexpression does not simply phenocopy loss of BMP receptors. Thus TDP-43 misexpression strongly reduces synaptic BMP signaling but has no detectable effect in our assays on nuclear BMP signaling or BMP receptor transcript levels.
FIGURE 2:

Misregulation of TDP-43 leads to defects in BMP signaling. (A) pMad levels are reduced at TDP-43–misexpressing NMJs. Images show representative maximum intensity projections of muscle 4 NMJs. Scale bar, 5 μm. Graph shows mean NMJ pMad intensity ± SEM per NMJ. n is the number of NMJs. (B) pMad levels are not significantly altered in TDP-43–misexpressing motor neuron cell bodies. Images show representative maximum intensity projections of ventral nerve cords. Scale bar, 20 μm. Quantification shows mean pMad/eve ratio ± SEM. n is the number of animals. (C) Expression of a twit reporter is not significantly altered in TDP-43–misexpressing motor neuron cell bodies. Images show representative maximum intensity projections of ventral nerve cords. Scale bar, 20 μm. Quantification shows mean pMad/eve ratio ± SEM per animal. Top, quantification of mean pMad or Twit::GFP normalized to the motor neuron marker eve. Bottom, histogram of Twit::GFP/eve intensity distributions. n is the number of animals. (D) Left, misexpression of hTDP-43 does not affect mRNA levels of the BMP receptors wit or tkv or the BMP target twit in larval brains. Graph depicts mean qPCR log fold change (relative to rpl32) normalized to wild type ± SD. Statistical significance was calculated separately for each primer set. Right, misexpression of hTDP-43 does not affect Wit protein levels. Image shows representative immunoblot of larval brain lysates. Statistical significance was calculated using one-way ANOVA (A), Kruskal–Wallis tests (B, D), or two-way ANOVA (C, bottom) and Student’s t tests (C, right)
We next tested for genetic interactions between TDP-43 and the BMP pathway. We first examined whether BMP signaling could be restored in TDP-43–overexpressing NMJs by mutating the negative regulator daughters against decapentaplegic (dad), which antagonizes Mad. Loss-of-function mutations in dad cause synaptic overgrowth (Sweeney and Davis, 2002), and we found that they suppressed hTDP-43–induced undergrowth, including bouton number, NMJ area, and NMJ branch length (Figure 3, A–D). Finally, whereas removing one copy of dad had little effect on NMJ morphology in otherwise wild-type NMJs (Figure 3, B–D; O’Connor-Giles et al., 2008), it significantly suppressed the increased proportion of small boutons in TDP-43–overexpressing NMJs (Figure 3F). These results suggest that synaptic growth defects caused by TDP-43 misexpression arise at least in part from loss of BMP signaling.
FIGURE 3:

TDP-43 genetically interacts with the BMP pathway. (A) hTDP-43-induced synaptic growth defects are rescued by mutation of dad. Representative maximum intensity projections of muscle 4 NMJs. Scale bar, 20 μm. (B) Quantification of mean type Ib bouton number and main axis (excluding satellite) bouton number ± SEM. (C) Quantification of mean α-HRP–positive type 1b NMJ area ± SEM. (D) Quantification of mean length of longest NMJ branch ± SEM. (E) Heterozygosity for dad suppresses the TDP-43–induced increase in small-bouton ratio. Quantification of mean fraction of boutons <2 μm in diameter/total main axis boutons ± SEM. (B–E) Wild-type and hTDP-43 controls are identical to Figure 6, B–D. Statistical significance was calculated using Kruskal–Wallis tests (B–D) or one-way ANOVA (E). n is the number of NMJs.
Altered TDP-43 levels disrupt BMP receptor endosomal localization but not axonal transport
Neuronal growth factor signaling is heavily dependent on intracellular trafficking. Long-range axonal transport is required to conduct trophic signals from the nerve terminal to the cell body, whereas local endosomal trafficking of signaling complexes at the synapse modulates the intensity and duration of signaling (Cosker and Segal, 2014; Deshpande and Rodal, 2015). To test whether TDP-43 misexpression altered either local or axonal trafficking of BMP receptors, we generated transgenic flies expressing Tkv-mCherry under the control of the GAL4/UAS system. Neuronally driven Tkv-mCherry did not affect bouton number and localized similarly to a previously characterized rescuing Tkv-GFP transgene (Dudu et al., 2006; Figure 4A), suggesting that these expression levels and the mCherry tag do not disrupt synaptic growth.
FIGURE 4:

TDP-43 misexpression does not cause strong defects in axonal transport of Tkv-containing particles. (A) Tkv-mCherry colocalizes at the NMJ with a previously described rescuing Tkv-GFP transgene (Dudu et al., 2006). Left, representative maximum intensity projection of confocal Z-stack. Scale bar, 5 μm. Tkv-mCherry expression does not affect synaptic growth. Right, mean bouton number on muscle 6/7 ± SEM; n is the number of NMJs. (B) Representative kymograph of Vglut>Tkv-mCherry axonal transport. Scale bar, 10 μm. (C) Quantification of mean particle velocity. Data represent mean particle velocity/axon imaged per animal ± SEM. n is the number of animals. (D) Quantification of mean stall length (for stalls >3 s). Data represent mean stall duration ± SEM. n is the number of stalls. Wild type: Vglut-GAL4/w or Y; UAS-Tkv-mCherry/+ (seven animals; 16 axons; 505 retrograde particles; 505 anterograde particles; 111 stalls). tbph∆23: Vglut-GAL4/w or Y; tbph∆23 UAS-Tkv-mCherry/tbph∆23 (seven animals; 16 axons; 492 retrograde particles; 437 anterograde particles; 136 stalls). Vglut>hTDP-43: Vglut-GAL4/w or Y; UAS-Tkv-mCherry/+; UAS-TDP-43-IIId/+ (eight animals; 16 axons; 411 retrograde particles; 326 anterograde particles; 208 stalls). Statistical significance was calculated using Student’s t test (A) or Kruskal–Wallis test (C, D).
We initially examined fast axonal transport, which is a common point of dysfunction in neurodegenerative disease (Maday et al., 2014), including in Drosophila models (Torroja et al., 1999; Gunawardena and Goldstein, 2001; Gunawardena et al., 2003; Fuger et al., 2012; Lloyd et al., 2012; Kang et al., 2014; Butzlaff et al., 2015). Motor neuron–driven Tkv-mCherry exhibited rapid bidirectional axonal transport, with occasional particle stalls and reversals, and few stationary particles (Figure 4B). On hTDP-43 overexpression, we observed a small but significant increase in the rate of retrograde transport of Tkv-mCherry (Figure 4C), whereas both overexpression and tbph loss of function caused a small decrease in mean Tkv-mCherry stall duration (Figure 4D). Anterograde transport of Tkv-mCherry was not altered in either hTDP-43–overexpressing or tbph loss-of-function animals. These subtle phenotypes indicate modestly enhanced rather than grossly disrupted traffic and suggest that altered axonal transport of BMP receptors is unlikely to account for the reduction in BMP signaling and synaptic growth that we observed. This prompted us to examine local intracellular transport defects as the underlying cause.
To test whether endosomal trafficking of BMP receptors might lead to signaling defects caused by TDP-43 misexpression, we next examined the subcellular localization of Tkv at the NMJ (Figure 5, A–D). We found that the fraction of Tkv-mCherry puncta colocalizing with Rab5-positive early endosomes was markedly reduced in larvae overexpressing or underexpressing TDP-43 (Figure 5E). Further, we observed a concomitant increase in the fraction of Tkv puncta colocalizing with Rab11-positive recycling endosomes (Figure 5E). Overexpression of hTDP43 also caused a decrease in colocalization of Tkv with the early/late endosome marker SNX16-GFP (Rodal et al., 2011), whereas loss of tbph slightly increased SNX16 colocalization (Figure 5E). These results again suggest that more severe defects arise from gain than from loss of function of TDP-43. We corroborated our colocalization results by measuring Pearson’s r for Tkv-mCherry and GFP-Rab5 (which was most suitable for this analysis due its high signal-to-noise ratio) and found a similar TDP-43–induced decrease in overlap (Figure 5F). Of note, mean levels of Tkv at the NMJ were unaffected by TDP-43 misexpression, suggesting that Tkv is not simply targeted for endolysosomal degradation in these NMJs (Figure 5G). To test whether the shift of Tkv from early to recycling endosomes was similar to a previously observed TDP-43–induced general disruption in autophagosome–lysosome traffic in mammalian cells and in Drosophila fat body and heads (Xia et al., 2016), we examined the expression and localization of the autophagy marker Atg8-GFP with Tkv-mCherry. Atg8-GFP was not altered in NMJs upon overexpression of TDP-43, suggesting that a general up-regulation of the autolysosomal pathway in NMJs is unlikely to account for misregulation of BMP signaling (Figure 5, D and H). Thus overexpression or underexpression of TDP-43 induced a specific shift of Tkv from early endosomes, which are signaling permissive, to recycling compartments, which are signaling nonpermissive (Rodal et al., 2011; Liu et al., 2014; West et al., 2015), in accordance with synaptic growth and BMP signaling phenotypes (Figures 1, C–E, and 2A).
FIGURE 5:

TDP-43 misexpression shifts Tkv localization from early to recycling endosomes. (A–D) NMJs expressing Tkv-mCherry and (A) endogenously tagged GFP-Rab5 (Fabrowski et al., 2013), (B) UAS-SNX16-GFP, (C) stained for endogenous Rab11, or (D) UAS-Atg8-GFP. Representative maximum intensity projections of muscle 4 NMJs. Arrows indicate colocalizing puncta; scale bars, 5 μm. (E) Mean percentage of colocalizing puncta/NMJ ± SEM. (F) Mean Pearson’s r ± SEM for GFP-Rab5 and Tkv-mCherry within the presynaptic terminal. Rab5 levels were not altered by TDP-43 misexpression (mean pixel intensity/μm ± SEM. Rab5: control, 45.4 ± 4.3; tbph∆23, 37.7 ± 3.6; hTDP43, 52.6 ± 3.6). (G) Tkv levels are not affected by TDP-43 misexpression. Mean NMJ Tkv intensity ± SEM for α-Rab11 data set. (H) NMJ autophagy is not induced by TDP-43 misexpression. Mean NMJ ATG8-GFP and Tkv intensities ± SEM. n is the number of NMJs. Statistical significance was calculated using one-way ANOVA (E–G) or Mann–Whitney tests (H).
Inhibition of Rab11 rescues TDP-43–induced synaptic growth and motor defects
We next considered whether rerouting of BMP receptors, via manipulation of the recycling compartment, might suppress the synaptic growth signaling and motor defects caused by TDP-43 misregulation. We focused on our TDP-43 overexpression model because it is cell autonomous to the neuron and produced a more severe synaptic growth phenotype than the tbph mutant. Previous reports showed that loss or inhibition of Rab11 in otherwise wild-type larvae leads to excessive synaptic growth by altering BMP signaling (Khodosh et al., 2006; Liu et al., 2014). We found that the TDP-43–induced synaptic growth defect (including bouton number and NMJ area) was partially suppressed by a dominant-negative mutant of Rab11 (Rab11DN; Figure 6, A–D) without any changes in hTDP-43 overexpression levels (Figure 6E). Further, Rab11DN significantly rescued synaptic pMad levels in both hTDP-43–overexpressing and tbph loss-of-function animals (Figure 6F). Thus Rab11-mediated suppression of Tkv traffic and synaptic growth correlated with restored BMP signaling.
FIGURE 6:

Manipulating Rab11 restores TDP-43–induced defects in synaptic growth and BMP signaling. (A) Rab11DN partially suppresses TDP-43–induced synaptic growth defects. Representative maximum intensity projections of α-HRP–labeled muscle 4 NMJs. Scale bar, 20 μm. (B) Quantification of mean type Ib bouton number and main axis (excluding satellite) bouton number ± SEM. (C) Quantification of mean α-HRP–positive type 1b NMJ area ± SEM. (D) Quantification of mean length of longest NMJ branch ± SEM. (E) TDP-43 overexpression levels are not affected by Rab11DN. Image shows representative immunoblot from 10 larval brains/genotype. Graph shows mean TDP-43/tubulin intensity ± SEM from three independent experiments. (F) Rab11DN suppresses TDP-43–induced BMP signaling defects. Images show representative sum intensity projections of confocal stacks. Blue outlines represent α-HRP–positive area. Scale bar, 5 μm. Graph shows quantification of mean NMJ pMad intensity ± SEM. (G) Rab11DN does not rescue reduced Futsch levels in hTDP-43-expressing NMJs. Top, representative maximum intensity projections of confocal stacks. Scale bar, 10 μm. Bottom, Mean α-Futsch intensity ± SEM. For B–D, wild-type and hTDP-43 controls are identical to those in Figure 3. Statistical significance was calculated using Kruskal–Wallis (B–G) or Mann–Whitney tests (E). n is the number of NMJs.
We next asked whether other synaptic growth–promoting mutants affecting membrane traffic could suppress TDP-43–induced defects. We previously showed that a dominant-active mutant of Snx16 (Snx163A) traps activated BMP receptors (TkvQ199D) at early endosomes, promoting synaptic growth and elevating pMad levels (Rodal et al., 2011). In contrast to Rab11DN suppression of TDP-43 phenotypes, we found that larvae coexpressing Snx163A, TkvQ199D, and hTDP-43 exhibited indistinguishable synaptic undergrowth from larvae overexpressing hTDP-43 alone (Figure 6, A and B). Similarly, inhibition of Rab5 using a dominant-negative mutant failed to rescue TDP-43–induced NMJ morphology defects (Figure 6, A and B). These results indicate that rescue of synaptic growth defects is specific to manipulation of Rab11.
TDP-43 affects the expression, splicing, transport, stability, and translation of a host of RNA targets, many of which could contribute to the membrane traffic and growth factor signaling defects that we observed. One particular TDP-43 target that could be relevant is the microtubule-associate protein Futsch, which is required for synaptic growth and is regulated by both TDP-43 and BMP signaling (Roos et al., 2000; Godena et al., 2011; Coyne et al., 2014). However, Rab11DN did not restore reduced Futsch levels in TDP-43–overexpressing larvae (Figure 6G) despite rescuing synaptic growth defects, indicating that Rab11DN suppresses TDP-43–induced defects via a Futsch-independent pathway. Thus further work is required to determine which of many TDP-43 targets (or combination of targets) could lead to BMP receptor trafficking defects.
Rab11DN reroutes BMP receptor traffic
To better understand how Rab11DN rescues synaptic defects caused by TDP-43 misexpression, we asked whether traffic of Tkv had been rerouted. Surprisingly, motor neuron expression of Rab11DN did not restore the fraction of colocalizing Tkv-mCherry– and GFP-Rab5–positive early endosomes in TDP-43–overexpressing NMJs (Figure 7, A and B), even though it partially rescued BMP signaling defects (Figure 6). Instead, Rab11DN altered the spatial distribution of Tkv-mCherry puncta within the nerve terminal. In wild-type NMJs, Tkv-mCherry was enriched in the terminal bouton of the NMJ arbor. This enrichment was significantly reduced in TDP-43–overexpressing larvae and rescued in larvae coexpressing TDP-43 and Rab11DN (Figure 7C), whereas mean Tkv-mCherry levels were unchanged (Figure 7D). Thus Rab11DN rescues the TDP-43–induced defect of Tkv redistribution away from terminal boutons.
FIGURE 7:

Rab11DN reroutes BMP receptor traffic at the NMJ. (A) Representative maximum intensity projections of muscle 4 NMJs expressing Tkv-mCherry and an endogenously tagged GFP-Rab5. Scale bar, 5 μm. (B) Rab11DN does not restore Tkv-Rab5 colocalization. Mean percentage of colocalizing puncta ±SEM. (C) Rab11DN rescues Tkv enrichment in terminal boutons. Mean terminal bouton intensity/total NMJ intensity ± SEM. (D) Rab11DN does not affect Tkv levels. Mean NMJ Tkv-mCherry intensity ± SEM. (E, F) Rab11DN suppresses TDP-43–induced Tkv mobility defects at the NMJ. TDP-43 has no effect on Snx16 mobility. Representative kymographs from time-lapse series of a single confocal slice of a muscle 4 NMJ. Scale bar, 10 μm. (G) Quantification of compartment mobility. Mean mobile particle index ± SEM; n is the number of NMJs (B–D) or animals (G). Control genotypes: (A–G) Vglut-GAL4/w or Y; UAS-Tkv-mCherry/+; (F) Vglut-GAL4/w or Y; UAS-Snx16-GFP/+. Statistical significance was calculated using one-way ANOVA (B–D, G) or Mann–Whitney tests (G). Also see Supplemental Movie S1.
To explore how manipulating Rab11 might restore the distribution of Tkv, we characterized the behavior of Tkv-mCherry compartments at the NMJ by live imaging (Figure 7, E–G). In wild-type NMJs, Tkv-mCherry particles exhibited small movements within a single bouton but rarely moved longer distances (>10 μm) between adjacent boutons (Figure 7, E and G, and Supplemental Movie S1). By contrast, TDP-43–overexpressing NMJs exhibited significantly more-frequent long-range bidirectional movements of Tkv-mCherry (Figure 7, E and G, and Supplemental Movie S1). This increased mobility was suppressed upon coexpression of Rab11DN (Figure 7, E and G, and Supplemental Movie S1). We were unable to test directly whether the fast-moving Tkv-mCherry compartment corresponded directly to the Rab11 compartment because GAL4/UAS-driven GFP-Rab11 is largely cytosolic (Rodal et al., 2011). However, SNX16-GFP puncta did not change mobility upon TDP-43 overexpression (Figure 7, F and G), suggesting that increased mobility is not a general phenotype for all compartments. Thus the mobility of the Tkv-positive compartment regulates its distribution among boutons, and enrichment in the terminal bouton correlates with increased levels of BMP signaling.
Rab11DN suppresses TDP-43–induced motor defects
Finally, we tested whether manipulation of Rab11 could suppress motor defects in larvae misexpressing TDP-43. Both overexpression of hTDP43 and loss of tbph result in strongly reduced rates of larval crawling (Figure 8, A and B), as previously described (Feiguin et al., 2009; Li et al., 2010; Lin et al., 2011; Diaper et al., 2013). Rab11DN partially rescued the larval crawling defects that arise from TDP-43 overexpression, whereas coexpression of Snx163A and TkvQ199D did not suppress these larval crawling defects (Figure 8, A and B). Of importance, loss of dad rescued hTDP-43-overexpression–induced larval crawling defects at a similar level to Rab11DN (Figure 8B). This suggests that the component of the crawling defect that is due to loss of BMP signaling can be restored by Rab11 manipulation. Finally, Rab11DN rescued larval crawling defects in tbph loss-of-function larvae (Figure 8B), suggesting that both gain and loss of function of TDP-43 are sensitive to Rab11 manipulation.
FIGURE 8:
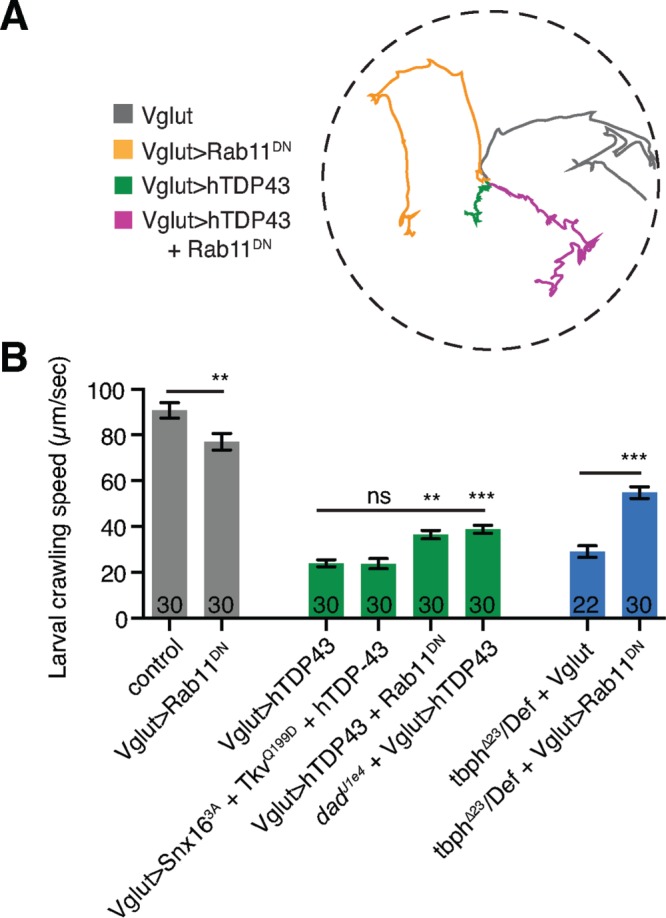
Rab11DN suppresses TDP-43-induced larval crawling defects. (A) Representative traces show larval displacement in a 2-min recording window. (B) Quantification of mean larval displacement per second ± SEM. Statistical significance was calculated using one-way ANOVA. n is the number of animals.
DISCUSSION
In this study, we found that misexpression of the ALS gene TDP-43 impairs synaptic growth and BMP signaling at the Drosophila larval NMJ. These growth defects correlate with altered endosomal localization, distribution, and mobility of BMP receptors and occur without receptor degradation or defects in long-range axonal transport. By genetically rerouting the recycling pathway, we were able to suppress TDP-43–induced defects in synaptic growth, BMP signaling, BMP receptor traffic, and larval crawling. These results pinpoint a likely cellular location of growth factor signaling defects in the TDP-43 ALS model and thus identify a new potential target of therapeutic intervention to restore signaling to a healthy state.
TDP-43 misexpression inhibits BMP signaling
The cellular mechanisms underlying ALS-associated growth factor signaling defects and their connection to motor neuron dysfunction have been elusive (Gould and Oppenheim, 2010; Tovar et al., 2014). Using Drosophila TDP-43 models, we found that disrupted NMJ growth and motor neuron function correlate with reduced BMP signaling and that restoring BMP signaling (either genetically using dad mutants or by rerouting traffic of BMP receptors) partially rescues defects in synaptic growth and larval crawling. BMP signaling is required early in larval development to drive synaptic growth and continuously throughout larval development to support synaptic function (Berke et al., 2013). Similarly, tbph is required for synaptic growth and function throughout larval development (Romano et al., 2014), suggesting that it may acutely control BMP signaling rather than regulating a developmental function.
Where in the BMP signaling cascade does TDP-43 act? We found that the activating BMP receptor mutant TkvQ199D failed to rescue TDP-43-induced synaptic growth defects, suggesting that they occur downstream of receptor activation. Further, TDP-43 misregulation strongly disrupted synaptic BMP signaling but not nuclear BMP signaling. These synaptic growth– and synapse-specific pMad phenotypes are reminiscent of mutants in importin-β11, which localizes to synapses and is believed to retain a locally active BMP signal (Higashi-Kovtun et al., 2010). However, synaptic pMad signaling can be uncoupled from synaptic growth (Sulkowski et al., 2016), leaving uncertain the precise aspect of BMP pathway in which TDP-43 or importin-β11 act. Finally, we found that loss-of-function mutants in dad, a negative regulator of Mad, suppressed TDP-43-induced reductions in NMJ growth, suggesting that signaling defects occur upstream of Mad. Thus our experiments indicate that the point of pathological TDP-43 action lies between receptor activation and dad, at the level of intracellular traffic of the BMP receptors.
To test this hypothesis more specifically, we examined the traffic of the BMP receptor Tkv. We did not detect strong differences in axonal transport of Tkv upon TDP-43 misexpression in motor neurons. By contrast, a recent report found alterations in mitochondrial and dense-core vesicle axonal traffic in TDP-43–misexpressing peptidergic neurons in Drosophila, suggesting that axonal transport defects may be cargo or neuron-type specific (Baldwin et al., 2016). We did find significant defects in the endosomal localization, transport, and distribution of Tkv at the NMJ, correlating with TDP-43–induced reduction in BMP signaling. Finally, Rab11-dependent rerouting of BMP receptor traffic partially restored synaptic BMP signaling. Thus our data suggest a model in which TDP-43 perturbs BMP signaling by disrupting endocytic transport of synaptic BMP receptors.
TDP-43–induced endosomal trafficking defects impair BMP signaling
Previous studies showed that traffic through the recycling compartment is required to attenuate BMP signaling, as mutants affecting this recycling pathway (e.g., rab8, rab11, dASCL) produce synaptic overgrowth and accumulation of BMP receptors in early endosomes (Khodosh et al., 2006; Liu et al., 2014; West et al., 2015). We found that misregulation of TDP-43 caused the converse effect: a shift of BMP receptors from early to recycling endosomes, together with a loss of BMP signaling. Although previous reports suggested that TDP-43 misexpression can induce global dysregulation of endolysosomal traffic by increasing autophagolysosome pathway function (Xia et al., 2016), we did not observe this effect in NMJs. Further, increased autophagy in NMJs was previously reported to produce synaptic overgrowth rather than undergrowth (Shen and Ganetzky, 2009). Thus the trafficking defect we observed may be specific to the early-recycling endosome pathway. Finally, our data showing that dad mutants can rescue TDP-43–induced synaptic undergrowth suggest that altered BMP signaling plays a significant role in defects arising from altered recycling endosome traffic. However, the possibility remains that other signaling pathways previously shown to be regulated by recycling endosome traffic (e.g., JNK signaling; West et al., 2015) make additional contributions to TDP-43 phenotypes.
Our results show that neuronal expression of a dominant-negative mutant of Rab11 partially rescues TDP-43–induced BMP signaling, synaptic growth, and larval crawling defects. This nucleotide binding–defective mutant sequesters Rab11 regulators and effectors that control multiple facets of recycling endosome function, including motor-driven transport, endosome tubulation, and cargo sorting (Pasqualato et al., 2004). Our investigation of the Rab11DN suppression mechanism sheds light on the specific aspect of recycling endosome traffic that may be critical for TDP-43 phenotypes. We found that the ability of Rab11DN to rescue TDP-43–induced defects occurred without shifting BMP receptor localization back to early endosomes. Instead, Rab11DN specifically rescued defects in NMJ mobility and enrichment of Tkv in terminal boutons. These results suggest one of two possibilities. 1) Reduced early endosome localization of Tkv upon TDP-43 misexpression is merely correlated and not causative of synaptic growth defects. However, this model is inconsistent with many previous findings showing that early endosomal BMP receptors promote signaling. 2) Rab11DN suppresses synaptic growth defects by bypassing the need for early endosome localization of Tkv. In the second model, Rab11DN-induced accumulation of the Tkv compartment in terminal boutons might be sufficient to drive partial BMP signaling, which opens up interesting new questions about how BMP receptor distribution affects synaptic growth and function. In fact, previous studies showed that the strength of synaptic transmission follows a BMP receptor–dependent gradient, from weak transmission in proximal boutons to strong transmission in terminal boutons (Guerrero et al., 2005; Ball et al., 2015). Future studies are required to test whether Tkv enrichment in terminal boutons participates in modulating transmission and whether TDP-43–dependent motor neuron dysfunction arises in part from misregulation of the transmission gradient.
Our study also sheds light on the mechanisms of Tkv NMJ distribution. Until now, studies of BMP receptor trafficking at the NMJ have relied on imaging endosomal compartments in fixed tissue. By using live imaging, we found that the dynamics of receptors may play an important role in both their distribution and signaling levels. We found that loss of terminal bouton accumulation correlates with rapid movements of Tkv-positive compartments at the NMJ and that Rab11DN suppresses these rapid dynamics, restoring terminal bouton enrichment and BMP signaling. This aberrant mobility of the Tkv compartment is analogous to the normal trafficking pattern of dense-core vesicles, which are supplied evenly to boutons by a “circulate-and-capture” mechanism by which vesicles are first delivered to the terminal bouton and then distributed by recycling and sporadic capture throughout the nerve terminal (Wong et al., 2012). Our results suggest that Tkv-positive compartments, which normally accumulate in terminal boutons, convert into a more mobile dense-core vesicle–like trafficking pattern upon TDP-43 overexpression, resulting in redistribution among all boutons. In the future, it will be important to test whether receptor transport dynamics and distribution are points of regulation for growth factor signaling in healthy neurons in response to developmental cues or neuronal activity.
One other question that remains to be answered is which of the many cellular RNAs downstream of TDP-43 misexpression leads to the defects in receptor traffic through recycling compartments. Our study suggests that TDP-43 targets controlling early and recycling endosome traffic and dynamics should be considered as strong candidates for further investigation.
Implications for human neurological disease
To date, there are no treatments that halt or reverse ALS. Growth factor signaling pathways may be compelling therapeutic targets, particularly if methods can be found to modify their activities locally and specifically, for example, at the level of intracellular traffic of their receptors. Our results define endosomal traffic of BMP receptors as a point of dysfunction in Drosophila TDP-43 model of ALS, and several lines of evidence suggest that this is a promising target for treating human disease. First, endosomal trafficking mechanisms that regulate BMP signaling are similar to those that control ALS-implicated neurotrophin, IGF-1, and VEGF signaling pathways in mammals (Horowitz and Seerapu, 2012; Cosker and Segal, 2014; Morcavallo et al., 2014). Second, consistent with our observations for TDP-43–induced endosomal defects, early and recycling endosomes are sites of action for many ALS/FTD-linked genes, including Alsin, CHMP2B, UBQLN2, OPTN, and C9ORF72 (Devon et al., 2006; Hadano et al., 2006; Urwin et al., 2010; Farg et al., 2014; Osaka et al., 2015). Third, spinal cord neurons from C9ORF72 or sporadic ALS patients display an aberrant accumulation of early and recycling endosome markers (Farg et al., 2014; Sanhueza et al., 2015), suggesting that endosomal dysfunction is a common denominator in ALS. Thus our results indicate that rerouting receptor traffic by manipulating the endosomal machinery may be an effective strategy to restore growth factor signaling and motor neuron function in ALS.
MATERIALS AND METHODS
Drosophila strains
UAS-Tkv-mCherry constructs were generated from the Tkv-RD splice isoform using Gateway technology (Invitrogen, Carlsbad, CA) in pBI-UASC-mCherry (based on Wang et al., 2011) and injected into flies (Genetic Services, Cambridge, MA) using Φc381 recombinase at the Attp40 locus (Ni et al., 2008). Previously described fly stocks include UAS-Tkv-GFP (Dudu et al., 2006), UAS-SNX16-GFP and UAS-SNX163A-GFP (Rodal et al., 2011), UAS-TkvQ199D (TkvB3; Hoodless et al., 1996), GFP-Rab5KI (Fabrowski et al., 2013), tbph∆23 (Feiguin et al., 2009), Rab11DN (Rab11N124I; Satoh et al., 2005), and Vglut-GAL4(X) (Daniels et al., 2008). UAS-TDP-43 lines have also been previously described: TDP-43-IIA (Lu et al., 2009) was obtained from Fen-Biao Gao (University of Massachusetts Medical School, Worcester, MA). Lines TDP-43-IIb (TDP-43-L1 from Hanson et al., 2010), TDP-43-IIIa (TDP-43-L2 from Hanson et al., 2010), and TDP-43-IIIc (TDP-43-L3 from Hanson et al., 2010) were obtained from Randal Tibbetts (University of Wisconsin, Madison, WI). TDP-43-IIIc contains two P element insertions and was backcrossed five times to white flies to isolate TDP43-IIIb and TDP43-IIId. We used TDP-43-IIId for all remaining experiments. All other stocks (Appl-GAL4, Df(2R)BSC610 [used in-trans to tbph∆23], dadJ1E4, Mi{MIC}twit [MI06552], witA12, and witB11) were obtained from the Bloomington Drosophila Stock Center (Bloomington, IN).
Real-time PCR
Total RNA was isolated from larval brains using RNeasy Mini RNA isolation kit (Qiagen, Valencia, CA). All RNA samples were treated with DNase I (Invitrogen) to remove genomic DNA contamination, and purified with RNeasy columns to remove any traces of DNase enzyme or buffer. cDNA was prepared from 200 ng of total RNA using Superscript III reverse transcriptase and random primers (Invitrogen) from four independent larval brain samples (25 brains each) for each genotype. Real-time quantitative PCR (qPCR) was performed using following primer sets. For tkv: forward, TCCAAGATCATGCAGGAGTG, and reverse, TGGGCACATCGATTAGACAG. For twit: forward, ACAAATCAGATCCCGTGGAG, and reverse, AATGGATGCACCTGCTAAGG. For wit: forward, TCTGACAATGCGAACTCCAC, and reverse, TCATGCTCCAAGCAAGACAG. For rpl32: forward, ATCGGTTACGGATCGAACAA, and reverse, GACAATCTCCTTGCGCTTCT. The qPCRs were set up in triplicate for each of the two cDNA samples for each gene, using SYBR advantage qPCR premix (Clontech, Mountain View, CA), and run for 40 cycles (Tm = 60°C) on a Rotor-gene thermocycler. Fold changes in expression levels were calculated by the 2−∆∆CT method using rpl32 as a reference gene. Graphs depict mean log fold change ± SD for four biological replicates for each genotype, normalized to control (Vglut/w).
Antibodies
The antibodies α-Cpx (Troy Littleton, MIT, Cambridge, MA; Huntwork and Littleton, 2007) and α-phospho-Mad (PS1; Peter ten Dijke, Leiden University Medical Center, Leiden, Netherlands; Dan Vasiliauskas, Susan Morton, Tom Jessell, and Ed Laufer, Columbia University Medical Center, New York, NY; Persson et al., 1998) have been described previously. α-Rab11 antibodies were obtained from BD Biosciences, San Jose, CA (clone 47; Khodosh et al., 2006), α-TDP-43 antibodies (10782-2-AP) from Proteintech, Rosemont, IL, and α-tubulin antibodies (clone B-5-1-2) from Sigma-Aldrich, St. Louis, MO. Rhodamine Red-X– and Alexa 647-conjugated α–horseradish peroxidase (HRP) antibodies were obtained from Jackson ImmunoResearch, West Grove, PA. α-Dlg (4F3), α-Futsch (22c10), α-Wit (23C7), α-actin (JLA20), α-eve (2B8), and α-synapsin (3C11) antibodies were obtained from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). Secondary antibodies for imaging were conjugated to Dylight 488 or Rhodamine Red-X (Jackson ImmunoResearch). Immunoblots were imaged using infrared dye–conjugated secondary antibodies (Rockland Immunochemicals, Pottstown, PA) on a LI-COR Odyssey device.
Immunohistochemistry and analysis of NMJ morphology
For analysis of NMJ morphology and protein localization at the NMJ, flies were cultured at low density at 25ºC. Control genotypes were Vglut-GAL4/w or Y. Wandering third-instar larvae were dissected in calcium-free HL3.1 saline (Feng et al., 2004) and fixed for 30 min in HL3.1 containing 4% formaldehyde (Sigma-Aldrich) before antibody staining. For live imaging, wandering third-instar larvae were dissected in room temperature HL3.1, leaving the CNS and axons intact. Segmental nerve bundles were imaged in a single confocal slice ∼100 μm from the ventral ganglion at 2 frames/s. Focus was maintained manually during imaging, and larvae were imaged for a maximum of 45 min after dissection. All samples were imaged using a spinning-disk confocal system consisting of a Nikon Ni-E upright microscope, a Yokogawa CSU-W1 spinning-disk head, and an Andor iXon 897U electron-multiplying charge-coupled device camera. Fixed samples were imaged using 40× (numerical aperture [NA] 1.3), 60× (NA 1.4), or 100× (NA 1.4) oil immersion objectives. Live samples were imaged using a 60× (NA 1.0) water immersion objective.
Image analyses
All image analysis was performed in ImageJ. For quantification of signal intensity, measurements were made from sum intensity projections of confocal stacks, except for Twit::GFP/eve measurements, which were made from single confocal slices at the widest diameter of the motor neuron nucleus. Muscle or ventral ganglion background mean intensity was subtracted from neuronal measurements. For NMJs, mean intensities were measured within α-HRP–positive NMJs on muscle 4, segments A2–A4. For ventral ganglion, mean pMad intensities were measured within masks created by thresholding between 24 and 50 eve-positive nuclei/brain. Mean Twit::GFP/eve intensities were measured within masks created by manually tracing the cell bodies and nuclei of between 24 and 57 cells with eve-positive nuclei per brain.
Colocalization between Tkv and endosomal markers was analyzed from each slice of a Z-series from fixed muscle 4 NMJs. The total number of Tkv-mCherry puncta/NMJ and the number of Tkv puncta colocalized with the indicated markers were scored manually. Pearson’s r was calculated from each slice of this Z-series using the ImageJ Coloc-2 plug-in (Bolte and Cordelieres, 2006). Masks were created using thresholded Tkv-mCherry signal smoothed with a Gaussian blur of 2 pixels.
For analysis of NMJ morphology, α-HRP– and α-Dlg–labeled NMJs on muscle 4, segments A2–A4, or muscle 6/7, segment A3, were selected for analysis in ImageJ from two-dimensional projections of confocal stacks. Only type Ib innervation, delineated by extensive postsynaptic α-Dlg staining, was quantified. Main axis boutons were defined as all boutons excluding satellite boutons (strings of five or more boutons extending from the main axis of the NMJ). Only these main NMJ axis boutons were used for small-bouton ratio measurements. For NMJ branch length, each branch was measured from the point the axon entered into the muscle to its distal tip, using the segmented line tool in ImageJ, and the length of the longest branch was recorded. For NMJ area measurements, α-HRP signal from type 1b boutons (excluding regions of axon without NMJ boutons) was manually outlined.
For axonal transport quantification, a kymograph was generated using the ImageJ reslice tool by drawing a straight line from the proximal to the distal end of the in-focus region of the axon bundle. Particle speed was determined from these stacks by measuring the anterograde or retrograde displacement of particles over time. Stall duration was measured only for particles that stopped for at least 3 s and resumed movement within the recording interval. To avoid overrepresenting or underrepresenting axons with different in-focus lengths and best represent any animal-to-animal variance, particle velocities and stall durations were averaged per axon per animal.
For NMJ transport, type 1b synapses on muscle 4, segments A2–A3, were imaged at 2 frames/s in a single confocal slice, as before. A kymograph was generated from the longest NMJ branch in the imaging plane, beginning at the distal bouton of the NMJ and terminating at the axon entry point into the muscle. These kymographs ranged from 27 to 124 μm in length over 147–180.5 s in time. Particle motility index was defined as number of particles exhibiting >10-μm movements/number total particles per micrometer NMJ length per second and was averaged across 1–4 NMJs/animal; n in bar graphs is the number of animals.
Lifespan and crawling assays
To measure adult lifespan, 10–20 newly eclosed flies/vial (male and female) were incubated at 25ºC and transferred every 3–5 d to fresh vials. The number of surviving flies was recorded daily. To measure larval crawling speed, wandering third-instar larvae were briefly washed with distilled H2O and placed in the center of a 35-mm Petri dish of 2% agar (Genesee, San Diego, CA). Crawling behavior was recorded for 3 min at 2 frames/s using the time-lapse function of an iPhone 5 (Apple, Cupertino, CA). To account for the time required for acclimation, the first 30 s of the movie was excluded from the analysis. Total distance crawled and crawling speed were calculated using the manual tracking tool in ImageJ. Larvae from three independent crosses were analyzed for each genotype.
Statistical analyses
All errors shown are mean ± SEM. Statistical significance was calculated using GraphPad Prism software using one-way analysis of variance (ANOVA) followed by multiple Tukey’s comparisons (one-way ANOVA), Kruskal–Wallis tests followed by multiple Dunn comparisons (Kruskal–Wallis), Student’s t tests, Mann–Whitney tests, or two-way ANOVA with Sidak’s multiple comparisons (two-way ANOVA), as indicated in figure legends. *p < 0.05, **p < 0.01, and ***p < 0.001. Comparisons are to the leftmost genotype in each bar graph, unless indicated otherwise by horizontal bars.
Supplementary Material
Acknowledgments
We thank Stefano De Renzis, Fabian Feiguin, Peter ten Dijke, Fen-Biao Gao, Ed Laufer, Troy Littleton, Randal Tibbetts, and the Developmental Studies Hybridoma Bank for reagents; Dana Maxwell and ShiYu Wang for technical assistance; and Steve Del Signore, Bruce Goode, Kate Koles, Suzanne Paradis, and Rylie Walsh for helpful discussions and comments on the manuscript. This work was supported by a Blazeman Foundation for ALS Research Postdoctoral Fellowship to M.D. and National Institutes of Health Grant DP2 NS60947 to A.A.R.
Abbreviations used:
- ALS
amyotrophic lateral sclerosis
- BMP
bone morphogenetic protein
- Dad
daughters against decapentaplegic
- FTD
frontotemporal dementia
- Gbb
glass bottom boat
- GFP
green fluorescent protein
- IGF-1
insulin-like growth factor 1
- Mad
Mothers Against Decapentaplegic
- NMJ
neuromuscular junction
- Sax
Saxophone
- TDP-43
TAR DNA-binding protein 43
- Tkv
Thickveins
- Twit
target of wit
- VEGF
vascular endothelial growth factor
- Wit
Wishful Thinking
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-07-0519) on August 17, 2016.
REFERENCES
- Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhaes TR, Goodman CS. Wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron. 2002;33:545–558. doi: 10.1016/s0896-6273(02)00589-5. [DOI] [PubMed] [Google Scholar]
- Baldwin KR, Godena VK, Hewitt VL, Whitworth AJ. Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum Mol Genet. 2016:ddw105. doi: 10.1093/hmg/ddw105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball RW, Peled ES, Guerrero G, Isacoff EY. BMP signaling and microtubule organization regulate synaptic strength. Neuroscience. 2015;291:155–166. doi: 10.1016/j.neuroscience.2015.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball RW, Warren-Paquin M, Tsurudome K, Liao EH, Elazzouzi F, Cavanagh C, An BS, Wang TT, White JH, Haghighi AP. Retrograde BMP signaling controls synaptic growth at the NMJ by regulating trio expression in motor neurons. Neuron. 2010;66:536–549. doi: 10.1016/j.neuron.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Berke B, Wittnam J, McNeill E, Van Vactor DL, Keshishian H. Retrograde BMP signaling at the synapse: a permissive signal for synapse maturation and activity-dependent plasticity. J Neurosci. 2013;33:17937–17950. doi: 10.1523/JNEUROSCI.6075-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Butzlaff M, Hannan SB, Karsten P, Lenz S, Ng J, Vossfeldt H, Prussing K, Pflanz R, Schulz JB, Rasse T. Impaired retrograde transport by the Dynein/Dynactin complex contributes to Tau-induced toxicity. Hum Mol Genet. 2015;24:3623–3637. doi: 10.1093/hmg/ddv107. [DOI] [PubMed] [Google Scholar]
- Casci I, Pandey UB. A fruitful endeavor: modeling ALS in the fruit fly. Brain Res. 2015;1607:47–74. doi: 10.1016/j.brainres.2014.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BJ, Imlach WL, Jiao W, Wolfram V, Wu Y, Grbic M, Cela C, Baines RA, Nitabach MN, McCabe BD. Miniature neurotransmission regulates Drosophila synaptic structural maturation. Neuron. 2014;82:618–634. doi: 10.1016/j.neuron.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosker KE, Segal RA. Neuronal signaling through endocytosis. Cold Spring Harb Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a020669. a020669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne AN, Siddegowda BB, Estes PS, Johannesmeyer J, Kovalik T, Daniel SG, Pearson A, Bowser R, Zarnescu DC. Futsch/MAP1B mRNA is a translational target of TDP-43 and is neuroprotective in a Drosophila model of amyotrophic lateral sclerosis. J Neurosci. 2014;34:15962–15974. doi: 10.1523/JNEUROSCI.2526-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RW, Gelfand MV, Collins CA, DiAntonio A. Visualizing glutamatergic cell bodies and synapses in Drosophila larval and adult CNS. J Comp Neurol. 2008;508:131–152. doi: 10.1002/cne.21670. [DOI] [PubMed] [Google Scholar]
- Deshpande M, Rodal AA. The crossroads of synaptic growth signaling, membrane traffic, and neurological disease: insights from Drosophila. Traffic. 2015;17:87–101. doi: 10.1111/tra.12345. [DOI] [PubMed] [Google Scholar]
- Devon RS, Orban PC, Gerrow K, Barbieri MA, Schwab C, Cao LP, Helm JR, Bissada N, Cruz-Aguado R, Davidson TL. Als2-deficient mice exhibit disturbances in endosome trafficking associated with motor behavioral abnormalities. Proc Natl Acad Sci USA. 2006;103:9595–9600. doi: 10.1073/pnas.0510197103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaper DC, Adachi Y, Sutcliffe B, Humphrey DM, Elliott CJ, Stepto A, Ludlow ZN, Vanden Broeck L, Callaerts P, Dermaut B. Loss and gain of Drosophila TDP-43 impair synaptic efficacy and motor control leading to age-related neurodegeneration by loss-of-function phenotypes. Hum Mol Genet. 2013;22:1539–1557. doi: 10.1093/hmg/ddt005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudu V, Bittig T, Entchev E, Kicheva A, Julicher F, Gonzalez-Gaitan M. Postsynaptic mad signaling at the Drosophila neuromuscular junction. Curr Biol. 2006;16:625–635. doi: 10.1016/j.cub.2006.02.061. [DOI] [PubMed] [Google Scholar]
- Eaton BA, Davis GW. LIM Kinase1 controls synaptic stability downstream of the type II BMP receptor. Neuron. 2005;47:695–708. doi: 10.1016/j.neuron.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Estes PS, Boehringer A, Zwick R, Tang JE, Grigsby B, Zarnescu DC. Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum Mol Genet. 2011;20:2308–2321. doi: 10.1093/hmg/ddr124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrowski P, Necakov AS, Mumbauer S, Loeser E, Reversi A, Streichan S, Briggs JA, De Renzis S. Tubular endocytosis drives remodelling of the apical surface during epithelial morphogenesis in Drosophila. Nat Commun. 2013;4:2244. doi: 10.1038/ncomms3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, Halloran MA, Gleeson PA, Blair IP, Soo KY. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014;23:3579–3595. doi: 10.1093/hmg/ddu068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiguin F, Godena VK, Romano G, D’Ambrogio A, Klima R, Baralle FE. Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 2009;583:1586–1592. doi: 10.1016/j.febslet.2009.04.019. [DOI] [PubMed] [Google Scholar]
- Feng Y, Ueda A, Wu CF. A modified minimal hemolymph-like solution, HL3.1, for physiological recordings at the neuromuscular junctions of normal and mutant Drosophila larvae. J Neurogenet. 2004;18:377–402. doi: 10.1080/01677060490894522. [DOI] [PubMed] [Google Scholar]
- Fuger P, Sreekumar V, Schule R, Kern JV, Stanchev DT, Schneider CD, Karle KN, Daub KJ, Siegert VK, Flotenmeyer M. Spastic paraplegia mutation N256S in the neuronal microtubule motor KIF5A disrupts axonal transport in a Drosophila HSP model. PLoS Genet. 2012;8:e1003066. doi: 10.1371/journal.pgen.1003066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godena VK, Romano G, Romano M, Appocher C, Klima R, Buratti E, Baralle FE, Feiguin F. TDP-43 regulates Drosophila neuromuscular junctions growth by modulating Futsch/MAP1B levels and synaptic microtubules organization. PLoS One. 2011;6:e17808. doi: 10.1371/journal.pone.0017808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TW, Oppenheim RW. Motor neuron trophic factors: therapeutic use in ALS. Brain Res Rev. 2010;67:1–39. doi: 10.1016/j.brainresrev.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero G, Reiff DF, Agarwal G, Ball RW, Borst A, Goodman CS, Isacoff EY. Heterogeneity in synaptic transmission along a Drosophila larval motor axon. Nat Neurosci. 2005;8:1188–1196. doi: 10.1038/nn1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LS. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron. 2001;32:389–401. doi: 10.1016/s0896-6273(01)00496-2. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- Hadano S, Benn SC, Kakuta S, Otomo A, Sudo K, Kunita R, Suzuki-Utsunomiya K, Mizumura H, Shefner JM, Cox GA. Mice deficient in the Rab5 guanine nucleotide exchange factor ALS2/alsin exhibit age-dependent neurological deficits and altered endosome trafficking. Hum Mol Genet. 2006;15:233–250. doi: 10.1093/hmg/ddi440. [DOI] [PubMed] [Google Scholar]
- Hanson KA, Kim SH, Wassarman DA, Tibbetts RS. Ubiquilin modifies TDP-43 toxicity in a Drosophila model of amyotrophic lateral sclerosis (ALS) J Biol Chem. 2010;285:11068–11072. doi: 10.1074/jbc.C109.078527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi-Kovtun ME, Mosca TJ, Dickman DK, Meinertzhagen IA, Schwarz TL. Importin-beta11 regulates synaptic phosphorylated mothers against decapentaplegic, and thereby influences synaptic development and function at the Drosophila neuromuscular junction. J Neurosci. 2010;30:5253–5268. doi: 10.1523/JNEUROSCI.3739-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoodless PA, Haerry T, Abdollah S, Stapleton M, O’Connor MB, Attisano L, Wrana JL. MADR1, a MAD-related protein that functions in BMP2 signaling pathways. Cell. 1996;85:489–500. doi: 10.1016/s0092-8674(00)81250-7. [DOI] [PubMed] [Google Scholar]
- Horowitz A, Seerapu HR. Regulation of VEGF signaling by membrane traffic. Cell Signal. 2012;24:1810–1820. doi: 10.1016/j.cellsig.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntwork S, Littleton JT. A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nat Neurosci. 2007;10:1235–1237. doi: 10.1038/nn1980. [DOI] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kang MJ, Hansen TJ, Mickiewicz M, Kaczynski TJ, Fye S, Gunawardena S. Disruption of axonal transport perturbs bone morphogenetic protein (BMP)-signaling and contributes to synaptic abnormalities in two neurodegenerative diseases. PLoS One. 2014;9:e104617. doi: 10.1371/journal.pone.0104617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodosh R, Augsburger A, Schwarz TL, Garrity PA. Bchs, a BEACH domain protein, antagonizes Rab11 in synapse morphogenesis and other developmental events. Development. 2006;133:4655–4665. doi: 10.1242/dev.02650. [DOI] [PubMed] [Google Scholar]
- Kim NC, Marques G. The Ly6 neurotoxin-like molecule target of wit regulates spontaneous neurotransmitter release at the developing neuromuscular junction in Drosophila. Dev Neurobiol. 2012;72:1541–1558. doi: 10.1002/dneu.22021. [DOI] [PubMed] [Google Scholar]
- Landgraf M, Roy S, Prokop A, VijayRaghavan K, Bate M. Even-skipped determines the dorsal growth of motor axons in Drosophila. Neuron. 1999;22:43–52. doi: 10.1016/s0896-6273(00)80677-7. [DOI] [PubMed] [Google Scholar]
- Li Y, Ray P, Rao EJ, Shi C, Guo W, Chen X, Woodruff EA, 3rd, Fushimi K, Wu JY. A Drosophila model for TDP-43 proteinopathy. Proc Natl Acad Sci USA. 2010;107:3169–3174. doi: 10.1073/pnas.0913602107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MJ, Cheng CW, Shen CK. Neuronal function and dysfunction of Drosophila dTDP. PLoS One. 2011;6:e20371. doi: 10.1371/journal.pone.0020371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Huang Y, Hu W, Huang S, Wang Q, Han J, Zhang YQ. dAcsl, the Drosophila ortholog of acyl-CoA synthetase long-chain family member 3 and 4, inhibits synapse growth by attenuating bone morphogenetic protein signaling via endocytic recycling. J Neurosci. 2014;34:2785–2796. doi: 10.1523/JNEUROSCI.3547-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd TE, Machamer J, O’Hara K, Kim JH, Collins SE, Wong MY, Sahin B, Imlach W, Yang Y, Levitan ES. The p150(Glued) CAP-Gly domain regulates initiation of retrograde transport at synaptic termini. Neuron. 2012;74:344–360. doi: 10.1016/j.neuron.2012.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Ferris J, Gao FB. Frontotemporal dementia and amyotrophic lateral sclerosis-associated disease protein TDP-43 promotes dendritic branching. Mol Brain. 2009;2:30. doi: 10.1186/1756-6606-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S, Twelvetrees AE, Moughamian AJ, Holzbaur EL. Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron. 2014;84:292–309. doi: 10.1016/j.neuron.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques G, Bao H, Haerry TE, Shimell MJ, Duchek P, Zhang B, O’Connor MB. The Drosophila BMP type II receptor Wishful Thinking regulates neuromuscular synapse morphology and function. Neuron. 2002;33:529–543. doi: 10.1016/s0896-6273(02)00595-0. [DOI] [PubMed] [Google Scholar]
- Marques G, Zhang B. Retrograde signaling that regulates synaptic development and function at the Drosophila neuromuscular junction. Int Rev Neurobiol. 2006;75:267–285. doi: 10.1016/S0074-7742(06)75012-7. [DOI] [PubMed] [Google Scholar]
- McCabe BD, Marques G, Haghighi AP, Fetter RD, Crotty ML, Haerry TE, Goodman CS, O’Connor MB. The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron. 2003;39:241–254. doi: 10.1016/s0896-6273(03)00426-4. [DOI] [PubMed] [Google Scholar]
- Miskiewicz K, Jose LE, Yeshaw WM, Valadas JS, Swerts J, Munck S, Feiguin F, Dermaut B, Verstreken P. HDAC6 is a Bruchpilot deacetylase that facilitates neurotransmitter release. Cell Rep. 2014;8:94–102. doi: 10.1016/j.celrep.2014.05.051. [DOI] [PubMed] [Google Scholar]
- Morcavallo A, Stefanello M, Iozzo RV, Belfiore A, Morrione A. Ligand-mediated endocytosis and trafficking of the insulin-like growth factor receptor I and insulin receptor modulate receptor function. Front Endocrinol (Lausanne) 2014;5:220. doi: 10.3389/fendo.2014.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Ni JQ, Markstein M, Binari R, Pfeiffer B, Liu LP, Villalta C, Booker M, Perkins L, Perrimon N. Vector and parameters for targeted transgenic RNA interference in Drosophila melanogaster. Nat Methods. 2008;5:49–51. doi: 10.1038/nmeth1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor-Giles KM, Ho LL, Ganetzky B. Nervous wreck interacts with thickveins and the endocytic machinery to attenuate retrograde BMP signaling during synaptic growth. Neuron. 2008;58:507–518. doi: 10.1016/j.neuron.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osaka M, Ito D, Yagi T, Nihei Y, Suzuki N. Evidence of a link between ubiquilin 2 and optineurin in amyotrophic lateral sclerosis. Hum Mol Genet. 2015;24:1617–1629. doi: 10.1093/hmg/ddu575. [DOI] [PubMed] [Google Scholar]
- Pasqualato S, Senic-Matuglia F, Renault L, Goud B, Salamero J, Cherfils J. The structural GDP/GTP cycle of Rab11 reveals a novel interface involved in the dynamics of recycling endosomes. J Biol Chem. 2004;279:11480–11488. doi: 10.1074/jbc.M310558200. [DOI] [PubMed] [Google Scholar]
- Persson U, Izumi H, Souchelnytskyi S, Itoh S, Grimsby S, Engstrom U, Heldin CH, Funa K, ten Dijke P. The L45 loop in type I receptors for TGF-beta family members is a critical determinant in specifying Smad isoform activation. FEBS Lett. 1998;434:83–87. doi: 10.1016/s0014-5793(98)00954-5. [DOI] [PubMed] [Google Scholar]
- Piccioli ZD, Littleton JT. Retrograde BMP signaling modulates rapid activity-dependent synaptic growth via presynaptic LIM kinase regulation of cofilin. J Neurosci. 2014;34:4371–4381. doi: 10.1523/JNEUROSCI.4943-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14:459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson JM, Lee M, Kennedy EL, Selleck SB. Drosophila neuromuscular synapse assembly and function require the TGF-beta type I receptor saxophone and the transcription factor Mad. J Neurobiol. 2003;55:134–150. doi: 10.1002/neu.10189. [DOI] [PubMed] [Google Scholar]
- Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14:248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- Rodal AA, Blunk AD, Akbergenova Y, Jorquera RA, Buhl LK, Littleton JT. A presynaptic endosomal membrane trafficking pathway controls synaptic growth signaling. J Cell Biol. 2011;193:201–217. doi: 10.1083/jcb.201009052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano G, Klima R, Buratti E, Verstreken P, Baralle FE, Feiguin F. Chronological requirements of TDP-43 function in synaptic organization and locomotive control. Neurobiol Dis. 2014;71:95–109. doi: 10.1016/j.nbd.2014.07.007. [DOI] [PubMed] [Google Scholar]
- Roos J, Hummel T, Ng N, Klambt C, Davis GW. Drosophila Futsch regulates synaptic microtubule organization and is necessary for synaptic growth. Neuron. 2000;26:371–382. doi: 10.1016/s0896-6273(00)81170-8. [DOI] [PubMed] [Google Scholar]
- Rutherford NJ, Zhang Y-J, Baker M, Gass JM, Finch NA, Xu Y-F, Stewart H, Kelley BJ, Kuntz K, Crook RJP. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4:e1000193. doi: 10.1371/journal.pgen.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanhueza M, Chai A, Smith C, McCray BA, Simpson TI, Taylor JP, Pennetta G. Network analyses reveal novel aspects of ALS pathogenesis. PLoS Genet. 2015;11:e1005107. doi: 10.1371/journal.pgen.1005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh AK, O’Tousa JE, Ozaki K, Ready DF. Rab11 mediates post-Golgi trafficking of rhodopsin to the photosensitive apical membrane of Drosophila photoreceptors. Development. 2005;132:1487–1497. doi: 10.1242/dev.01704. [DOI] [PubMed] [Google Scholar]
- Schreij AM, Fon EA, McPherson PS. Endocytic membrane trafficking and neurodegenerative disease. Cell Mol Life Sci. 2016;73:1529–1545. doi: 10.1007/s00018-015-2105-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton CF, Cenik C, Kucukural A, Dammer EB, Cenik B, Han Y, Dewey CM, Roth FP, Herz J, Peng J, et al. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J Biol Chem. 2011;286:1204–1215. doi: 10.1074/jbc.M110.190884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Ganetzky B. Autophagy promotes synapse development in Drosophila. J Cell Biol. 2009;187:71–79. doi: 10.1083/jcb.200907109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulkowski MJ, Han TH, Ott C, Wang Q, Verheyen EM, Lippincott-Schwartz J, Serpe M. A novel, noncanonical BMP pathway modulates synapse maturation at the Drosophila neuromuscular junction. PLoS Genet. 2016;12:e1005810. doi: 10.1371/journal.pgen.1005810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney ST, Davis GW. Unrestricted synaptic growth in spinster—a late endosomal protein implicated in TGF-beta-mediated synaptic growth regulation. Neuron. 2002;36:403–416. doi: 10.1016/s0896-6273(02)01014-0. [DOI] [PubMed] [Google Scholar]
- Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Konig J, Hortobagyi T, Nishimura AL, Zupunski V. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14:452–458. doi: 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torroja L, Chu H, Kotovsky I, White K. Neuronal overexpression of APPL, the Drosophila homologue of the amyloid precursor protein (APP), disrupts axonal transport. Curr Biol. 1999;9:489–492. doi: 10.1016/s0960-9822(99)80215-2. [DOI] [PubMed] [Google Scholar]
- Tovar YRLB, Ramirez-Jarquin UN, Lazo-Gomez R, Tapia R. Trophic factors as modulators of motor neuron physiology and survival: implications for ALS therapy. Front Cell Neurosci. 2014;8:61. doi: 10.3389/fncel.2014.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urwin H, Authier A, Nielsen JE, Metcalf D, Powell C, Froud K, Malcolm DS, Holm I, Johannsen P, Brown J. Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum Mol Genet. 2010;19:2228–2238. doi: 10.1093/hmg/ddq100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JW, Brent JR, Tomlinson A, Shneider NA, McCabe BD. The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J Clin Invest. 2011;121:4118–4126. doi: 10.1172/JCI57883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West RJ, Lu Y, Marie B, Gao FB, Sweeney ST. Rab8, POSH, and TAK1 regulate synaptic growth in a Drosophila model of frontotemporal dementia. J Cell Biol. 2015;208:931–947. doi: 10.1083/jcb.201404066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MY, Zhou C, Shakiryanova D, Lloyd TE, Deitcher DL, Levitan ES. Neuropeptide delivery to synapses by long-range vesicle circulation and sporadic capture. Cell. 2012;148:1029–1038. doi: 10.1016/j.cell.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Q, Wang H, Hao Z, Fu C, Hu Q, Gao F, Ren H, Chen D, Han J, Ying Z. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J. 2016;35:121–142. doi: 10.15252/embj.201591998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.