

A model for generating highly polyploid cells through endoreplication shows that iECs have lower levels of HSET, overexpression of which rescues spindle multipolarity. The results provide insight into the mechanisms by which alternative cell cycles lead to polyploid cell division and genomic instability.
Abstract
Polyploid cancer cells exhibit chromosomal instability (CIN), which is associated with tumorigenesis and therapy resistance. The mechanisms that induce polyploidy and how these mechanisms contribute to CIN are not fully understood. Here we evaluate CIN in human cells that become polyploid through an experimentally induced endoreplication cycle. When these induced endoreplicating cells (iECs) returned to mitosis, it resulted in aneuploidy in daughter cells. This aneuploidy resulted from multipolar divisions, chromosome missegregation, and failure in cytokinesis. The iECs went through several rounds of division, ultimately spawning proliferative cells of reduced ploidy. iECs have reduced levels of the kinesin-14 HSET, which likely accounts for the multipolar divisions, and overexpression of HSET reduced spindle multipolarity. However, HSET overexpression had only mild effects on CIN, suggesting that additional defects must contribute to genomic instability in dividing iECs. Overall our results suggest that transient endoreplication cycles generate a diverse population of proliferative aneuploid cells that have the potential to contribute to tumor heterogeneity.
INTRODUCTION
Proper regulation of the cell cycle is critical for normal development and to prevent aneuploidy. Most cells undergo a canonical cell cycle in which rounds of DNA synthesis (S) and mitosis (M) occur with intervening gap (G) phases. However, some cells undergo alternative cell cycles composed of periodic S phases in the absence of a complete mitotic division, termed endoreplication, leading to polyploid cells (Fox and Duronio, 2013; Schoenfelder and Fox, 2015). One type of alternative cell cycle is called the endocycle, in which cells undergo rounds of G/S, and M phase is skipped. Alternatively, cells can become polyploid through endomitosis, in which cells enter but do not complete mitosis. Both types of endoreplication cycles are a normal part of development in some cell types—for example, hepatocytes and trophoblast giant cells (Zybina et al., 1975; Klisch et al., 2005; Celton-Morizur and Desdouets, 2010)—or can occur as an adaptation to stress responses (Fox and Duronio, 2013). Some cells that enter a natural endoreplication cycle are resistant to apoptosis, suggesting that endoreplication causes genotoxic stress that requires a shutdown of the apoptotic response to DNA damage (Mehrotra et al., 2008; Hassel et al., 2014; Zhang et al., 2014). Alternatively, it may be that high polyploidy itself is a buffer against stress.
For most naturally endoreplicating cells, the polyploid state is considered irreversible (Fox and Duronio, 2013). However, there are instances in which polyploid cells do divide. For example, Drosophila rectal papillae and mouse polyploid hepatocytes undergo polyploid mitoses, which lead to error-prone division and aneuploidy (Duncan et al., 2010; Fox et al., 2010). In addition, Drosophila induced endocycling cells, generated either by knockdown of cyclin A or overexpression of Cdh1, can return to mitosis (RTM) but undergo an error-prone division (Hassel et al., 2014). Several previous studies suggested that human or mouse cells can also switch to a developmental-like endoreplication cycle through cell cycle perturbation (Trakala et al., 2015), response to radiation (Erenpreisa et al., 2008), or drug treatment (Puig et al., 2008). In some instances, the divisions of these cells result in aneuploidy, which potentially is linked to tumorigenesis (Wheatley, 2008; Davoli et al., 2010; Davoli and de Lange, 2011, 2012; Vitale et al., 2011).
There are many examples of human cancers that contain polyploid cells, and polyploidy in tumors is associated with poor prognosis (Storchova and Pellman, 2004; Davoli and de Lange, 2011). What is not clear is the spectrum of mechanisms by which cells become polyploid in tumors and whether high levels of polyploidy arise from an inappropriate switch to endoreplication. There is growing evidence that genome amplification is correlated with aneuploidy and genomic instability and that most tumors have undergone genome doubling at some point in their lifetime (Zack et al., 2013; Dewhurst et al., 2014). Computational modeling and analysis of a large number of tumor samples demonstrated that many cancers evolve to a near-triploid state (Laughney et al., 2015), suggesting that higher ploidy may provide a selective advantage to tumor cells for their growth and survival.
A major question is whether human induced endoreplicating cells (iECs) that undergo RTM have higher levels of chromosomal instability (CIN). It has been demonstrated that tetraploid cells that arise from cytokinesis failure (Fujiwara et al., 2005) or from mitotic slippage caused by telomere erosion in p53− cells (Davoli and de Lange, 2012) have a high propensity for CIN, which generates diverse aneuploidy and promotes tumorigenesis (Davoli and de Lange, 2011). In contrast, polyploid (≥8C) mouse hepatocytes can go through genomic reductive divisions to generate genetic diversity that is favorable in response to nutritional stresses (Duncan et al., 2010). This suggests that RTM after transient endoreplication represents a bona fide alternative cell cycle oscillator that is fundamentally different from mitotic slippage and may represent an alternative cell proliferation strategy that can generate massive aneuploidy and yet maintain cell survival. However, important unanswered questions are whether RTM in polyploid endoreplicating cells leads to sustainable proliferation and what the genome content and fates are of these individual daughter cells.
A major challenge for RTM in polyploid cells is that they often contain supernumerary centrosomes, which lead to the formation of multipolar spindles and potential multipolar divisions. In normal diploid cells and even in tetraploid cells, multipolar divisions usually result in cell death because the genomic material is distributed among three or more daughter cells, causing a detrimental loss of genome content. To overcome this problem, tetraploid cells cluster centrosomes to bypass the transient multipolar spindle stage and form bipolar spindles that divide into only two daughter cells (Ganem et al., 2009). In contrast, polyploid mouse hepatocytes undergo multipolar divisions that quarter or halve their genome content (Duncan et al., 2010), which is postulated to avoid aneuploidy and cell death. These findings suggest that there may be a fundamental difference in the divisions of tetraploid cells versus higher-ploidy cells. Another major question is how cells undergoing polyploid mitosis avoid the detrimental effects of multipolar divisions and apparently tolerate massive aneuploidy.
Here we investigate RTM of polyploid (≥8C) cultured human cells after induction of transient endoreplication and the resulting effects on genomic stability and cell fate. We demonstrate that these polyploid cells have frequent multipolar spindles and error-prone chromosome segregation, resulting in aneuploidy. Only a fraction of iEC daughter cells can survive and proliferate and gradually return to a near-triploid genome content over multiple divisions. The kinesin-14 HSET, which is important in centrosome clustering (Kwon et al., 2008; Ganem et al., 2009), is down-regulated in iECs, and overexpression of HSET partially rescues spindle multipolarity and reduces genomic heterogeneity in the iEC daughter cells. These findings support the idea that RTM after transient endoreplication may be an underappreciated aspect of tumorigenesis and provide a novel inroad to identification of new therapeutic strategies.
RESULTS
Establishing an efficient system to induce polyploidy through endoreplication
To evaluate the consequences of RTM for polyploid endoreplicating cells, we sought to generate a robust system to induce cells to undergo transient endoreplication. Previous studies demonstrated that multiple cell cycle kinase inhibitors were able to induce polyploidy in vitro (Usui et al., 1991; Verdoodt et al., 1999; Lannutti et al., 2005; Gizatullin et al., 2006; Zhu et al., 2011). We incubated HeLa cells for various amounts of time with a series of kinase inhibitors and measured DNA content by fluorescence-activated cell sorting (FACS) to identify inhibitors that could induce polyploidy efficiently and consistently (Figure 1). Note that HeLa cells normally have 75–82 chromosomes, which is a near-triploid DNA content, but which in the literature is referred to as 2C/4C DNA content. Flow cytometry analysis on HeLa cells stained with propidium iodide revealed that addition of dimethyl sulfoxide (DMSO) did not alter the DNA content profile, whereas addition of RO3306, a Cdk1 inhibitor (Vassilev et al., 2006), or SU6656, a Src kinase inhibitor (Blake et al., 2000) and a potent Aurora B inhibitor (Bain et al., 2007), induced many polyploid (≥8C) cells (Figure 1, A and B). Many cells with high ploidy also have a larger nuclear size (Comai, 2005; Zhu et al., 2011). We used high-throughput microscopy to visualize nuclear morphology in HeLa cells after drug treatment. Consistent with the FACS data, a quantitative analysis of nuclear size demonstrated that the average nuclear size in RO3306- or SU6656-treated HeLa cells increased significantly compared with control HeLa cells (Figure 1, C and D).
FIGURE 1:

Antimitotic drugs induce polyploidy. (A) Representative FACS profiles showing the DNA content of propidium iodide–stained cells with the indicated treatments. (B) Quantification of FACS data from three independent experiments showing the percentage of cells with different ploidy under the indicated treatments. For each experiment, 10,000 cells were analyzed for DNA content by propidium iodide staining, and the mean ± SD is graphed. *p < 0.05, **p < 0.01, ***p < 0.001. (C) Micrographs of HeLa cells stained with Hoechst to visualize DNA after the indicated treatments. Scale bar, 20 μm. (D) Dot plots showing the quantification of the nuclear size from three independent experiments in which at least 300 cells were scored per experiment. Bar and whiskers indicate the mean and SD. ***p < 0.001.
Three general mechanisms of polyploidization have been described: cell fusion, mitosis/cytokinesis failure, and endoreplication (Fox and Duronio, 2013). Because RO3306 and SU6656 are kinase inhibitors that may prevent proper mitosis and FACS revealed quantum doublings of genome content, we postulated that drug-treated cells underwent endoreplication (Zhu et al., 2011). To test this idea, we synchronized HeLa cells with thymidine to arrest the cells in S phase and then imaged live cells for ∼30 h after drug addition to visualize morphological changes during polyploidization (Figure 2A). At the drug concentrations used in this study, there was a low level of cell death (<1% for RO3306 and 9% for SU6656), consistent with the small <2C population in the FACS data. Analysis of the behavior of individual cells showed that unlike control cells, cells treated with RO3306 or SU6656 did not divide (Figure 2, B–D, and Supplemental Video S1). In RO3306-treated HeLa cells, we never saw mitotic rounding, the nuclear envelope never broke down, and the nuclei were larger and oval shaped, all features indicative of the endocycle (Supplemental Video S1). In contrast, most SU6656-treated HeLa cells did round up, a hallmark of mitosis, but then reflattened without dividing. After flattening, the nuclei were multilobed or multinucleated in these cells (Figure 2, C and D, and Supplemental Video S1), consistent with previous studies (Zhu et al., 2011).
FIGURE 2:
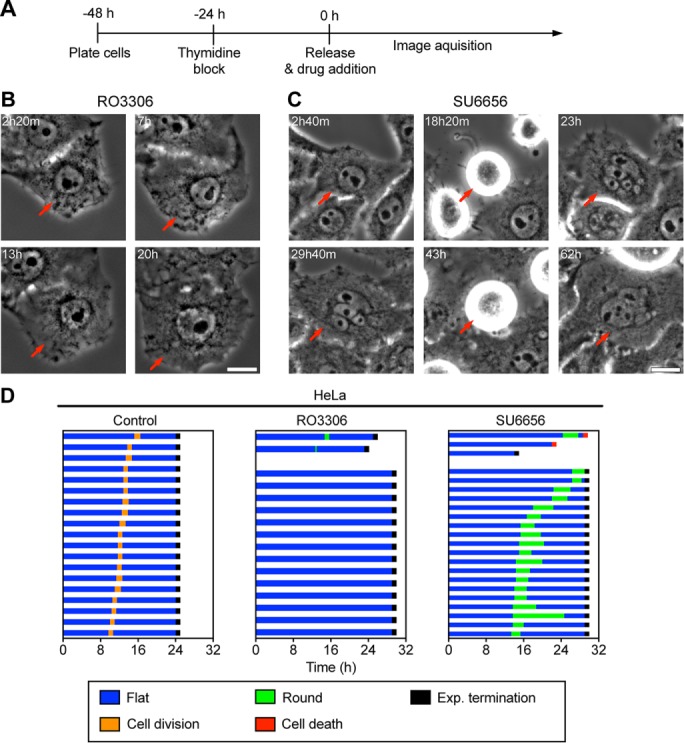
Antimitotic drugs induce polyploidy through endocycle/endomitosis. (A) Time line showing experimental procedure; t = 0 indicates the time of drug addition and corresponds to the timing in B–D. (B, C) Representative time-lapse images of HeLa cells treated with 6 μM RO3306 (B) or 6 μM SU6656 (C) for 24 or 40 h, respectively. Time is marked in the upper left, and the red arrow marks the same cell at different time points. Scale bar, 10 μm. (D) Cell fate profiles of HeLa cells exposed to indicated treatment; t = 0 corresponds to time of drug addition.
To further evaluate whether SU6656-treated HeLa cells showed mitotic features, we categorized cell morphology during time-lapse microscopy and located the time window for which most SU6656-treated cells were in a rounded state (Figure 2D). Immunofluorescence detection of DNA and microtubules revealed that control DMSO-treated HeLa cells showed all stages of mitosis, whereas SU6656-treated HeLa cells showed exclusively early mitotic stages from prophase to metaphase (Figure 3, A and B), supporting the idea that most SU6656-treated HeLa cells enter mitosis but do not undergo the metaphase/anaphase transition or cytokinesis. These results indicate that SU6656-treated HeLa cells undergo endomitosis, a specific form of endoreplication in which cells enter but do not complete mitosis or divide. To confirm this observation, we used time-lapse microscopy to track chromosome behavior in HeLa cells expressing fluorescently labeled histones (Figure 3C). In control DMSO-treated cells, the chromosomes condensed, aligned at the spindle equator, and then segregated to the daughter cells within ∼1.5 h. In contrast, in SU6656-treated HeLa cells, chromosomes condensed and eventually decondensed without chromosome disjunction or cell division (Figure 3C and Supplemental Video S2), confirming that SU6656 treatment induces HeLa cells to enter endomitosis. We found that high concentrations of RO3306 induced endocycles, whereas lower concentrations induced endomitosis (Supplemental Figure S1), consistent with the idea that Cdk1 is an important mediator in the choice between endocycle and endomitosis. These drug-induced phenotypes were not limited to HeLa cells, as similar results were obtained with the breast cancer cell line MDA-MB-231 (Supplemental Figure S2 and Supplemental Video S3). Furthermore, treatment of the p53+ diploid HCT-116 cell line with either drug also induced the accumulation of polyploid cells (Supplemental Figure S3A), albeit with less efficiency. This effect was not limited to tumor-derived cells, as treatment of telomerase-immortalized RPE-1 cells with either RO3306 or SU6656 also induced the accumulation of cells with ≥8C DNA content (Supplemental Figure S3B); however, there was also a larger population of <2C cells in this cell line, especially when treated with 6 μM SU6656 (unpublished data), suggesting that non–tumor-derived cells are more likely to apoptose. Taken together, our results show that we have established an efficient system to induce switching to an alternative cell cycle that undergoes repeated genome duplication without cell division, providing us with a model with which to explore the consequences of cell division in these iECs. Because treatment of cells with SU6656 reproducibly resulted in the highest percentage of endoreplicating cells, we used this treatment for the studies described here.
FIGURE 3:
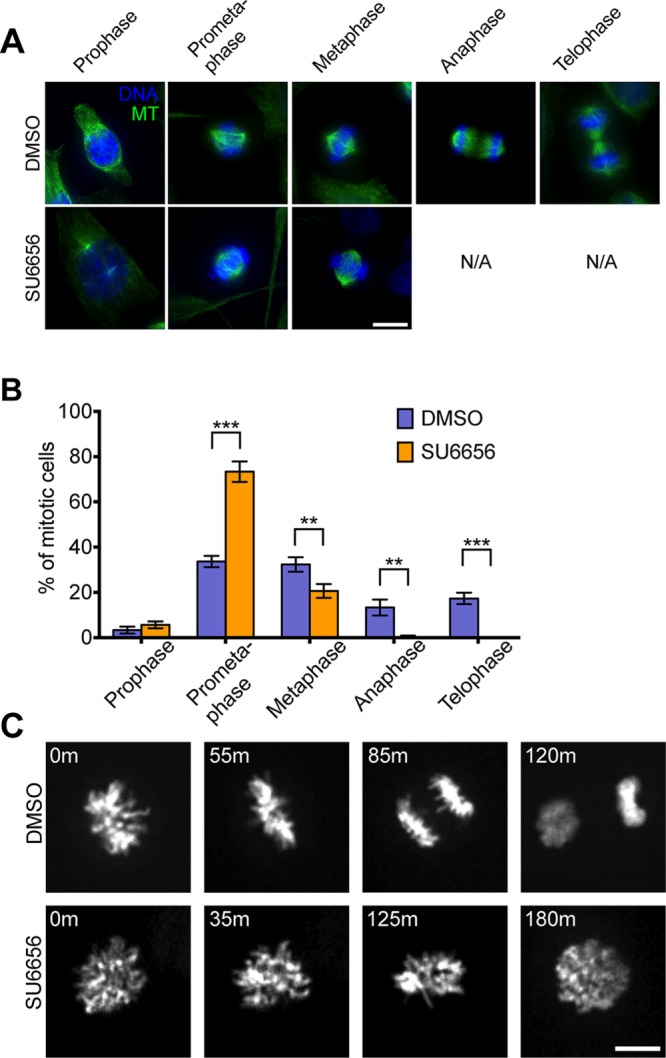
SU6656 induces polyploidy through endomitosis. (A) Control DMSO or SU6656 treated cells were fixed at ∼14 h after drug addition and processed for immunofluorescence to visualize MTs (green) and DNA (blue). Scale bar, 10 μm. (B) Quantification of the percentage of mitotic cells at different mitotic stages from the experiment in A. Data are mean ± SD from three independent experiments in which 900 cells were scored per condition. **p < 0.01, ***p < 0.001. (C) Representative time-lapse images of HeLa cells expressing GFP-labeled histone H2B treated with DMSO or SU6656 treatment. Time is marked in the upper left. Scale bar, 10 μm.
Return to mitosis is coupled with mitotic defects and genomic instability
Previous work in Drosophila showed that polyploid iECs could resume mitosis after the induction signal was removed (Hassel et al., 2014). To test whether human iECs can return to mitotic divisions, we synchronized cells with thymidine, treated them with SU6656 for 48 h, and then used time-lapse phase contrast imaging to examine cells after drug washout (Figure 4, A and B, and Supplemental Video S4). We found that HeLa iECs could return to mitosis and that the resulting iEC daughter cells can further divide (Figure 4, B and C). We then used high-throughput microscopy to monitor the morphological change of iECs at 7–16 h after drug washout, and we found that ∼32% of iECs entered mitosis but only 10% successfully divided. The majority of successful divisions (87%) resulted in two daughter cells of unequal size, suggesting that they underwent asymmetric cell division (Figure 4D). Of the cells that failed division, approximately one-half flattened without undergoing cell division, which we scored as mitotic failure, one-third arrested in mitosis, and the rest underwent cell death (Figure 4D). Many of these cells died after attempting cell division, as evidenced by cell rounding, suggesting that they underwent mitotic catastrophe.
FIGURE 4:
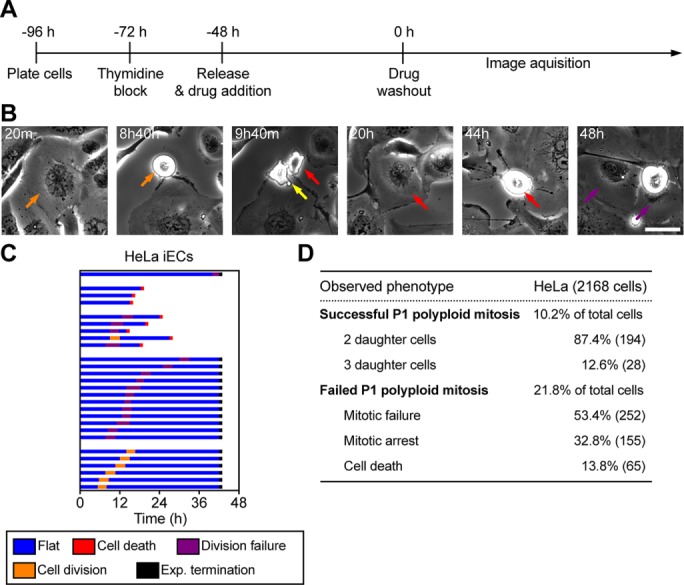
RTM in polyploid cells results in multipolar spindles and asymmetric cell division. (A) Time line showing experimental procedure. (B) Representative images of HeLa cells after SU6656 washout (t = 0). Time after washout is marked in the upper left corner. The orange arrow represents one cell that divides into two cells (yellow and red arrows). The purple arrow represents the two daughters from the previous division of the cell indicated by the red arrow. Scale bar, 20 μm. (C) Cell morphology profiles of HeLa cells; t = 0 indicates the time of drug washout. (D) Cell fate analysis of HeLa iECs from high-throughput imaging from 7 to 16 h after drug washout. HeLa cells were synchronized, treated with SU6656 for 48 h, and then imaged at 5-min intervals starting from 9 to 16 h after drug washout. A total of 2168 cells were analyzed, 694 of which entered mitosis. The number in parentheses represents the total number of mitotic cells with the indicated cell fate.
To gain insight into the underlying causes of the asymmetric cell divisions, we used superresolution microscopy to examine spindle structure and kinetochore distribution in the daughter cells. We found that all mitotic iECs had multipolar spindles in prometaphase (Figure 5A), and many of the anaphase cells had lagging chromosomes (Figure 5B), which likely evolved into the chromosome bridges that we observed during telophase (Figure 5C). Quantification of kinetochore numbers in iEC daughter cells showed that chromosome segregation was highly asymmetric, resulting in a wide variation in chromosome number among daughter cells (Figure 5, C–F). These cells also had micronuclei, which is another hallmark of CIN (unpublished data). These findings support the idea that RTM after transient endoreplication is highly error-prone and results in asymmetric distributions of chromosomes, leading to aneuploid daughter cells. The prevalence of multipolar spindles in early mitosis and multiple anaphase/telophase structures in late mitosis suggests that iECs undergo multipolar division during RTM. However, these observations apparently contradicted our high-throughput data showing that most iECs divided into only two daughter cells.
FIGURE 5:
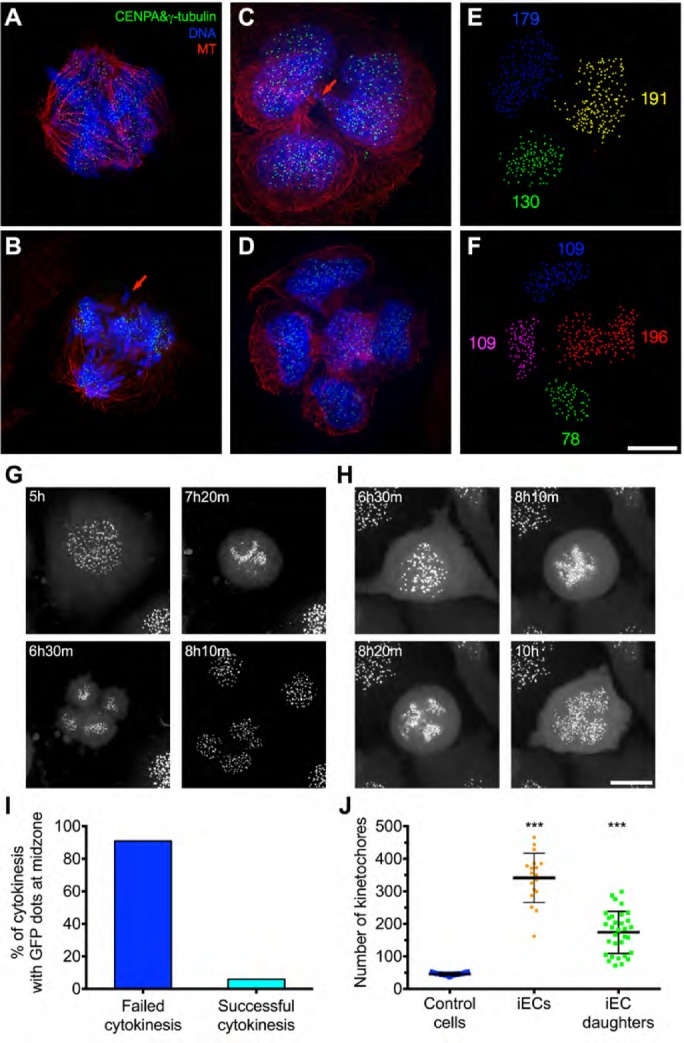
RTM in highly polyploid cells is error-prone and results in aneuploidy. (A–D) HeLa cells expressing GFP-CENP-A and GFP-γ-tubulin were fixed at 14 h after drug washout, labeled to visualize kinetochore/spindle poles (green), DNA (blue), and microtubules (red), and imaged by superresolution microscopy. The red arrows indicate abnormally located chromosomes during anaphase (B) or cytokinesis (C). (E, F) IMARIS 3D software was used to detect and quantify the number of GFP-CENP-A–labeled kinetochores from images in C and D. Clusters of individual kinetochores in different daughter cells are indicated by different colors, and the number of kinetochores detected is indicated by the number with the corresponding color. Scale bar, 20 μm. (G, H) HeLa cells expressing GFP-CENP-A and GFP-γ-tubulin were synchronized, treated with SU6656 for 48 h, and then imaged at 5-min intervals starting from 5 h after drug washout. Selected frames are shown, and the time is marked in the upper left corner. Scale bar, 20 μm. One example iEC divided into three daughter cells (G) and another failed in division (H). (I) Quantification of the percentage of cells with failed or successful cytokinesis among all anaphase/telophase cells with GFP-Cenp-A dots in the midzone; 62 cells. (J) Dot plot showing the quantification of the number of kinetochores in each individual cell taken from the time-lapse images. The dots represent the number of kinetochores, and the bar and whiskers indicate the mean ± SD; 18 cells. ***p < 0.001.
To resolve this contradiction, we used time-lapse confocal microscopy to image mitotic HeLa iECs expressing green fluorescent protein (GFP)–CENP-A and GFP-γ-tubulin during RTM (Figure 5, G and H, and Supplemental Video S5). Whereas all mitotic iECs initiated multipolar division, the majority (∼90%) of these divisions ultimately resulted in only two daughter cells. We did not observe frequent clustering of centrosomes into bipolar spindles. Instead, cells initially divided into multiple daughter cells, but frequent cytokinesis failure and furrow regression resulted in only two daughter cells of uneven chromosome number. There was a positive correlation between cells that failed cytokinesis and the presence of GFP-CENP-A dots near the spindle midzone (Figure 5I), suggesting that chromosomes in the midzone signal the no-cut checkpoint that contributes to cytokinesis failure (Agromayor and Martin-Serrano, 2013). Consistent with our fixed analysis of cells, time-lapse imaging also revealed an unequal and highly variable distribution of kinetochores between daughter cells, suggesting that these polyploid divisions result in a high level of CIN (Figure 5J). Together these findings show that iEC divisions often result in two daughter cells of unequal sizes due to a failure in cytokinesis.
To quantitatively evaluate chromosome missegregation rate, we used fluorescence in situ hybridization (FISH) to analyze the distributions of chromosome 2 and 7 in daughter cells that were in either late anaphase/telophase or early G1 (Figure 6, A and B). In control daughter cells, the mean number of chromosome 2 or 7 was three, consistent with HeLa cells being near triploid. In contrast, in iEC daughter cells, the mean number of chromosome 2 or 7 was seven, and the iEC daughter cells had a much broader range of copy numbers for these chromosomes (Figure 6C). In addition, we calculated the chromosome missegregation rate by determining the percentage of cells that had an unequal distribution of these chromosomes in the daughter nuclei at telophase/early G1. In control cells, only ∼5% of the cells had unequal numbers of chromosomes (Figure 6D), whereas in the polyploid cells, >95% of the daughter cells had unequal numbers of chromosomes. Together these results show that polyploid (≥8C) iECs can resume mitotic divisions after drug withdrawal but that these divisions are aberrant and result in daughter cells with high levels of aneuploidy.
FIGURE 6:
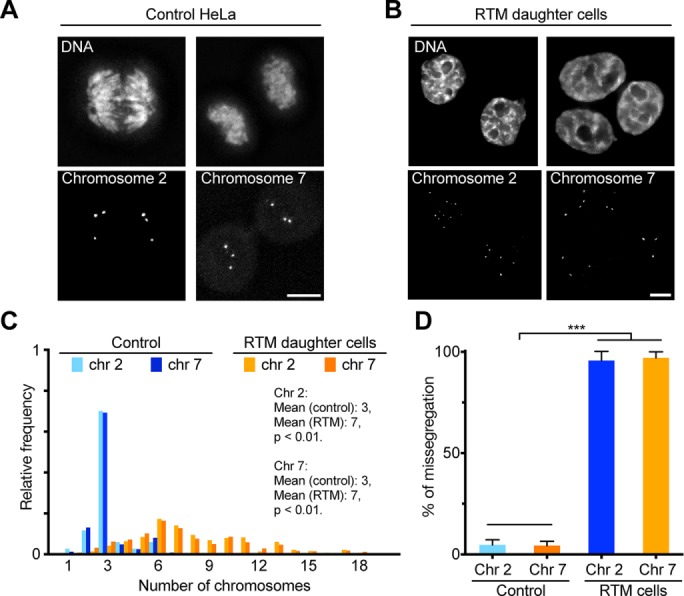
RTM in polyploid cells correlates with high missegregation rate. (A, B) Representative micrographs of HeLa cells labeled with FISH probes for chromosome 2 and 7. Cells were synchronized, treated with DMSO (A) or SU6656 (B) for 48 h, and then cultured in drug-free medium for 20 h. Cells were fixed and hybridized with probes specific for centrometric satellite DNA of chromosome 2 or 7 and stained with Hoechst to visualize DNA. Scale bar, 10 μm. (C) Histogram showing the relative frequency of cells with the indicated number of chromosomes (>480 cells). (D) Percentage of anaphase/telophase cells that have an unequal distribution of the indicated chromosomes, represented as mean ± SD from three independent experiments (200 cells total). ***p < 0.001.
iEC daughter cells proliferate and reduce genome content over successive divisions
High CIN during polyploid mitosis results in massive aneuploidy in the daughter cells. One prevailing theory is that polyploid cells can return to a diploid state after only one cell division, a process called ploidy reversal (Duncan et al., 2010). To test whether iECs that resumed mitosis reduced their ploidy, we used FACS to isolate polyploid iECs (≥8C) and then recultured them in drug-free medium. The iECs resumed cell division, and their daughters also continued to divide, although they grew more slowly than control cells (Figure 7A). Over these subsequent divisions, the cells reduced their ploidy (Figure 7B), and by day 10–14, their DNA profile was similar to that of control HeLa cells (Figure 1B), indicating that the ploidy of iEC daughter cells continues to decline over successive cell divisions. One concern was that a small percentage of cells with 2C/4C DNA content might be present in the 8C population after sorting and that these cells grew while the polyploid cells died. To evaluate this possibility, we used automated microscopy to analyze colonies of cells by imaging the cells daily over a 9-d period. We found that 42% of the polyploid cells divided and grew into individual colonies (Figure 7C). Together our results suggest that 8C iECs and their daughters can proliferate, and, contrary to previous models, iECs reverse their ploidy gradually over multiple divisions.
FIGURE 7:

Polyploid cells can resume cell proliferation and reduce their genome content over time. (A, B) HeLa cells were treated with SU6656 for 48 h and then cultured in drug-free medium for various amounts of time before the cells were counted or processed for FACS. (A) Quantification of cell density at selected days after drug washout. Each point represents mean ± SD from four independent experiments. (B) Quantification of FACS data from four independent experiments showing the percentage of cells with different ploidy after drug washout on the indicated days. For each experiment, 5000 cells were measured for DNA content, and the mean ± SD is graphed. (C, D) HeLa GFP–CENP-A cells were synchronized, treated with SU6656 for 48 h, and imaged at 1-d intervals (C) or fixed and stained every other day (D); t = 0 is time after drug washout. Selected frames are shown, and the time is marked in the upper left corner. Scale bar, 25 μm. (D) The dot plot shows the quantification of kinetochore number in individual cells on selected days after drug washout. The dots represent the number of kinetochores. The bars and whiskers indicate the mean ± SD; >120 total cells counted for controls and >240 for drug-treated cells. (E) Dot plot showing the quantification of the number of kinetochores in individual cells within a single colony of cells. The dots represent the number of kinetochores, and the bar and whiskers indicate the mean ± SD. **p < 0.01, ***p < 0.001.
To further address the process of ploidy reversal, we fixed cells on different days after drug withdrawal and quantified kinetochore number (Figure 7D). Consistent with our DNA content analysis, we found that the average kinetochore number in iEC daughter cells gradually decreased over successive divisions from 366 to 120 kinetochores/cell. We further examined the heterogeneity of this ploidy reduction by quantifying kinetochore number per cell among sister cells in single colonies. This analysis showed that sister cells from the same iEC mother cell have a large variance in the numbers of kinetochores (Figure 7E), further indicating that iEC return to mitosis results in high levels of unequal genome content among daughter cells.
HSET overexpression reduces the extent of mitotic defects in iECs
In Drosophila endocycling cells, transcription is down-regulated for many mitotic genes, including spindle assembly factors, such as the Drosophila kinesin-14 orthologue non-claret disjunctional (ncd; Maqbool et al., 2010). The human kinesin-14, HSET, is important for clustering of multiple centrosomes in cells that have undergone centrosome amplification (Kwon et al., 2008). We hypothesized that down-regulation of HSET in iECs could be responsible for the increase in the observed multipolar spindles. To ask whether HSET protein is down-regulated in mitotic iECs, we synchronized HeLa cells with a single thymidine block, treated cells with DMSO or SU6656 for 48 h, released the cells into medium containing nocodazole, incubated for an additional 14 h to accumulate cells in mitosis, and performed Western blot analysis. HSET is highest during mitosis, as evidenced by the increased HSET signal in nocodazole-treated cells, but its levels were much lower in nocodazole-treated HeLa iECs (Figure 8A). To ask whether overexpression of HSET can rescue spindle multipolarity during RTM, we used a HeLa cell line that overexpresses GFP-HSET to various levels. We found that mitotic polyploid cells with overexpressed HSET had fewer poles than did control mitotic iECs (Figure 8B), suggesting that HSET overexpression reduces the extent of spindle multipolarity. Moreover, immunofluorescence analysis of GFP-HSET expression in mitotic iECs revealed that HSET protein levels were negatively correlated with the number of spindle poles (Figure 8, C and D). Consistent with this observation, levels of HSET during RTM in nocodazole-treated iEC daughter cells were restored to levels in control nocodazole-treated cells (Supplemental Figure S4A). These results suggest that deficits in HSET contribute to high levels of multipolar spindles during iEC RTM.
FIGURE 8:

HSET overexpression partially rescues mitotic defects during RTM in polyploid cells. (A) Western blot of HeLa cells treated with DMSO (control) or SU6656 with washout for RTM and probed with anti-HSET (top) or anti-tubulin (bottom) antibodies. (B) Quantification of the number of spindle poles in either HeLa control or GFP-HSET–overexpressing iECs at 12 h after drug washout. Bars indicate the mean ± SD from three independent experiments in which at least 300 cells were scored in each condition. (C) Representative micrographs showing the inverse relationship between GFP-HSET intensity and number of spindle poles in different cells. Microtubule (MT) labeling (top) and level of GFP-HSET (bottom). Scale bar, 20 μm. (D) Average fluorescence intensity of GFP-HSET relative to number of poles from 116 cells from three independent experiments. (E, F) HeLa cells or GFP-HSET–overexpressing HeLa cells were synchronized, treated with SU6656 for 48 h, and cultured in drug-free medium for 20 h. Cells were then fixed and stained with probes specific for centrometric satellite DNA of chromosome 2 or 7 and stained with Hoechst to visualize DNA. (E) Histogram showing the relative frequency of the number of chromosomes (>450). (F) Percentage of anaphase/telophase cells that have an unequal distribution of the indicated chromosomes represented as mean ± SD from three independent experiments (200 total cells counted). *p < 0.05, **p < 0.01, ***p < 0.001.
To address whether HSET overexpression reduces aberrant divisions in polyploid cells, we used time-lapse microscopy to visualize cell division of HeLa iECs expressing GFP-HSET after drug washout (Supplementary Figure 4B and Supplemental Video S6). To follow the fate of a larger number of cells, we used high-throughput microscopy of HeLa iECs expressing GFP-HSET at 9–16 h after drug washout, and which showed that ∼50% of these cells entered mitosis. Of those cells, 28% successfully completed division, with the majority of them (85%) dividing into two daughter cells (Supplemental Figure S4C), similar to what we previously observed for iEC RTM. The distribution of fates of the cells that failed division was also similar between polyploid HeLa cells that did or did not overexpress GFP-HSET (Supplemental Figure S3C).
It was shown previously that multipolar spindles that cluster their centrosomes cause an increase in merotelic attachments, leading to defects in chromosome segregation (Ganem et al., 2009; Thompson and Compton, 2011). To ask whether HSET overexpression altered chromosome missegregation, we used FISH to analyze the distribution of chromosomes 2 and 7 (Supplemental Figure S4, D and E). We found that there was no significant difference in the mean number of these chromosomes per daughter cell between GFP-HSET–expressing and control HeLa cells during RTM (Figure 8E). Surprisingly, there was a small but statistically significant reduction (p < 0.01) in the heterogeneity of the distribution of these two chromosomes among daughter cells (Figure 8E). When looking at the distribution of chromosomes between pairs of daughters from the same division, we found that HSET overexpression resulted in a small but statistically significant reduction in the percentage of cells with missegregated chromosomes 2 and 7 (Figure 8F, p < 0.01). Taken together, these results support the idea that overexpression of HSET partially rescues multipolarity and chromosome missegregation defects during iEC RTM but that down-regulation of other genes in iECs must also contribute to aneuploidy generation in daughter cells, since GFP-HSET overexpression was not sufficient to fully restore spindle bipolarity or chromosome segregation to control levels.
DISCUSSION
Our results show that transient endoreplication generates polyploid (≥8C) cells that can return to error-prone division, which can cause severe genome instability by spawning proliferative aneuploid cells. Although polyploid iECs assemble multipolar spindles that begin to undergo multipolar anaphase during RTM, frequently there are lagging chromosomes in the cytokinetic furrow, which result in a failure in cytokinesis. This can lead to two daughter cells with very different genome contents. iECs returning to mitosis had lower levels of HSET, and its overexpression partially rescued spindle multipolarity in dividing iECs, consistent with its known role in centrosome clustering. However, HSET expression led to only a modest reduction in CIN, which suggests that additional down-regulated genes in iECs likely contribute to CIN during RTM. Taken together, our studies provide a model for understanding the mechanisms by which alternative cell cycles lead to the development of polyploidy and how these error-prone divisions ultimately lead to genomic instability.
Cell cycle perturbation can induce polyploidy
The responses of cells to cell cycle perturbation are highly heterogeneous, including cell cycle arrest, mitotic slippage, or cell death. For example, DNA-damaging agents, cell cycle kinase inhibitors, and antimicrotubule agents can induce polyploidization or mitotic slippage (Roberts et al., 1990; Verdoodt et al., 1999; Erenpreisa et al., 2005, 2008; Puig et al., 2008). It is clear from our work that cells can be induced into variant cell cycles, resulting in high ploidy with a low level of cell death. Recent studies indicate that mitotic failure is usually followed by p53-dependent cell cycle arrest or cell death through Hippo pathway activation (Ganem and Pellman, 2012; Ganem et al., 2014). In contrast, we found that p53+ HCT-116 cells underwent endoreplication upon RO3306 or SU6656 treatment rather than becoming arrested at a tetraploid state, indicating that endoreplication may be an alternative by which cells avoid mitotic catastrophe. In addition, our data demonstrate that most cell death occurs after iECs return to mitosis rather than upon the initial treatment with the drug. Endocycling cells can have a higher apoptosis threshold (Zheng et al., 2012) and tolerate high levels of DNA damage (Ullah et al., 2008, 2011; Davoli and de Lange, 2011). In Drosophila, naturally occurring and induced endocycling cells do not apoptose in response to genotoxic stress, because they repress the p53 pathway (Mehrotra et al., 2008; Hassel et al., 2014; Zhang et al., 2014). Furthermore, the DNA damage response network is up-regulated to prevent damage responses in induced endoreplicating mouse embryonic fibroblasts, which may also explain how endocycling cells can have a higher apoptotic threshold (Zheng et al., 2012). Together these studies support the idea that variant cell cycles, such as those that occur with endoreplication, may be an important but underappreciated contributor to cell survival and genomic instability.
One unanswered question is how cells choose to undergo the endocycle versus endomitosis. In some cases, this is a developmental choice. For example, trophoblast giant cells in mammals and salivary gland cells in Drosophila both strongly repress CDK1 function and have oscillations of Cdk2/cyclin E alternating with APC/Cdh1 (Palazón et al., 1998; Hattori et al., 2000; Edgar and Orr-Weaver, 2001). In contrast, megakaryocytes become polyploid through endomitosis (Jackson, 1990) due, at least in part, to lowered levels of Cdk1/cyclin B (Ravid et al., 2002). Our finding that high concentrations of RO3306 induced endocycles whereas lower concentrations induced endomitosis suggests that Cdk1 is an important mediator in the choice between endocycle and endomitosis. Together these results support the idea that endocycle and endomitosis are variations on a continuum with different degrees of repression of the mitotic kinase oscillators.
Down-regulation of key mitotic players may lead to defective chromosome segregation and cytokinesis
The kinesin-14 HSET is essential for centrosome clustering in cells with centrosome amplification (Kwon et al., 2008). In tetraploid cells, clustering centrosomes into two poles leads to defects in cell division due to improper kinetochore–microtubule attachments that form on the transient multipolar spindles (Ganem et al., 2009). Our data support the idea that polyploid cells have increased levels of multipolarity and a failure in centrosome clustering compared with dividing tetraploid cells, potentially amplifying the extent of kinetochore–microtubule malattachments (Fujiwara et al., 2005). Consistent with this idea, we found that HSET was down-regulated in iECs and that HSET overexpression reduced the extent of spindle multipolarity. Furthermore, our observations suggest that the increase in malattached kinetochores in polyploid cells may lead to increased lagging chromosomes that result in cytokinesis failure. Indeed, the kinesin-14 ncd was among the genes down-regulated in Drosophila endocycling cells, and our present findings support the idea that the human kinesin-14, HSET, may also be an important player in the fidelity of chromosome segregation during RTM of polyploid cells. Previous studies also showed that a similar group of mitotic genes are down-regulated in Drosophila and mouse endocycling cells (Maqbool et al., 2010; Chen et al., 2012; Pandit et al., 2012). An attractive model is that this lower level of mitotic gene expression contributes to the multiple defects in spindle formation, chromosome segregation, and cytokinesis when endoreplicating cells return to mitosis.
One interesting observation is that upon RTM, the iECs return to a chromosomal number similar to that of the initial starting population, suggesting that these polyploid cells undergo divisions that reduce genome content. Such reductive genomic divisions were first described in tetraploid fibroblasts and have subsequently been found in various cell types (Duncan et al., 2010; Fox et al., 2010; Hassel et al., 2014; Schoenfelder et al., 2014). Previous studies concluded that multipolar cell divisions in mouse polyploid hepatocytes resulted in one-step ploidy reversal to progeny with halved or quartered genome content (Duncan et al., 2010). Consistent with this observation, we found that our iECs feature multipolar spindles and undergo a multipolar anaphase. However, due to cytokinesis failure, iEC RTM usually resulted in two or three daughter cells of different sizes and DNA contents. Kinetochore analysis of the first-generation progeny in these cells showed highly variable levels of chromosomes in the resulting daughter cell nuclei, suggesting that ploidy reversal is not a one-step process in the iECs. In addition, our FACS data suggest that ploidy reversal occurs over successive cell divisions, ultimately resulting in a DNA content that is similar to that in control cells. Of interest, it was recently shown in an analysis of tumors with numerical CIN that both diploid and tetraploid chromosomal populations converged on a near-triploid DNA content, suggesting that there is likely some type of selective advantage to maintaining a certain level of aneuploidy (Laughney et al., 2015).
Consequence of CIN after transient endoreplication
A potential shortcoming of genome reductive mitosis is that mitotic cells do not have enough genome content to buffer the deleterious effects of the massive aneuploidy that occurs in these divisions. Multipolar cell divisions in cells with excess centrosomes typically result in low viability of the progeny (Ganem et al., 2009). In contrast, the kinetochore distribution analysis of our polyploid cells suggests that progeny from these divisions have a larger number of kinetochores and thus likely have a higher probability to have at least one complete set of chromosomes. In addition, the high cytokinesis failure rate during RTM in polyploid cells results in only two or three daughter cells, decreasing the probability of cells containing incomplete chromosomal complements incompatible with life. Our studies also show that iEC daughters undergo multiple rounds of division to ultimately reduce the genome, but the fraction of cells that ultimately survive is not known. A future goal will be to assess the precise mechanisms by which polyploid cells return to a near-triploid state.
Aneuploidy after RTM in polyploid cells after transient endoreplication may lead to karyotype evolution and selective cellular advantage. Our FISH analysis of chromosomes 2 and 7 demonstrated that the chromosomes are not evenly distributed into daughter cells during polyploid divisions, suggesting that these cells have high levels of CIN and a different chromosome composition from the original cell population. It has been proposed that high levels of CIN allow for selective advantages in the subsequent daughter cells due to the selection of certain favorable chromosomal complements (Duncan, 2013). In contrast, error-prone divisions in tetraploid cells usually lead to loss or gain of only a few chromosomes in daughter cells, resulting in subclones with accumulated mutations (Storchova and Pellman, 2004; Fujiwara et al., 2005). A more striking example is that the aneuploid daughters that arise after polyploid divisions in mouse hepatocytes quickly adapt to external stress (Duncan et al., 2012). A future goal will be to understand whether and how these polyploid divisions lead to favorable karyotypes and the underlying molecular mechanisms that govern this selection.
Polyploid mitosis could contribute to cancer relapse
CIN followed by selection of aneuploid cells is proposed to play a major role in tumorigenesis (Storchova and Pellman, 2004; Fujiwara et al., 2005; Weaver et al., 2007; Williams et al., 2008; Davoli and de Lange, 2011; Varetti and Pellman, 2012; Silk et al., 2013). Our data suggest that transient iECs are a potent contributor to CIN and aneuploidy, suggesting that transient iECs may contribute to tumorigenesis. It is well known that many cancers are polyploid and that polyploidy often correlates with poor prognosis (Davoli and de Lange, 2011), but it is not clear what the mechanistic link is between these observations. Some studies suggest that polyploidy invariably leads to senescence or apoptosis (Verdoodt et al., 1999), consistent with our observation that a fraction of polyploid cell divisions result in cell death. It is well established that many chemotherapeutic agents target the cell cycle, resulting in a failure in cell cycle progression and a triggering of apoptosis. However, we propose that a fraction of the cells escape apoptosis and instead enter the alternative cell cycle state of endocycle or endomitosis (Figure 9). When cells undergo endoreplication, they are resistant to genotoxic stress, such as would be caused by radiation therapy. Thus a transient switch to endoreplication could provide a mechanism for therapy resistance. On discontinuation of the chemotherapy, these transient iECs would be competent to resume mitotic divisions, which are highly error-prone. In this sense, error-prone RTM would elevate rates of genome instability, with microselection of proliferative aneuploid daughters contributing to tumor regrowth. Taken together, our findings support the idea that a return to mitosis by polyploid iECs may be an underappreciated aspect of tumorigenesis and provide a novel inroad into the identification of new therapeutic strategies.
FIGURE 9:

Model. After treatments that perturb cell cycle progression, a fraction of the cells escape apoptosis and instead enter the alternative cell cycle state of endoreplication (endocycle/endomitosis). Switching to endoreplication may provide a mechanism for therapy resistance. On discontinuation of the perturbation, many cells die, but a fraction of these polyploid cells resume mitotic divisions, which are highly error-prone, contributing to tumor heterogeneity and leading to cancer relapse.
MATERIALS AND METHODS
Cell culture and drug treatment
Breast cancer cells (MDA-MB-231), cervical cancer cells (HeLa), colon carcinoma cells (HCT-116), and human RPE-1 cells (hTERT-RPE-1) were grown in DMEM-complete (DMEM [Invitrogen, Carlsbad, CA] supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and 50 mg/ml penicillin/streptomycin [Invitrogen]) at 37°C and 5% CO2. HeLa cells expressing GFP–CENP-A and GFP–γ-tubulin, HeLa cells expressing GFP-H2B, and HeLa cells expressing GFP-HSET were constructed as described previously (Cai et al., 2009).
The cell cycle kinase inhibitors RO3306 (Invitrogen) and SU6656 (Cayman Chemical, Ann Arbor, MI) were dissolved in tissue culture–grade DMSO and used at a final concentration of 3 or 6 μM, with the final concentration of DMSO in the medium at no more than 0.15%. Thymidine (Sigma-Aldrich, St. Louis, MO) was used at a final concentration of 2 mM for thymidine block, and monastrol was used at a final concentration of 100 μM overnight.
Live-cell imaging
For analyzing the response of cells to drug treatments, HeLa cells, HeLa cells expressing GFP-HSET, or MDA-MB-231 cells were plated at cell densities of 4 × 104 cells/ml in MatTek plates (MatTek, Ashland, MA) and incubated at 37°C and 5% CO2 for 24 h to allow the cells to attach to the plate. Cells were synchronized with 2 mM thymidine for 24 h and then released into fresh medium containing 0.15% DMSO, 6 μM RO3306, or 6 μM SU6656. The MatTek plates were placed in a stage-top incubator with both temperature and CO2 control (Tokai Hit, Fujinomyiya-shi, Japan) and imaged by phase contrast microscopy at 20-min intervals overnight starting at the time of drug addition. To visualize chromosome behavior in HeLa GFP-H2B cells with SU6656 treatment, cells were plated and treated as described, but then the MatTek plates were placed in stage-top incubator (Tokai Hit) and imaged with a spinning-disk confocal at 5-min intervals overnight starting at 12 h after drug addition.
To visualize cells during RTM, HeLa cells or HeLa cells expressing GFP-HSET were plated at cell densities of 4 × 104 cells/ml in MatTek plates and incubated at 37°C and 5% CO2 for 24 h. Cells were then synchronized with 2 mM thymidine for 24 h, released into fresh medium with 0.15% DMSO, 6 μM RO3306, or 6 μM SU6656, and incubated for 48 h, rinsed twice in drug-free DMEM-complete, and then incubated with drug-free DMEM-complete. The MatTek plate was then placed in a stage-top incubator and imaged using phase contrast microscopy as described. For long-term imaging, cells were treated as before, except that they were plated at 4000 cells/ml and then imaged at a 4- to 6-h intervals using the EVOS FL Auto Digital Inverted Fluorescence System for 12–14 d.
For high-throughput live-cell microscopy, HeLa GFP-H2B cells, HeLa GFP-HSET, or MDA-MB-231 GFP-H2B cells were plated in 96-well Falcon imaging plates (BD, Bedford, MA) at 4000 cells/well and incubated at 37°C and 5% CO2 for 24 h. Cells were then synchronized with 2 mM thymidine for 24 h, incubated with 0.15% DMSO or 6 μM SU6656 for 48 h, rinsed twice in drug-free DMEM-complete, and incubated with drug-free DMEM-complete. At 7 h after drug washout, cells were imaged with a BD Pathway 855 at 5- or 10-min intervals overnight for 9 h. Cell fate profiles were analyzed using the BD AttoVision software by recording cell morphology from frame to frame.
To visualize kinetochore dynamics during RTM, HeLa cells expressing GFP–CENP-A and GFP–γ-tubulin were plated in 35-mm MatTek plates at 4 × 104 cells/ml and incubated at 37°C and 5% CO2 for 24 h. Cells were then synchronized with 2 mM thymidine for 24 h, incubated in DMEM-complete with 0.15% DMSO or 6 μM SU6656 for 48 h, rinsed twice in drug-free DMEM-complete, and incubated with drug-free DMEM-complete. At 7 h after drug washout, cells were imaged at 5-min intervals using a CV1000 (Olympus Life Science, Waltham, MA) for ∼10 h. For quantification of kinetochore number, the kinetochores were counted after anaphase/telophase when the cells reflattened so that there was better resolution of individual kinetochores. In addition, note that the number of z-sections captured does not encompass the total cell volume, so that the overall kinetochore numbers are less than those quantified from fixed-cell images. For the control cells and for iEC daughter cells, the kinetochores were counted in daughter cells immediately after division. For the iEC kinetochore number, we summed the total number of kinetochores from the two daughter cells.
Immunofluorescence and Western blot analysis
For mitotic stage analysis, HeLa cells, HeLa GFP-HSET cells, or MDA-MB-231 cells were plated on six-well plates with coverslips at 4 × 104 cells/ml and incubated at 37°C and 5% CO2 for 24 h. Cells were synchronized with 2 mM thymidine for 24 h, treated with 6 μM SU6656 for 12–14 h, and then processed for immunofluorescence. Cells were rinsed with 37°C phosphate-buffered saline (PBS; 12 mM phosphate, 137 mM NaCl, 3 mM KCl, pH 7.4) and then fixed in −20°C methanol for 5 min at room temperature and rehydrated in TBS-Tx (20 mM Tris, 150 mM NaCl, pH 7.5, 0.1% Triton X-100) for 2 min. Cells were then blocked in Abdil-Tx (TBS-TX, 2% bovine serum albumin, and 0.1% NaN3) for 30 min at room temperature or overnight at 4°C. Cells were stained with antibodies against α-tubulin DM1α (1:1000; Sigma-Aldrich) and diluted in Abdil-Tx for 30 min at room temperature. Cells were subsequently stained with 1.0 μg/ml donkey anti-rabbit Alexa Fluor 488 (Invitrogen) or donkey anti-mouse Texas Red (Jackson ImmunoResearch Laboratories, West Grove, PA). Fixed cells were stained with 2 μg/ml Hoechst (Sigma-Aldrich) for 5 min in TBS-TX to visualize DNA, mounted in ProLong Gold (Invitrogen), sealed with nail polish, and stored at −20°C until use.
To visualize spindle structures during return to mitosis, HeLa GFP-CENP-A and GFP-γ-tubulin cells were plated on six-well plates with coverslips at 4 × 104 cells/ml and incubated at 37°C and 5% CO2 for 24 h. Cells were synchronized with 2 mM thymidine for 24 h, treated with 6 μM SU6656 for 48 h, washed twice with drug-free DMEM, and incubated in drug-free DMEM-complete for 12 h. At 12 h after drug washout, cells were rinsed with MTSB buffer (4 M glycerol, 100 mM 1,4-piperazinediethanesulfonic acid, pH 6.8, 1 mM ethylene glycol tetraacetic acid, 5 mM MgCl2) for 2 min, extracted with MTSB plus 0.5% Triton X-100 for 2 min, and rinsed with MTSB buffer again for 2 min. Cells were then fixed with 1% glutaraldehyde in PBS for 10 min at room temperature and quenched by two rinses of 10 min each in freshly prepared 0.5 mg/ml NaBH4 in PBS buffer and processed for immunofluorescence as described.
For Western blot, HeLa cells were plated at cell density of 4 × 104 cells/ml in 10-cm culture dishes and incubated at 37°C and 5% CO2 for 24 h to allow the cells to attach to the plate bottom. Cells were then incubated with fresh medium with 0.15% DMSO or 6 μM SU6656 and incubated for 48 h. To collect unsynchronized/mitotic cells, cells were rinsed twice in drug-free DMEM and incubated with drug-free DMEM-complete or treated with DMEM-complete with 100 ng/ml nocodazole overnight. To collect unsynchronized/mitotic iEC daughters, cells were rinsed in drug-free DMEM twice, incubated with drug-free DMEM-complete for 7 d, and then treated with 100 ng/ml nocodazole overnight. HeLa cells were collected, counted, lysed in RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1.0% NP-40, 0.1% SDS, 0.1% Triton X-100, phenylmethylsulfonyl fluoride, and 10 μg/ml LPC [10 μg/ml leupeptin, pepstatin, and chymostatin]), and sonicated to shear the DNA. Lysed cells were then mixed with 2× sample buffer (4% SDS, 20% glycerol, 0.12 M Tris, pH 6.8, 1% bromophenol blue, and 4% β-mercaptoethanol) at a final cell density of 2500 cells/μl and then loaded on a 10% SDS–polyacrylamide gel and transferred to BioTrace-NT nitrocellulose (Pall Life Sciences, New York, NY). The blots were blocked with 5% nonfat dry milk in TBS-T (20 mM Tris, 150 mM NaCl, pH 7.5, 0.1% Tween-20), probed with anti-HSET (2.5 μg/ml; Cai et al., 2009) and DM1α (1:10,000; Sigma-Aldrich) in AbDil-T (20 mM Tris, 150 mM NaCl, pH 7.5, 2% bovine serum albumin, 0.1% Tween-20, 0.1% NaN3), followed by a mixture of sheep anti-mouse immunoglobulin G (IgG) horseradish peroxidase (HRP)–linked whole antibody (1:10,000; Amersham, Piscataway, NJ) and donkey anti-rabbit IgG HRP-linked whole antibody (1:5000; Amersham) in 5% nonfat dry milk in TBS-T and detected with SuperSignal West Pico enhanced chemiluminescence substrates (Thermo Fisher Scientific, Waltham, MA) according to standard protocols. Alternatively, the blots were first probed with anti-HSET (2.5 μg/ml), followed by donkey anti-rabbit IgG HRP-linked whole antibody (1:5000), and detected as described above. The blots were then stripped with Restore Western Blot Stripping Buffer (Thermo Fisher Scientific) and probed with DM1α (1:10,000) and sheep anti-mouse IgG HRP-linked whole antibody (1:5000) and detected as described.
Nuclear size measurements
To measure nuclear size, HeLa or MDA-MB-231 cells were plated in 96-well imaging plates (BD Falcon, San Jose, CA) at 4000 cells/well and incubated at 37°C and 5% CO2 for 24 h to allow the cells to attach to the wells. Cells were then incubated with 0.15% DMSO or 6 μM SU6656 for 48 or 72 h. Cells were then washed with PBS twice and stained with DRAQ5 (20 μM in sterile H2O). Cells were imaged with a BD Pathway 855 high-content bioimager at 37°C and 5% CO2.
Flow cytometry
Cells were plated in 10-cm culture dishes (BD Biosciences) at 4 × 104 cells/ml and incubated at 37°C and 5% CO2 for 24 h to allow the cells to attach to the plate bottom. Cells were then incubated with 0.15% DMSO, 3/6 μM RO3306, or 3/6 μM SU6656 for 48 or 72 h. To harvest cells for flow cytometry, cells were released with 0.25% trypsin-EDTA (Thermo Fisher Scientific) and rinsed with cold PBS. Cells were then fixed in 70% cold ethanol and stained with freshly prepared PI stain (20 μg/ml propidium iodide, 1% Triton X-100, and 0.2 mg/ml DNase-free RNase A in PBS). DNA content in fixed HeLa, MDA-MB-231, HCT-116, and RPE-1 cells was evaluated by standard methods with a BD Calibur flow cytometer (BD Biosciences). The percentage of cells at a certain ploidy was quantified as percentage of cells in the area under the individual peaks. Although most cells fall into discrete bins of 2C, 4C, 8C, and so on, there are cells with intermediate DNA content, which are not included in the data plotted as bar graphs.
FISH
For analyzing chromosome segregation, HeLa cells or HeLa cells expressing GFP-HSET were plated at a cell density of 4 × 104 cells/ml on coverslips in a six-well plate and incubated at 37°C and 5% CO2 for 24 h to allow the cells to attach to the coverslips. Cells were then synchronized with 2 mM thymidine for 24 h and released into fresh medium with 0.15% DMSO or 6 μM SU6656 for 48 h. The cells were washed with drug-free DMEM twice and incubated in DMEM-complete for ∼12 h. Cells on coverslips were washed twice with PBS, fixed with 3:1 methanol:acetic acid for 10 min, and washed with fix buffer for 2 min. Coverslips were then pretreated with 2× SSC (15 mM Na3 citrate, 150 mM NaCl), pH 7.0, and 0.5% NP-40 at 37°C for 15 min. Coverslips were dehydrated by sequential washes in 70, 85, and 100% ethanol at 2 min per wash. Coverslips were hybridized with centromeric FISH probes specific for chromosome 2 or 7 (1:10 dilution in hybridization buffer; Cytocell, Tarrytown, NY), adhered to a fresh slide with nail polish, incubated in a hybridization oven at 75°C for 10 min, and transferred to a 37°C humidified incubator for overnight incubation. The next day, the nail polish was removed and the coverslips were washed with wash buffer II (2× SSC, 0.4% NP-40) for 2 min at room temperature, with wash buffer I (0.4× SCC, 0.3% NP-40) for 2 min at 72°C, and with wash buffer II for 1 min at room temperature. Coverslips were then dehydrated with 70, 85, and 100% ethanol washes at 1 min per wash. After the coverslips dried, the cells were stained with 4′,6-diamidino-2-phenylindole (DAPI; 2 μg/ml in TBS-Tx) for 1 min, mounted with ProLong Gold (Invitrogen), and sealed with nail polish.
Imaging and statistical analysis
Images for kinetochore distribution analysis, HSET intensity analysis, and FISH were acquired on a Nikon Eclipse 90i, using a 40× (numerical aperture [NA] 1.0) Plan Apo or 100× (NA 1.4) Plan Apo VC objective and a CoolSnap HQ charge-coupled device (CCD) camera (Photometrics, Tucson, AZ). The camera and filters were controlled by MetaMorph (Molecular Devices, Sunnyvale, CA). Image stacks were collected at 0.2-μm steps through the whole cell volume and then deconvolved using Autodeblur (Autoquant Imaging, Troy, NY) for 10 or 20 iterations.
To quantify kinetochore number in individual cells, the image stacks obtained from microscopy were imported into Imaris three-dimensional (3D) imaging software (Bitplane Scientific, Concord, MA). The number of kinetochores was detected after using the intensity threshold, which was set individually for each image. This process was repeated for at least 50 cells per experiment for three experiments. Data from Imaris were exported to Excel (Microsoft, Redmond, WA).
To measure the fluorescence intensity of GFP-HSET, after 3D reconstruction in FIJI, an equivalently sized box was drawn in the cytoplasm as the background and on the spindle for measurement. Background-subtracted average fluorescence intensity was correlated with the number of spindle poles and plotted as a scatter plot. This process was repeated for at least 50 cells per experiment for three experiments.
FISH signals were counted in mitotic cells at anaphase or telophase to determine the chromosome distribution and calculate missegregation rate. At least 100 cells were counted per experiment for three experiments. The histograms of the chromosome distribution were fit with a Gaussian distribution by Prism (GraphPad Software, La Jolla, CA).
Fixed mitotic iEC daughters for detailed spindle structure were imaged using the OMX 3D-SIM Super-Resolution system (GE Healthcare Life Sciences, Pittsburgh, PA) controlled by DV-OMX software (GE Healthcare Life Sciences). Images were captured at 0.125-μm step size with a UNIPLANAPO 100× (NA 1.4) objective, using NA 1.516 immersion oil. The 405-nm images were acquired for 40 ms at 1% laser strength, 488-nm images for 30–50 ms at 10% laser strength, and 561-nm images for 60 ms at 10% laser strength. Images were processed using SoftWoRx (GE Healthcare Life Sciences, Marlborough, MA) and IMARIS 3D imaging software.
High-throughput microscopy was performed on a BD Pathway 855 high-content bioimager with a Hamamatsu Orca ER cooled CCD camera controlled by BD AttoVision software. The distributions of nuclear sizes were determined by ImageJ from three independent experiments (at least 500 cells per experiment) and graphed using Prism.
Chromosome behavior in HeLa GFP-H2B cells after SU6656 treatment was imaged on a Nikon TE2000U inverted microscope with a 60× (1.4 NA) objective equipped with a Yokogawa CSU-10 spinning-disk confocal head controlled by Nikon Elements. Images were collected on a Cascade-II electron-multiplying CCD camera (Photometrics).
All images were assembled in Adobe Illustrator CS equivalently for control and experimental results. Statistical significance was determined with Student’s t test performed in Excel.
Supplementary Material
Acknowledgments
We thank Suozhi Qi, Michael Rotelli, Hailing Zong, and Stephanie Ems-McClung for comments on the manuscript and Jim Powers for assistance with the imaging. We also thank Christopher Laucius and Duane Compton for advice on the FISH experiments and Kimberly Freeman at Olympus Microscopes for assistance with the CV1000. Support for this project was provided by the Office of the Vice Provost for Research at Indiana University, the Indiana University Clinical Translational Sciences Institute, and National Institutes of Health Grants GM590618 to C.E.W. and GM113107 to B.R.C. and C.E.W. The Indiana University Light Microscopy Imaging Facility is supported in part by the Office of the Vice Provost for Research and by the College of Arts and Sciences.
Abbreviations used:
- CIN
chromosomal instability
- iEC
induced endoreplicating cell
- RTM
return to mitosis.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-03-0159) on August 3, 2016.
REFERENCES
- Agromayor M, Martin-Serrano J. Knowing when to cut and run: mechanisms that control cytokinetic abscission. Trends Cell Biol. 2013;23:433–441. doi: 10.1016/j.tcb.2013.04.006. [DOI] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake RA, Broome MA, Liu X, Wu J, Gishizky M, Sun L, Courtneidge SA. SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol Cell Biol. 2000;20:9018–9027. doi: 10.1128/mcb.20.23.9018-9027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai S, Weaver LN, Ems-McClung SC, Walczak CE. Kinesin-14 family proteins HSET/XCTK2 control spindle length by cross-linking and sliding microtubules. Mol Biol Cell. 2009;20:1348–1359. doi: 10.1091/mbc.E08-09-0971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celton-Morizur S, Desdouets C. Polyploidization of liver cells. In: Poon RYC, editor. Polyploidization and Cancer. New York: Springer; 2010. pp. 123–135. [DOI] [PubMed] [Google Scholar]
- Chen HZ, Ouseph MM, Li J, Pécot T, Chokshi V, Kent L, Bae S, Byrne M, Duran C, Comstock G, et al. Canonical and atypical E2Fs regulate the mammalian endocycle. Nat Cell Biol. 2012;14:1192–1202. doi: 10.1038/ncb2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comai L. The advantages and disadvantages of being polyploid. Nat Rev Genet. 2005;6:836–846. doi: 10.1038/nrg1711. [DOI] [PubMed] [Google Scholar]
- Davoli T, de Lange T. The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol. 2011;27:585–610. doi: 10.1146/annurev-cellbio-092910-154234. [DOI] [PubMed] [Google Scholar]
- Davoli T, de Lange T. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell. 2012;21:765–776. doi: 10.1016/j.ccr.2012.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Denchi EL, de Lange T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell. 2010;141:81–93. doi: 10.1016/j.cell.2010.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewhurst SM, McGranahan N, Burrell RA, Rowan AJ, Grönroos E, Endesfelder D, Joshi T, Mouradov D, Gibbs P, Ward RL, et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Disc. 2014;4:175–185. doi: 10.1158/2159-8290.CD-13-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan AW. Aneuploidy, polyploidy and ploidy reversal in the liver. Semin Cell Dev Biol. 2013;24:347–356. doi: 10.1016/j.semcdb.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Duncan AW, Hanlon Newell AE, Bi W, Finegold MJ, Olson SB, Beaudet AL, Grompe M. Aneuploidy as a mechanism for stress-induced liver adaptation. J Clin Invest. 2012;122:3307–3315. doi: 10.1172/JCI64026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan AW, Taylor MH, Hickey RD, Newell A. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 2010;467:707–710. doi: 10.1038/nature09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar BA, Orr-Weaver TL. Endoreplication cell cycles. Cell. 2001;105:297–306. doi: 10.1016/s0092-8674(01)00334-8. [DOI] [PubMed] [Google Scholar]
- Erenpreisa J, Ivanov A, Wheatley SP, Kosmacek EA, Ianzini F, Anisimov AP, Mackey M, Davis PJ, Plakhins G, Illidge TM. Endopolyploidy in irradiated p53-deficient tumour cell lines: persistence of cell division activity in giant cells expressing Aurora-B kinase. Cell Biol Int. 2008;32:1044–1056. doi: 10.1016/j.cellbi.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erenpreisa J, Kalejs M, Cragg M. Mitotic catastrophe and endomitosis in tumour cells: An evolutionary key to a molecular solution. Cell Biol Int. 2005;29:1012–1018. doi: 10.1016/j.cellbi.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Fox DT, Duronio RJ. Endoreplication and polyploidy: insights into development and disease. Development. 2013;140:3–12. doi: 10.1242/dev.080531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox DT, Gall JG, Spradling AC. Error-prone polyploid mitosis during normal Drosophila development. Genes Dev. 2010;24:2294–2302. doi: 10.1101/gad.1952710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–1047. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- Ganem NJ, Cornils H, Chiu S-Y, O’Rourke KP, Arnaud J, Yimlamai D, Thery M, Camargo FD, Pellman D. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell. 2014;158:833–848. doi: 10.1016/j.cell.2014.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. doi: 10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Pellman D. Linking abnormal mitosis to the acquisition of DNA damage. J Cell Biol. 2012;199:871–881. doi: 10.1083/jcb.201210040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gizatullin F, Yao Y, Kung V, Harding MW, Loda M, Shapiro GI. The Aurora kinase inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer Res. 2006;66:7668–7677. doi: 10.1158/0008-5472.CAN-05-3353. [DOI] [PubMed] [Google Scholar]
- Hassel C, Zhang B, Dixon M, Calvi BR. Induction of endocycles represses apoptosis independently of differentiation and predisposes cells to genome instability. Development. 2014;141:112–123. doi: 10.1242/dev.098871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori N, Davies TC, Anson-Cartwright L, Cross JC. Periodic expression of the cyclin-dependent kinase inhibitor p57(Kip2) in trophoblast giant cells defines a G2-like gap phase of the endocycle. Mol Biol Cell. 2000;11:1037–1045. doi: 10.1091/mbc.11.3.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CW. Megakaryocyte endomitosis: a review. Int J Cell Cloning. 1990;8:224–226. doi: 10.1002/stem.5530080405. [DOI] [PubMed] [Google Scholar]
- Klisch K, Bevilacqua E, Olivera L. Mitotic polyploidization in trophoblast giant cells of the alpaca. Cells Tissues Organs. 2005;181:103–108. doi: 10.1159/000091099. [DOI] [PubMed] [Google Scholar]
- Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, Pellman D. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008;22:2189–2203. doi: 10.1101/gad.1700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannutti BJ, Blake N, Gandhi MJ, Reems JA, Drachman JG. Induction of polyploidization in leukemic cell lines and primary bone marrow by Src kinase inhibitor SU6656. Blood. 2005;105:3875-3878. doi: 10.1182/blood-2004-10-3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughney AM, Elizalde S, Genovese G, Bakhoum SF. Dynamics of tumor heterogeneity derived from clonal karyotypic evolution. Cell Rep. 2015;12:809–820. doi: 10.1016/j.celrep.2015.06.065. [DOI] [PubMed] [Google Scholar]
- Maqbool SB, Mehrotra S, Kolpakas A, Durden C, Zhang B, Zhong H, Calvi BR. Dampened activity of E2F1-DP and Myb-MuvB transcription factors in Drosophila endocycling cells. J Cell Sci. 2010;123:4095–4106. doi: 10.1242/jcs.064519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrotra S, Maqbool SB, Kolpakas A, Murnen K, Calvi BR. Endocycling cells do not apoptose in response to DNA rereplication genotoxic stress. Genes Dev. 2008;22:3158–3171. doi: 10.1101/gad.1710208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazón LS, Davies TJ, Gardner RL. Translational inhibition of cyclin B1 and appearance of cyclin D1 very early in the differentiation of mouse trophoblast giant cells. Mol Hum Reprod. 1998;4:1013–1020. doi: 10.1093/molehr/4.11.1013. [DOI] [PubMed] [Google Scholar]
- Pandit SK, Westendorp B, Nantasanti S, van Liere E, Tooten PCJ, Cornelissen PWA, Toussaint MJM, Lamers WH, de Bruin A. E2F8 is essential for polyploidization in mammalian cells. Nat Cell Biol. 2012;14:1181–1191. doi: 10.1038/ncb2585. [DOI] [PubMed] [Google Scholar]
- Puig P-E, Guilly M-N, Bouchot A, Droin N, Cathelin D, Bouyer F, Favier L, Ghiringhelli F, Kroemer G, Solary E. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol Int. 2008;32:1031–1043. doi: 10.1016/j.cellbi.2008.04.021. [DOI] [PubMed] [Google Scholar]
- Ravid K, Lu J, Zimmet JM, Jones MR. Roads to polyploidy: the megakaryocyte example. J Cell Physiol. 2002;190:7–20. doi: 10.1002/jcp.10035. [DOI] [PubMed] [Google Scholar]
- Roberts JR, Allison DC, Donehower RC, Rowinsky EK. Development of polyploidization in taxol-resistant human leukemia cells in vitro. Cancer Res. 1990;50:710–716. [PubMed] [Google Scholar]
- Schoenfelder KP, Fox DT. The expanding implications of polyploidy. J Cell Biol. 2015;209:485–491. doi: 10.1083/jcb.201502016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder KP, Montague RA, Paramore SV, Lennox AL, Mahowald AP, Fox DT. Indispensable pre-mitotic endocycles promote aneuploidy in the Drosophila rectum. Development. 2014;141:3551–3560. doi: 10.1242/dev.109850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silk AD, Zasadil LM, Holland AJ, Vitre B, Cleveland DW, Weaver BA. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc Natl Acad Sci. 2013;110:E4134–E4141. doi: 10.1073/pnas.1317042110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol. 2004;5:45–54. doi: 10.1038/nrm1276. [DOI] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. Chromosome missegregation in human cells arises through specific types of kinetochore-microtubule attachment errors. Proc Natl Acad Sci. 2011;108:17974–17978. doi: 10.1073/pnas.1109720108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trakala M, Rodríguez-Acebes S, Maroto M, Symonds CE, Santamaría D, Ortega S, Barbacid M, Méndez J, Malumbres M. Functional reprogramming of polyploidization in megakaryocytes. Dev Cell. 2015;32:155–167. doi: 10.1016/j.devcel.2014.12.015. [DOI] [PubMed] [Google Scholar]
- Ullah Z, de Renty C, DePamphilis ML. Checkpoint kinase 1 prevents cell cycle exit linked to terminal cell differentiation. Mol Cell Biol. 2011;31:4129–4143. doi: 10.1128/MCB.05723-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullah Z, Kohn MJ, Yagi R, Vassilev LT, DePamphilis ML. Differentiation of trophoblast stem cells into giant cells is triggered by p57/Kip2 inhibition of CDK1 activity. Genes Dev. 2008;22:3024–3036. doi: 10.1101/gad.1718108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui T, Yoshida M, Abe K, Osada H, Isono K, Beppu T. Uncoupled cell cycle without mitosis induced by a protein kinase inhibitor, K-252a. J Cell Biol. 1991;115:1275–1282. doi: 10.1083/jcb.115.5.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varetti G, Pellman D. “Two” much of a good thing: telomere damage-induced genome doubling drives tumorigenesis. Cancer Cell. 2012;21:712–714. doi: 10.1016/j.ccr.2012.05.033. [DOI] [PubMed] [Google Scholar]
- Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, Chen L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci USA. 2006;103:10660–10665. doi: 10.1073/pnas.0600447103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdoodt B, Decordier I, Geleyns K, Cunha M, Cundari E, Kirsch-Volders M. Induction of polyploidy and apoptosis after exposure to high concentrations of the spindle poison nocodazole. Mutagenesis. 1999;14:513–520. doi: 10.1093/mutage/14.5.513. [DOI] [PubMed] [Google Scholar]
- Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385–392. doi: 10.1038/nrm3115. [DOI] [PubMed] [Google Scholar]
- Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Wheatley D. Growing evidence of the repopulation of regressed tumours by the division of giant cells. Cell Biol Int. 2008;32:1029–1030. doi: 10.1016/j.cellbi.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703–709. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang CZ, Wala J, Mermel CH, et al. Pan-cancer patterns of somatic copy number alteration. Nat Gen. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Mehrotra S, Ng WL, Calvi BR. Low levels of p53 protein and chromatin silencing of p53 target genes repress apoptosis in Drosophila endocycling cells. PLoS Genet. 2014;10:e1004581. doi: 10.1371/journal.pgen.1004581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Dai H, Zhou M, Li X, Liu C, Guo Z, Wu X, Wu J, Wang C, Zhong J, et al. Polyploid cells rewire DNA damage response networks to overcome replication stress-induced barriers for tumour progression. Nat Commun. 2012;3:815. doi: 10.1038/ncomms1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Lee CY, Johnson RL, Wichterman J, Huang R, DePamphilis ML. An image-based, high-throughput screening assay for molecules that induce excess DNA replication in human cancer cells. Mol Cancer Res. 2011;9:294–310. doi: 10.1158/1541-7786.MCR-10-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zybina DEV, Kudryavtseva MV, Kudryavtsev BN. Polyploidization and endomitosis in giant cells of rabbit trophoblast. Cell Tissue Res. 1975;160:525–537. doi: 10.1007/BF00225769. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.