

Abstract
Background & Aims
Interactions between mucosal cell types, environmental stressors, and intestinal microbiota contribute to pathogenesis in inflammatory bowel disease (IBD). Here, we applied metaproteomics of the mucosal–luminal interface to study the disease-related biology of the human colonic mucosa.
Methods
We recruited a discovery cohort of 51 IBD and non-IBD subjects endoscopically sampled by mucosal lavage at 6 colonic regions, and a validation cohort of 38 no-IBD subjects. Metaproteome data sets were produced for each sample and analyzed for association with colonic site and disease state using a suite of bioinformatic approaches. Localization of select proteins was determined by immunoblot analysis and immunohistochemistry of human endoscopic biopsy samples.
Results
Co-occurrence analysis of the discovery cohort metaproteome showed that proteins at the mucosal surface clustered into modules with evidence of differential functional specialization (eg, iron regulation, microbial defense) and cellular origin (eg, epithelial or hemopoietic). These modules, validated in an independent cohort, were differentially associated spatially along the gastrointestinal tract, and 7 modules were associated selectively with non-IBD, ulcerative colitis, and/or Crohn’s disease states. In addition, the detailed composition of certain modules was altered in disease vs healthy states. We confirmed the predicted spatial and disease-associated localization of 28 proteins representing 4 different disease-related modules by immunoblot and immunohistochemistry visualization, with evidence for their distribution as millimeter-scale microgeographic mosaic.
Conclusions
These findings suggest that the mucosal surface is a microgeographic mosaic of functional networks reflecting the local mucosal ecology, whose compositional differences in disease and healthy samples may provide a unique readout of physiologic and pathologic mucosal states.
Keywords: Inflammatory Bowel Disease, Mucosal, Networks, Ecology, Metaproteomics
Abbreviations used in this paper: ANOVA, analysis of variance; CD, Crohn’s disease; HBD, human β-defensin; HD5, human alpha defensin 5; HNP, human neutrophil peptide; HPLC, high-performance liquid chromatography; IBD, inflammatory bowel disease; IHC, immunohistochemistry; MALDI, matrix-assisted laser desorption/ionization; MFN, mucosal functional network; MLI, mucosal–luminal interface; MS/MS, tandem mass spectrometry; NLME, nonlinear mixed-effect model; PVCA, principal variance component analysis; TOF, time of flight; UC, ulcerative colitis; WGCNA, weighted correlation network analysis
Summary.
By using metaproteomics of the human colonic mucosal surface, we found evidence for a proteomic ecology of millimeter-scale protein networks distinguished by functional specialization and cell source predominance, and their relative abundance across colonic regions and in health vs quiescent inflammatory bowel disease.
The intestinal mucosa plays diverse and critical roles in nutrient uptake, host defense, and local and systemic endocrinology.1, 2, 3, 4 The functional state of the mucosa in health and disease can be affected profoundly by its interplay with environmental metabolic stressors and luminal intestinal microbiota.4, 5 Studying the mucosal–luminal interface (MLI) and how it is changed in disease is difficult because of the many dimensions of complexity of this ecosystem.6, 7 The analytic challenge is central to the pathogenesis of inflammatory bowel disease (IBD), which is a multifactorial process involving genetic susceptibility, environmental factors, and microbiota.8, 9
Metaproteomics is an emerging technology to address this challenge. The metaproteome of the mucosal surface is a composite of human and microbial products, skewed for proteins devoted to translation, energy, carbohydrate metabolism, and antimicrobial defense.10, 11, 12, 13, 14, 15, 16 Focusing on the metaproteome recovered by lavage from the MLI of healthy human subjects, we previously observed that the predominant feature is intersubject variation, with an additional significant intestinal segmental pattern of the surface metaproteome distinguishing the distal and proximal colon.10 A similar segmental difference was observed in foundational studies of the mucosal bacterial composition.17, 18 In view of emerging concepts of intestinal ecology, this suggests that the metaproteome reflects the human contribution to the mucosal habitat.19 With this foundation, this study applies metaproteomic analysis to characterize the disease-related features of the MLI in ulcerative colitis (UC) and Crohn’s disease (CD).
Materials and Methods
Mucosal Lavage Sample Collection and Analysis
The overall study design is shown in the flowchart in Figure 1. The demographics of the study population and the sample characteristics are summarized in Table 1. All enrolled subjects at both Cedars Sinai Medical Center and the University of California Los Angeles Ronald Reagan Medical Centers were consented to participate in research studies approved by the Institutional Review Board. CD and UC subjects were recruited from those undergoing surveillance colonoscopy; lavage samples were taken from mucosal sites that were endoscopically normal. Non-IBD control subjects were recruited from patients undergoing colonoscopy for colorectal screening. For each patient, 6 colonic regions were sampled and collected by endoscopic lavage.
Figure 1.

Flowchart of metaproteomic analytic pipeline.
Table 1.
Summary of Sample Collection and Clinical Characteristics
| Normal | UC | CD | |
|---|---|---|---|
| Total subjects | 17 | 13 | 21 |
| Total mucosal lavage samples | 81 | 75 | 101 |
| Sex | |||
| Female | 34 (42%) | 35 (47%) | 36 (36%) |
| Male | 47 (58%) | 40 (53%) | 65 (65%) |
| Age, median ± SD, y | 56 ± 13 | 60 ± 10 | 40 ± 10 |
| Region | |||
| Cecum | 14 (17%) | 12 (16%) | 8 (8%) |
| Ascending | 14 (17%) | 13 (17%) | 16 (16%) |
| Transverse | 14 (17%) | 13 (17%) | 19 (19%) |
| Descending | 15 (19%) | 11 (16%) | 19 (19%) |
| Sigmoid | 12 (15%) | 13 (17%) | 19 (19%) |
| Rectum | 12 (15%) | 13 (17%) | 20 (19%) |
All sample collections and processing followed the pre-analytic proteomic pipeline previously detailed.10 The demography of an independent control data set of 205 mucosal lavage samples from 38 non-IBD subjects, used as a validation cohort for this study, was reported previously.10
Protein Identification
Protein identification was performed by an in silico search, followed by two strategies: immunoprecipitation matrix-assisted laser desorption/ionization (MALDI) (when the antibodies against the target proteins were available), or high-performance liquid chromatography (HPLC) isolation followed by tandem mass spectrometry (MS/MS) fragmentation. An in silico search for putative protein identities was performed using the Empirical Proteomics Ontology Knowledge Base.20 When a specific antibody was available, the in silico identification was validated by immunoprecipitation of the original clinical isolate using an antibody specific to the protein of interest, and magnetic beads conjugated with either protein A or G (Invitrogen, Carlsbad, CA). After washing and desalting, specifically bound peptides were extracted with formic acid and mixed with matrix for spotting on a MALDI target. Protein identification was performed by confirming the expected m/z of the original peptide extracted by antibody, as well as examining the immunodepleted clinical sample to see if the same peptide/protein mass was reduced quantitatively.
In the second strategy, MALDI MS–defined peptides of interest were fractionated and isolated by HPLC. Purified peptides smaller than 10 kilodaltons were fragmented directly by LC-MS/MS coupled to a Nano2DLC pump (Eksigent, Dublin, CA) and LTQ-Orbitrap (Thermo Fisher Scientific, Waltham, MA), or by Ultraflex MALDI-TOF/TOF (time of flight) with LIFT technology (Bruker, Billerica, MA). Purified peptides larger than 10 kilodaltons were digested with trypsin in solution, or in a gel plug after running a sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and the fragments were analyzed by LC-MS/MS. Proteins were identified by searching against the SwissProt database using Mascot (Matrix Science Inc, Boston, MA), and only proteins with a P value less than .05 were included in the results.
Immunoblotting
Ten mucosal lavage samples from each patient group were selected randomly, including 5 from proximal and 5 from distal regions, and 50 μg protein was immunoblotted to ensure equal loading. A Tris-glycine gel system with 0.2-μm nitrocellulose membranes was used for proteins greater than 5 kilodaltons, and a tricine system with 0.1-μm Immobilon-PSQ membranes (followed by 25% glutaraldehyde fixation) was used for smaller proteins/peptides (Millipore, Billerica, MA; Invitrogen, Carlsbad, CA). Primary antibodies included rabbit anti–human neutrophil peptides (HNPs)1–3, rabbit anti-human alpha defensin 5 (HD5), rabbit anti–human β-defensin (HBD)1, rabbit anti-HBD2, rabbit antihepcidin (all gifts from Dr Tomas Ganz’s laboratory at the University of California Los Angeles). Purchased antibodies included mouse anti-Peptidase M20 Domain Containing 1 (PM20D1) (ab70916; Abcam, Cambridge, United Kingdom), and rabbit anti-transferrin (ab30525; Abcam). Secondary antibodies were horseradish peroxidase–conjugated goat anti-rabbit or goat anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA) developed with enhanced chemiluminescence (ECL) substrate (Pierce, IL), or alkaline phosphatase–conjugated goat anti-rabbit IgG antibody (Jackson ImmunoResearch) developed with BCIP (5-bromo-4-chloro-3-indolyl-phosphate)/NBT (nitro blue tetrazolium) substrate (MP Biomedicals, Santa Ana, CA). For quantitation, blots were digitized and pixels were quantitated by Adobe Photoshop (Adobe, San Jose, CA). Each pixel count was normalized by dividing it with the background pixel count.
Immunohistochemistry
To examine the cross-sectional histology of human mucosa, microtome sections of paraffin tissues were obtained from an independent non-IBD human cohort, and stained by immunohistochemistry with primary antibody and developed by VECTASTAIN Elite ABC Kit (Vector Lab, Burlingame, CA) as previously described.21 The same antibodies used in immunoblotting also were used in immunohistochemistry (IHC), with the exception that the antihepcidin antibody was replaced by an antiprohepcidin antibody (gifts from Dr Tomas Ganz's laboratory). To examine whole-mounts of intestinal mucosa, 3 cm2 human intestinal samples were processed as previously described,22 and reacted with biotin-conjugated primary antibodies using EZ-link Sulfo-NHS-Biotin (Thermo Fisher Scientific). Detection was accomplished with horseradish peroxidase–conjugated streptavidin antibody (Jackson Lab, Bar Harbor, ME) and 3’-diaminobenzidine metal peroxide substrate.
Data Analysis
All analyses were conducted using R software (available from: www.r-project.org). The preprocessing procedures of proteomics data have been described in detail previously.10 Here, we focused on assembling a bioinformatics pipeline using readily available statistical tools to resolve unique challenges in analyzing proteomic data and distill useful and biologically relevant information.
Because of multiple sources of variance in the metaproteome data set, we first used the principal variance component analysis (PVCA) R package23 to evaluate the intersubject and intrasubject variance. Sources of variance included in our analysis were subject, collection site, colonic region, diagnosis, sex, and age (Table 1). The PVCA integrates 2 methods: principal components analysis (which finds low-dimensional linear combinations of data with maximal variability) and variant component analysis (which attributes and partitions variability into known sources via a classic random-effects model). The significance of the difference from the attributed variance was evaluated with a nonlinear mixed-effect model (NLME), and a permutation test was used to further evaluate the biogeographic feature by comparing two colonic sites at a time. NLME modeling accounted for both fixed effects (eg, diagnosis and biogeographic) and random effects (eg, subject), and thus was a good fit for analyzing this data set. Each NLME model was evaluated using analysis of variance (ANOVA), and the P values were plotted for visualization of the significance of the effect of interest. For the permutation test, the data set was divided into 6 sub–data sets based on the colonic region. The mean distance was calculated between two sub–data sets containing different colonic regions (referred to as the actual distance). The two sub–data sets then were permutated randomly 1000 times and the mean distance was calculated after every permutation. If the difference between the two regions was not significant, the permutation should have a random effect on the mean distance. After each permutation test, we calculated the probability using the following formula: (the number of permutated mean distances that are larger than the actual mean distance)/1000. If the probability was less than 0.05, the interpretation was that the chance of observing such difference between the two colonic regions being a random event is less than 5%.
To help distill the number of variables for further validation, weighted correlation network analysis (WGCNA) was used to construct protein modules (defined as branches of a hierarchical cluster tree based on the topologic overlaps).24, 25 A signed, weighted adjacency protein network was defined by applying a soft threshold of 6, chosen with the scale-free topology criterion.24 Once the network was constructed, modules then were defined by applying the dynamic tree cut method from the package dynamicTreeCut with a minimal module size of 10 protein members, and similar modules were merged with a height cut of 0.4, corresponding to a correlation of 0.6.
There is extensive literature comparing clustering procedures, including simple k-means, partitioning around medoid, hierarchical clustering, message passing, and model-based methods.25, 26, 27 Here, different clustering procedures (hierarchical clustering, partitioning around medoid clustering) and dissimilarity measures (Topological Overlap Matrix (TOM) dissimilarity, adjacency matrix, or correlation) were used to produce clusterings on the same IBD data set for comparison with those produced by WGCNA. The degree of agreement between different cluster assignments was measured using the Rand28 index. WGCNA-based clusters were compared with hierarchical clustering resulting from adjacency matrix-based dissimilarity (Supplementary Table 1, top 2 tables), partitioning around medoids clustering with adjacency (Supplementary Table 1, middle 2 tables), and partitioning around medoids clustering with a correlation matrix-based dissimilarity (Supplementary Table 1, bottom 2 tables). We found high pairwise Rand28 indices (0.796, 0.858, and 0.861, respectively). This analysis indicated that the protein modules uncovered by WCGNA could be reproduced with any of these alternative clustering procedures.
To test the reproducibility of the identified modules across independent data sets, a module preservation test was performed using two independent data sets: the IBD data set and a normal data set published previously.10 The two data sets were compared, and reduced to include only common peaks shared by both data sets. Two functions, the Z-summary test and the median rank test were implemented in the WGCNA package.29 An advantage of the Z-summary statistic is that it allows for significance thresholds: a Z-summary < 2 indicates no significant module preservation; 2 < Z-summary < 10 indicates moderate preservation; and a Z-summary > 10 indicates strong preservation. The median rank statistic, another network-based module-preservation statistic, allows modules to be ranked with respect to preservation. Statistical advantages of the median rank statistic include computational speed and the result is independent of module size. However, because the median rank statistic does not make use of a permutation test, it cannot be used to assign significance thresholds. Alternative statistics are available to assess the quality and reproducibility of clusters among data sets.30, 31, 32, 33 Our previous work outlined 7 simulation scenarios in which we compared WGCNA's module preservation statistics with the hitherto best-performing alternative approach, and the Z-summary statistic had distinct advantages when it came to studying the preservation of network modules.29, 34 Detailed discussions of the pros and cons of these statistics were presented recently.25, 35
To summarize the profiles of each module, we calculated the weighted average value (defined as the left singular vector that explains the highest amount of the underlying variation), referred to as the eigenprotein of each module. The Kruskal–Wallis test was used to determine whether the eigenprotein represented intestinal segments and/or disease-related features. To examine the significance of differences observed in immunoblotting, ANOVA was used for quantified data, whereas the Fisher exact test was used for cross-tabulation data.
All authors had access to the study data and reviewed and approved the final manuscript.
Results
Profiling the Mucosal Metaproteome in IBD Patients
MALDI-TOF mass spectrometry was used to analyze each lavage sample in duplicate or triplicate using a rigorous pipeline to ensure reproducible intersample comparisons. A total of 599 protein/peptide features (peaks) from 677 spectra were selected for subsequent analysis (Supplementary Table 2). In our previous study,10 we used a shotgun proteomic method (MS/MS fragmentation and high-probability matching to the nonredundant Uniprot database (www.uniprot.org) indexed for bacteria and human proteins) to characterize the metaproteome composition of the soluble component of the mucosal lavage compartment (the biospecimen used in the present study). Sixty-three percent of the proteins were human proteins and 30% were bacterial. Among the bacterial proteins, 48% belonged to Bacteroidetes, the most abundant phylum of human intestinal microbiota. We also observed proteins from other bacterial phyla, including Proteobacteria, Acidobacteria, Firmicutes, Chlorobi, and Cyanobacteria. These findings showed that mucosal lavage proteins represent a mixture of host and microbial products.
An analysis of the region- and disease-related effects on the human mucosal metaproteome is presented in Figure 2 using PVCA. The first component (PC1) accounted for 16.6% of the total variance in the data (Figure 2A), and the intersubject (individual) factor contributed the majority of the variance (57.5%). Consistent with our previous study, the relative contributions to the metaproteome were intersubject variability (57.5%), disease-related feature (21.5%), and colonic segmental feature (15%) (Figure 2B), which explains why we could not observe a clear separation on the principle component plots of PC1 and PC2 (Figure 2C and D).
Figure 2.

Features of IBD mucosal metaproteome. (A) Principal components analysis of the metaproteome. (C) Samples colored by colonic regions plotted against PC1 and PC2. C, cecum; A, ascending; T, transverse; D, descending; S, sigmoid; R, rectum. (D) Samples colored by diseases plotted against PC1 and PC2. N, non-IBD; U, ulcerative colitis; C, Crohn’s disease. (B) Variance components analysis of the principal component 1. (E) Frequency plot of region-related P value for each protein feature from NMLE analysis. (F) Frequency plot of disease-related P value for each protein feature from NMLE analysis. (G) P values between colon regions from the permutation test analysis. Only significant P values (<.05) are shown.
To further examine these factors, we used NLME modeling to account for individual variations, and to calculate the significance of the biologically relevant features for each peak. A substantial fraction of the proteins were detected differentially among the 6 different colonic regions (Figure 2E and G), and 3 disease groups (Figure 2F). At an uncorrected significance threshold of 0.05, 191 peaks (31.9%) were significantly different for the biogeographic regional feature and 146 peaks (24.4%) were significant as a disease-related feature. To account for multiple comparisons, we also used a false-discovery rate threshold of 0.1, which resulted in 166 peaks that were significant for regions and 85 peaks that were significant for disease (Supplementary Table 3).
Definition and Preservation of Protein Modules at the Mucosal Surface
To explore which proteins contributed to the segment- or disease-related features at the mucosal surface, we assessed the formation of protein modules, where proteins within the same module share connectivity through similar quantitative expression and localization features.36 We hypothesized that such protein modules exist at the MLI, and that some may show segment- or disease-related features. We tested this hypothesis using weighted correlation network analysis (implemented in the WGCNA R package)24, 25 to construct mucosal protein modules from metaproteomic data, calculate eigenprotein values to quantitate the representation of the modules among samples, and define intramodular hub proteins.37
By using this approach, 9 distinct protein modules were identified in the IBD data set (Figure 3A). Because module detection does not make use of prior biological knowledge about the protein, the biological meaning of each module initially is unknown and hence modules initially are assigned a color label (Table 2). To determine the statistical robustness of these protein modules, we used 3 different clustering and dissimilarity methods on the same IBD data set, and compared the resulting clustering with those produced by WGCNA. We found that clusters were highly robust across the 3 independent methodologies (Supplementary Table 1).
Figure 3.

Definition and preservation of protein modules at the mucosal surface. (A) Dendrogram of 9 protein modules for 599 protein features from the IBD data set. (B) Dendrogram of 7 protein modules for 438 protein features from the normal data set. Each module was designated by a different color. (C) Preservation of normal modules in the IBD metaproteomic data set. Upper color panel: dendrogram of overlapped peaks generated by WGCNA in the reduced IBD data set. Lower color panel: the corresponding module color of the same peak in the reduced normal data set. (D) Preservation of IBD modules in the normal metaproteomic data set. Upper color panel: dendrogram of overlapped peaks generated by the WGCNA in the reduced normal data set. Lower color panel: the corresponding module color of the same peak in the reduced IBD data set. (E) Preservation Z-summary test of IBD modules. (F) Preservation Z-summary test of normal modules. Less than 0, no preservation; 0–2, weak preservation; 2–10, moderate preservation; and more than 10, high preservation. (G) Preservation median rank test of IBD modules. (H) Preservation median rank test of normal modules. Each module was ranked based on its degree of preservation.
Table 2.
Association of Protein Modules With Intestinal Segments and IBD Diagnosis
| Module | Members, n | Notable proteins | IBD-related P valuea | Disease association | Region-related P valuea | Regional association |
|---|---|---|---|---|---|---|
| Cyan | 41 | ENPP7 | .46 | N/A | .89 | N/A |
| Green | 32 | HBD | .038 | Increased in CD only | .98 | N/A |
| Orange | 15 | N/A | .39 | N/A | .64 | N/A |
| Black | 31 | N/A | <.001 | Reduced in both UC and CD | .94 | N/A |
| Brown | 17 | HD5 | <.001 | .099 | N/A | |
| Pink | 76 | Elastase | .45 | N/A | .72 | N/A |
| Yellow | 34 | HNP | .01 | Increased in both UC and CD | <.001 | Increased in distal colon |
| Red | 111 | N/A | .03 | Increased in UC only | .003 | |
| Blue | 226 | Hepcidin | <.001 | .005 |
P values were generated using the Kruskal–Wallis test. P < .05 is considered statistically significant.
Next, we tested the preservation of the modules between independent data sets: the present IBD data set, and a previously published normal dataset,10 an independent metaproteomic data set of 205 mucosal lavage samples from 38 non-IBD subjects. By using the same WGCNA method, analysis of the normal data set discovered 6 distinct protein modules (Figure 3B). To compare protein peaks from the 2 data sets, we smoothed the spectra at 5 m/z to minimize the isotopic effect on peak ascertainment. This yielded 346 shared peaks, shared in both data sets and selected for module preservation analysis. The reduced common set of shared peaks yielded the same module organization: 9 modules in the IBD data set (Figures 3C, upper color panel IBD), and 6 modules in the normal data set (Figure 3D, upper color panel normal ).
When the protein memberships of the modules from the 2 data sets were compared (Table 3 and Figure 3C and D, lower color panel), 4 modules were concordant: cyan, green, black, and brown. The membership of the largest module detected in the control data set (yellow) was divided into 4 submodules in the IBD data set (the pink, yellow, red, and blue modules). The diversification of the yellow module indicates that the detailed protein composition of the module changes in disease compared with normal conditions, which is reflected in altered module formation.
Table 3.
Counts of Overlapped Peaks Between IBD and Normal Modules
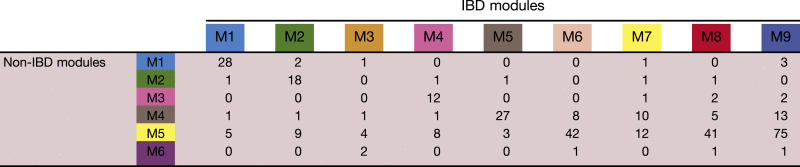 |
To evaluate quantitatively the degree of module preservation across independent data sets, we used the Z-summary (Figure 4E and F) and median rank test (Figure 4G and H) statistics.29 As expected, all modules except one from the normal data set were highly preserved in the IBD data set; only the small purple module was absent in the IBD data set. Conversely, except for the orange and yellow modules in the IBD data set, all other IBD modules also were well preserved in the normal data set. Thus, it appeared that there were 4 core modules (cyan, green, black, and brown) shared in both normal and IBD data sets.
Figure 4.

Characterization of the yellow module. (A) The expression of the yellow module represented by its eigenproteins in normal and IBD disease states. (B) Beanplots of representative protein levels detected in MALDI segregated by regions. (C) Immunoblot detection of HNPs in mucosal lavage samples. (D) IHC detection of HNPs in human colonic biopsy specimens (100×). NM, normal.
Association of Protein Modules With Intestinal Segments and IBD Diagnosis
To test if any IBD modules showed colonic segment- or disease-related features, we compared the eigenprotein of each module in different colon regions or diseases using the Kruskal–Wallis test. Three modules showed a significant segment-related feature (yellow, blue, and red modules), which each were more abundant in the distal colon. Six modules were significantly disease-related: the green module was increased in CD; the expression of the brown and black modules was reduced in both UC and CD, the yellow module (distal colon-related) was increased in both UC and CD, and the blue and red modules (distal colon-related) were increased in UC (Table 2).
Characterizing Individual Protein Modules
To characterize individual modules, we used several strategies to obtain protein identities of module-associated peaks. The identified protein peaks are listed in Table 4, representing 6 of the 9 IBD modules. We then selected 4 modules for detailed analysis, based on the following criteria: biologically relevant annotation, availability of detection antibodies, and association with disease-associated modules (Table 2; Figures 4, 5, 6, and 7).
Table 4.
List of Identified Proteins/Peptides
| Peak m/z | Module | Protein/peptide ID |
|---|---|---|
| 2478.37 | Cyan | Ectonucleotide pyrophosphatase/phosphodiesterase 7 (Homo sapiens) |
| 3935.44 | Cyan | α-1 antitrypsin (H sapiens) |
| 4135.65 | Cyan | α-1 antitrypsin (H sapiens) |
| 2490.45 | Green | Immunoglobulin heavy-chain variable region (Homo sapiens) or KIAA0297 (H sapiens) |
| 2980.79 | Green | Peptidase M20 domain containing 1 (H sapiens) |
| 3222.84 | Green | Pancreatic amylase α 2A (H sapiens) |
| 3336.94 | Green | Dynamin-like protein (Acaryochloris marina MBIC11017) |
| 3992.28 | Green | β-defensin 1 (H sapiens) |
| 4317.93 | Green | β-defensin 2 (H sapiens) |
| 3583 | Brown | α-defensin5 (H sapiens) |
| 2139.78 | Pink | Elastase 2A preproprotein (H sapiens) |
| 2334.36 | Pink | ASAH2 protein (H sapiens) |
| 3281.32 | Pink | Elastase 2A preproprotein (H sapiens) |
| 3309.33 | Pink | Chymotrypsin-like elastase family member 2A (H sapiens) |
| 2313.27 | Yellow | Guanosine triphosphatase activating Rap/RanGAP domain-like 1 isoform 1 (H sapiens) |
| 2402.33 | Yellow | Tetratricopeptide TPR_2 repeat protein (Flavobacteria bacterium) |
| 3107.69 | Yellow | hCG2042445 (H sapiens) |
| 3253.69 | Yellow | Tyrosine-protein kinase (Flavobacteria bacterium BAL38) |
| 3371.75 | Yellow | α-defensin 2 (H sapiens) |
| 3442.69 | Yellow | α-defensin 1 (H sapiens) |
| 3486.84 | Yellow | α-defensin 3 (H sapiens) |
| 2052.13 | Blue | Haptoglobin (H sapiens) or anti–tumor necrosis factor-α antibody heavy-chain Fab fragment (H sapiens) |
| 2159.68 | Blue | Chymotrypsin C preproprotein (H sapiens) |
| 2197.11 | Blue | Procarboxypeptidase B (H sapiens) |
| 2607.54 | Blue | Procarboxypeptidase B (H sapiens) |
| 2721.59 | Blue | Transferrin (H sapiens) |
| 2781.92 | Blue | Hepcidin (H sapiens) |
| 11606.9 | Blue | Serum amyloid protein (H sapiens) |
Figure 5.

Characterization of the blue module. (A) The expression of the blue module represented by its eigenproteins in normal and IBD disease states. (B) Beanplots of representative protein levels detected in MALDI segregated by regions: upper panel, hepcidin; lower panel, transferrin. (C) Immunoblot detection of hepcidin and transferrin in mucosal lavage samples. (D) IHC detection (100×) of prohepcidin and transferrin in human colonic biopsy specimens (insets, 400×). NM, normal control.
Figure 6.

Characterization of the green module. (A) The expression of the green module represented by its eigenproteins in normal and IBD disease states. (B) Beanplots of representative protein levels detected in MALDI segregated by regions (C) Immunoblot detection of proteins in mucosal lavage samples. left panel, HBD1; middle panel, HBD2; right panel: PM20D1. (D) IHC detection (100×) of HBD1, HBD2, and PM20D1 in human colonic biopsy specimens (insets, 400×). NM, normal.
Figure 7.

Expression of HD5 in colonic mucosal lavage and biopsy samples. (A) Immunoblot detection of HD5 in mucosal lavage samples. (B) Quantitation of HD5 immunoblots. (C) IHC detection (100×) of HD5 in human colonic biopsy specimens. NM, normal.
We first plotted the yellow module eigenprotein vs disease status (Figure 4A). The eigenprotein had a significantly higher value in both UC and CD samples compared with normal, indicating that the proteins of this module are more abundant in diseased individuals. The yellow module had both segmental and disease-related features. Therefore, we separated the samples according to these locations, and visualized the samples from cecum and sigmoid separately (Figure 4B). An in silico search and immunoprecipitation followed by MALDI MS established their identity as α-defensins, including HNP 1 (3442.69 m/z), HNP2 (3771.75 m/z), and HNP3 (3486.84 m/z). When we visualized each protein level using beanplots, they all shared a similar expression pattern, with increased levels in both UC and CD patients (Figure 4B). Immunoblotting showed that 8 of 10 CD samples and 6 of 10 UC samples were positive (Figure 4C, top panel). When the immunoblot data were quantitated by digitization and normalization, a significant difference between disease and normal samples was observed (Figure 4C, lower panel), formally established by ANOVA of disease and normal groups (P = .02). As expected, tissue expression of HNP1-3 by IHC staining established that these proteins were associated with sites of tissue neutrophil infiltration (Figure 4D), and such sites were strongly positive in all UC and CD biopsy samples that were evaluated.
The blue module was the largest protein network, containing 180 proteins. Its eigenprotein was significantly lower in CD subjects compared with both normal and UC subjects (Figure 5A). We were able to identify 2 peaks in this module using HPLC followed by LC-MS/MS. The peak at 2781.92 m/z was hepcidin and the peak at 2721.85 m/z was a transferrin protein fragment. When we plotted these 2 proteins, we noticed that both were increased slightly in the cecal samples from UC patients (Figure 5B). Immunoblotting of both proteins also showed higher expression in UC samples (Figure 5C), but only transferrin reached significance by the Fisher exact test (P = .01). To test the cellular origins of the intestinal hepcidin and transferrin, we performed IHC staining on biopsy tissues from human colons. The major cellular source of hepcidin was epithelial cells, because these cells expressed the presecretory prohepcidin molecule in a perinuclear vesicular site (Figure 5D). On rare occasions, we also observed some sporadic lamina propria hepcidin-positive cells, which might represent macrophages or monocytes that are known to express hepcidin.38 Normal or CD tissues uniformly showed weak or no signal for prohepcidin, but all UC samples had moderate to high signal levels. For transferrin, a gradient expression pattern was observed in 4 of the UC tissues, which showed higher signals toward the bottom of the crypts but lower signals toward the mucosal surface. Transferrin was localized to the lamina propria interstitium and diffuse intracellular uptake, apparently reflecting a vascular leak of plasma transferrin and local cellular uptake (Figure 5D, box). In this small sampling, normal and CD tissues had minimal signal for transferrin, with the exception of 1 CD sample with very high transferrin localization.
The green module eigenprotein was significantly lower in CD compared with both normal and UC (Figure 6A), which indicates that these proteins are greatly diminished in CD patients. Two abundant peaks of the green module (3992.28 m/z and 4317.93 m/z) were identified successfully through a combination of in silico search and IP-MALDI as HBD1 and HBD2. When we plotted the intensity of each protein on a beanplot, both were increased in CD patients, but decreased in UC patients (Figure 6B). We confirmed this trend by immunoblotting (Figure 6C), and ANOVA of the digitized immunoblot data established a significant increase of HBD1 and HBD2 (P = .04). To explore the cellular origins of the intestinal HBD1 and HBD2, we performed IHC on biopsy samples from human colon. We tested 5 biopsy specimens in each group, and found both HBD1 and HBD2 were expressed exclusively in colonic epithelial cells (Figure 6D, top and middle panels). For HBD1, 3 of 5 normal and all UC tissues showed weak staining, but all 5 CD tissues were strongly positive. For HBD2, all CD samples were very strongly stained; in contrast, UC samples had weak or no signal, and normal tissues uniformly showed a moderate signal. Another peptide also identified within the green module was a fragment of PM20D1, which shared the same expression pattern as HBD1 and HBD2, with the highest expression in CD patients (Figure 6B and C). By using both immunoblotting and IHC, we also found high levels of PM20D1 in CD only (Figure 6D, bottom panel).
The brown module was notable for the presence of α-defensin 5 (HD5) (Table 4). HD5 is secreted specifically by Paneth cells that classically reside in the small intestine.39, 40 Its presence in lavage samples may have reflected distal luminal migration to the colon from the small intestine. However, IHC staining showed that specific HD5-positive cells were readily detectable in the characteristic crypt base location in colon biopsy specimens (Figure 7C). Therefore, local colonic production of HD5 appeared to be a significant source of the colonic HD5 signal.
Assessment of Microgeographic Distribution of Module Proteins
We used mucosal whole-mount immunohistochemistry to attempt to visualize protein networks. Mucosal specimens from surgical colonic resections of non-IBD and IBD patients were evaluated with antibodies for the epithelial-derived protein members of the yellow and pink modules. In most specimens, only low expression was detected, as expected for minimal constitutive expression levels of these proteins (data not shown). However, some specimens showed intense protein expression that occurred as an intermittent mosaic of sites of small dimensions (diameter, 1–4 mm) (Figure 8A). This suggests that module proteins are expressed intermittently at the mucosal surface in a microgeographic pattern.
Figure 8.

Visualization of MFNs. (A) Whole-mount immunohistochemistry of non-IBD mucosa detected with anti-HNP1 (left) or anti-elastase 2a (right) antibodies. Scale bars: 1 mm. (B) MFN hypothesis. The human mucosal surface is a mosaic of MFNs. Host and microbial cells and their products directly interact over a distance of millimeters within its MFN, and reach a metastable composition and state of activity. Certain MFNs carry disease- or region-related features.
Discussion
General Features of Mucosal Metaproteome in IBD
Concordant with our previous study of the mucosal–luminal interface metaproteome, this independent data set confirmed that its two major properties are individual variation, and a characteristic segmental feature distinguishing the proximal (cecum through descending) and distal (rectum and sigmoid) colon. A recent independent study of the microbial-associated metaproteome has shown distinct abundance patterns of microbial proteins among 6 regions of the gastrointestinal tract.14 The present study adds two new observations regarding the mucosal–luminal metaproteome. First, it presents evidence that disease state also significantly alters composition of this metaproteome. Second, it uncovers a set of protein modules of the MLI metaproteome, validated bioinformatically and confirmed by direct visualization using tissue-based cellular immunohistochemistry and immunoblot analysis.
Each protein module had a distinct segmental and disease association. The expression of three modules (Yellow, Red, and Blue) was associated selectively with both the colonic regions and diseases. Two modules were overexpressed in UC, and one module was overexpressed in both UC and CD. Among the remaining modules, three show only disease association, but no colonic segmental feature. The green module was overexpressed in CD compared with both non-IBD and UC, and both the black and brown modules were underexpressed in UC and CD. There were also apparent differences in the module structure between the IBD and normal data sets. Four modules were highly conserved in both data sets (cyan, green, brown, and black modules). These modules may represent biologically resilient networks, relatively unaffected in composition in non-IBD and IBD states, even though the abundance of one such module (green) was disease-associated (increased in CD). Other modules were present in both data sets, but with different hierarchical branches indicating module reorganization. For instance, the yellow module of the normal data set consisted of a single large module of 270 peaks, but was split into 4 smaller submodules (pink, yellow, blue, and red modules) in the IBD data sets. Such reorganization of protein networks, presumably reflecting the altered coordination of source cell types at the mucosal surface, may provide fresh clues to these physiologic and disease states of the mucosa.
Functional Features of the Index Module Proteins
Neutrophil infiltration of the mucosa is a common occurrence in IBD, and thus provides a context for the increased abundance of the neutrophil products HNP1–3, and their associated yellow module, in the mucosal metaproteome of UC and CD. It is notable that their abundance was observed in endoscopically quiescent sites, and was increased in the distal colon of both normal and IBD patients. Thus, the yellow module may reflect an underappreciated level of neutrophil engagement in the subclinical physiologic inflammation of the distal colon.
HD5 is an α-defensin produced by ileal Paneth cells, and its production is impaired in a subset of ileal Crohn’s disease as a result of Paneth cell dysfunction associated with Nucleotide-Binding Oligomerization Domain Containing 2 (NOD2) and Autophagy Related 16-Like 1(ATG16L1).39, 40 UC and CD patients commonly acquire Paneth cell metaplasia, however, we also found HD5-positive cells in normal colon biopsy specimens. This indicates that by the sensitive detection of HD5 IHC, Paneth cell metaplasia also occurs in clinically normal colonic mucosa, and may play an antimicrobial role in such sites of the distal intestine.
HBD1 and HBD2, predominantly produced by epithelial cells, are minimally expressed in normal mucosa, but are induced in active CD and UC.41, 42 HBD2 is up-regulated by a NOD2- or toll-like receptor-dependent nuclear factor-κB pathway,43, 44 but its intestinal expression may be independent of NOD2 mutation status in CD patients.45 In this study, both HBD1 and HBD2 were increased in CD patients compared with normal and UC; this distinctive pattern may reflect the antimicrobial peptide features of the colonic mucosal surface compartment.
Transferrin and hepcidin are two important regulators of iron homeostasis, and both are linked to innate immune function and inflammation.38 Hepcidin regulates iron homeostasis by binding to the sole iron exporter ferroportin and promoting its internalization and degradation. Hepcidin expression is abundant in hepatocytes. Our finding of prohepcidin expression in the colonic epithelium supports this novel local origin for hepcidin. Transferrin also is expressed primarily by the liver, and is an abundant serum protein that regulates iron homeostasis by binding tissue-derived ferric ions and internalization by ubiquitous cellular transferrin receptors. We found abundant levels of transferrin in the lamina propria, probably owing to such cellular uptake; its vesicular intracellular localization is consistent with this idea. Because both proteins reduce tissue ferric ions, overexpression of both hepcidin and transferrin in UC indicate a more active antimicrobial state. We show that colonic expression of hepcidin and transferrin are associated with IBD.
It is not surprising that the most abundantly identified proteins in our mucosal surface samples were proteases and protease inhibitors because these are major intestinal epithelial products in support of alimentary enzymatic digestion and metabolism. Curiously, the 6 proteins in this category are associated with several distinct modules, suggesting local factors that differentially affect their production. Several host and microbial-derived proteases have been associated with IBD,46, 47 and increased concentrations of proteases and proteolytic activities have been reported in human fecal and colonic biopsy samples. The mechanism of their differential control and their contribution to the IBD disease state are open issues, but may relate to their participation in the well-studied inflammatory tissue metalloprotease pathway.48
Mucosal Functional Networks
There is increasing evidence that the mucosal surface is an integrated ecology formed by the interplay of host cell types, environmental and dietary biomolecules, and microbes.4, 12, 13, 14, 15, 16, 17, 19, 49 Based on the present metaproteomic study, we hypothesize that the human mucosal surface is not homogeneous, but instead consists of local mucosal functional networks (MFNs) (Figure 8B). Our bioinformatic and immunohistochemistry data suggest that there may be a few types of networks that are distributed in a mosaic of microgeographic (millimeter-range) scale. We speculate that these may form as host and microbial cells, and their products may interact directly over a distance of microns to millimeters, and in this physical range reach a metastable composition and state of activity. Consistent with this idea, recent work from Lee et al13 suggested that specific bacteria may colonize unique areas of the gut, as shown by specific localization of Bacteroides fragilis deep within crypt channels. Some MFNs also are likely to represent a habitat for host responses such as inflammation, which may contribute to preclinical or clinical disease states.
One limitation of the present study was that because of the metaproteomics method used, our study mostly detected the human peptides of the mucosal metaproteome. Accordingly, the relationship of the MFNs to microbial interaction is uncertain. Recent advances in detection and bioinformatics methods are enriching the identification of mucosal microbial proteins, and their use in assessing microbial functional in the mucosal ecology.10, 11, 12, 13, 14, 15 These microbial findings, and the present human study, suggest that integrated host-microbial metaproteomics will be a promising approach to biochemically and functionally link the local interaction of host, microbiota, and environmental integration of the mucosal state.
Acknowledgments
The authors thank the University of California Los Angeles Translational Pathology Core Laboratory for providing us with sufficient human tissue samples and processing them for subsequent analysis, and Dr Tomas Ganz’s laboratory for the generous gifts of the antibodies. The High-Throughput Clinical Proteomics Core performed the MALDI mass spectrometry and the Molecular Instrumentation Center assisted with LC-MS/MS and protein identification. The authors acknowledge Dr Tania Kamphaus for her critical editorial input. The authors are grateful to the patients and physicians at the University of California Los Angeles and Cedars-Sinai Medical Center, whose participation made this study possible. This paper is a memorial to our colleague and friend, Dr Lee Goodglick.
Footnotes
Conflicts of interest The authors disclose no conflicts.
Funding This study was supported by National Institutes of Health grants DK46763 (J.B.), AI078885 (J.B.), and AI28697 (University of California Los Angeles Center for Acquired Immune Deficiency Syndrome Research); National Center for Research Resources (NCRR) and National Center for Advancing Translational Sciences (NCATS) grants UL1TR000124 (J.B.) and DK64798 (R.D.N.); the Crohn’s and Colitis Foundation of America grants 156 (L.G.) and 3153 (J.B.); the Ursula Mandel Student Fellowship and the University of California Los Angeles Dissertation Year Fellowships (X.L.); and the Burroughs Wellcome Fund Inter-school Program in Metabolic Diseases Fellowship (M.T.).
Supplementary Material
References
- 1.Blikslager A.T., Moeser A.J., Gookin J.L. Restoration of barrier function in injured intestinal mucosa. Physiol Rev. 2007;87:545–564. doi: 10.1152/physrev.00012.2006. [DOI] [PubMed] [Google Scholar]
- 2.Sansonetti P.J. War and peace at mucosal surfaces. Nat Rev Immunol. 2004;4:953–964. doi: 10.1038/nri1499. [DOI] [PubMed] [Google Scholar]
- 3.Badman M.K., Flier J.S. The gut and energy balance: visceral allies in the obesity wars. Science. 2005;307:1909–1914. doi: 10.1126/science.1109951. [DOI] [PubMed] [Google Scholar]
- 4.Lewis J.D., Chen E.Z., Baldassano R.N. Inflammation, antibiotics, and diet as environmental stressors of the gut microbiome in pediatric Crohn’s disease. Cell Host Microbe. 2015;18:489–500. doi: 10.1016/j.chom.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dethlefsen L., McFall-Ngai M., Relman D.A. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature. 2007;449:811–818. doi: 10.1038/nature06245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costello E.K., Lauber C.L., Hamady M. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morgan X.C., Huttenhower C. Meta'omic analytic techniques for studying the intestinal microbiome. Gastroenterology. 2014;146:1437–1448 e1. doi: 10.1053/j.gastro.2014.01.049. [DOI] [PubMed] [Google Scholar]
- 8.McGovern D.P., Kugathasan S., Cho J.H. Genetics of inflammatory bowel diseases. Gastroenterology. 2015;149:1163–1176 e2. doi: 10.1053/j.gastro.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee D., Albenberg L., Compher C. Diet in the pathogenesis and treatment of inflammatory bowel diseases. Gastroenterology. 2015;148:1087–1106. doi: 10.1053/j.gastro.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li X., LeBlanc J., Truong A. A metaproteomic approach to study human-microbial ecosystems at the mucosal luminal interface. PLoS One. 2011;6:e26542. doi: 10.1371/journal.pone.0026542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verberkmoes N.C., Russell A.L., Shah M. Shotgun metaproteomics of the human distal gut microbiota. ISME J. 2009;3:179–189. doi: 10.1038/ismej.2008.108. [DOI] [PubMed] [Google Scholar]
- 12.Lichtman J.S., Marcobal A., Sonnenburg J.L. Host-centric proteomics of stool: a novel strategy focused on intestinal responses to the gut microbiota. Mol Cell Proteomics. 2013;12:3310–3318. doi: 10.1074/mcp.M113.029967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee S.M., Donaldson G.P., Mikulski Z. Bacterial colonization factors control specificity and stability of the gut microbiota. Nature. 2013;501:426–429. doi: 10.1038/nature12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lichtman J.S., Alsentzer E., Jaffe M. The effect of microbial colonization on the host proteome varies by gastrointestinal location. ISME J. 2016;10:1170–1181. doi: 10.1038/ismej.2015.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lichtman J.S., Ferreyra J.A., Ng K.M. Host-microbiota interactions in the pathogenesis of antibiotic-associated diseases. Cell Rep. 2016;14:1049–1061. doi: 10.1016/j.celrep.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Earle K.A., Billings G., Sigal M. Quantitative imaging of gut microbiota spatial organization. Cell Host Microbe. 2015;18:478–488. doi: 10.1016/j.chom.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eckburg P.B., Bik E.M., Bernstein C.N. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frank D.N., St Amand A.L., Feldman R.A. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Costello E.K., Stagaman K., Dethlefsen L. The application of ecological theory toward an understanding of the human microbiome. Science. 2012;336:1255–1262. doi: 10.1126/science.1224203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lustgarten J.L., Kimmel C., Ryberg H. EPO-KB: a searchable knowledge base of biomarker to protein links. Bioinformatics. 2008;24:1418–1419. doi: 10.1093/bioinformatics.btn125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar A.R., Li X., Leblanc J.F. Proteomic analysis reveals innate immune activity in intestinal transplant dysfunction. Transplantation. 2011;92:112–119. doi: 10.1097/TP.0b013e31821d262b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lorenz R.G., Chaplin D.D., McDonald K.G. Isolated lymphoid follicle formation is inducible and dependent upon lymphotoxin-sufficient B lymphocytes, lymphotoxin β receptor, and TNF receptor I function. J Immunol. 2003;170:5475–5482. doi: 10.4049/jimmunol.170.11.5475. [DOI] [PubMed] [Google Scholar]
- 23.Scherer A. J. Wiley; Chichester, UK: 2009. Batch effects and noise in microarray experiments: sources and solutions. [Google Scholar]
- 24.Zhang B., Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4 doi: 10.2202/1544-6115.1128. Article17. [DOI] [PubMed] [Google Scholar]
- 25.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nugent R., Meila M. An overview of clustering applied to molecular biology. Methods Mol Biol. 2010;620:369–404. doi: 10.1007/978-1-60761-580-4_12. [DOI] [PubMed] [Google Scholar]
- 27.Jay J.J., Eblen J.D., Zhang Y. A systematic comparison of genome-scale clustering algorithms. BMC Bioinformatics. 2012;13(Suppl 10):S7. doi: 10.1186/1471-2105-13-S10-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rand W.M. Objective criteria for the evaluation of clustering methods. J Am Stat Assoc. 1971;66:846–850. [Google Scholar]
- 29.Langfelder P., Luo R., Oldham M.C. Is my network module preserved and reproducible? PLoS Comput Biol. 2011;7:e1001057. doi: 10.1371/journal.pcbi.1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Handl J., Knowles J., Kell D. Computational cluster validation in post-genomic data analysis. Bioinformatics. 2005;21:3201–3212. doi: 10.1093/bioinformatics/bti517. [DOI] [PubMed] [Google Scholar]
- 31.Garge N.R., Page G.P., Sprague A.P. Reproducible clusters from microarray research: whither? BMC Bioinformatics. 2005;6(Suppl 2):S10. doi: 10.1186/1471-2105-6-S2-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giancarlo R., Scaturro D., Utro F. Computational cluster validation for microarray data analysis: experimental assessment of Clest, Consensus Clustering, Figure of Merit, Gap Statistics and Model Explorer. BMC Bioinformatics. 2008;9:462. doi: 10.1186/1471-2105-9-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smolkin M., Ghosh D. Cluster stability scores for microarray data in cancer studies. BMC Bioinformatics. 2003;4:36. doi: 10.1186/1471-2105-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kapp A., Tibshirani R. Are clusters found in one dataset present in another dataset? Biostatistics. 2007;8:9–31. doi: 10.1093/biostatistics/kxj029. [DOI] [PubMed] [Google Scholar]
- 35.Horvath S. Weighted Network Analysis: Applications in Genomics and Systems Biology, Berlin, Germany: Springer 2011.
- 36.Han J., Bertin N., Hao T. Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature. 2004;430:88–93. doi: 10.1038/nature02555. [DOI] [PubMed] [Google Scholar]
- 37.Horvath S., Dong J. Geometric interpretation of gene coexpression network analysis. PLoS Comput Biol. 2008;4:e1000117. doi: 10.1371/journal.pcbi.1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nemeth E., Ganz T. Regulation of iron metabolism by hepcidin. Annu Rev Nutr. 2006;26:323–342. doi: 10.1146/annurev.nutr.26.061505.111303. [DOI] [PubMed] [Google Scholar]
- 39.Salzman N.H., Hung K., Haribhai D. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol. 2010;11:76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cadwell K., Patel K.K., Maloney N.S. Virus-plus-susceptibility gene interaction determines Crohn's disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. doi: 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wehkamp J., Fellermann K., Herrlinger K.R. Human beta-defensin 2 but not beta-defensin 1 is expressed preferentially in colonic mucosa of inflammatory bowel disease. Eur J Gastroenterol Hepatol. 2002;14:745–752. doi: 10.1097/00042737-200207000-00006. [DOI] [PubMed] [Google Scholar]
- 42.Fahlgren A., Hammarstrom S., Danielsson A. Increased expression of antimicrobial peptides and lysozyme in colonic epithelial cells of patients with ulcerative colitis. Clin Exp Immunol. 2003;131:90–101. doi: 10.1046/j.1365-2249.2003.02035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Voss E., Wehkamp J., Wehkamp K. NOD2/CARD15 mediates induction of the antimicrobial peptide human beta-defensin-2. J Biol Chem. 2006;281:2005–2011. doi: 10.1074/jbc.M511044200. [DOI] [PubMed] [Google Scholar]
- 44.Vora P., Youdim A., Thomas L.S. Beta-defensin-2 expression is regulated by TLR signaling in intestinal epithelial cells. J Immunol. 2004;173:5398–5405. doi: 10.4049/jimmunol.173.9.5398. [DOI] [PubMed] [Google Scholar]
- 45.Simms L.A., Doecke J.D., Walsh M.D. Reduced α-defensin expression is associated with inflammation and not NOD2 mutation status in ileal Crohn’s disease. Gut. 2008;57:903–910. doi: 10.1136/gut.2007.142588. [DOI] [PubMed] [Google Scholar]
- 46.Steck N., Mueller K., Schemann M. Bacterial proteases in IBD and IBS. Gut. 2011 doi: 10.1136/gutjnl-2011-300775. [DOI] [PubMed] [Google Scholar]
- 47.Cleynen I., Jüni P., Bekkering G.E. Genetic evidence supporting the association of protease and protease inhibitor genes with inflammatory bowel disease: a systematic review. PLoS One. 2011;6:e24106. doi: 10.1371/journal.pone.0024106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ravi A., Garg P., Sitaraman S.V. Matrix metalloproteinases in inflammatory bowel disease: boon or a bane? Inflamm Bowel Dis. 2007;13:97–107. doi: 10.1002/ibd.20011. [DOI] [PubMed] [Google Scholar]
- 49.Martens E.C., Chiang H.C., Gordon J.I. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe. 2008;4:447–457. doi: 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.