

Abstract
Tormentic acid (TA) is a triterpene isolated from the stem bark of the plant Vochysia divergens and has been reported to exhibit anticancer, anti-inflammatory and anti-atherogenic properties. However, the functions of TA in hydrogen peroxide (H2O2)-induced oxidative stress and inflammation in rat vascular smooth muscle cells (RVSMCs) remain unclear. Therefore, the present study aimed to investigate whether TA suppressed H2O2-induced oxidative stress and inflammation in RVSMCs, and to determine its molecular mechanisms. The present study demonstrated that TA inhibited reactive oxygen species (ROS) generation, induced H2O2 in RVSMCs, and inhibited H2O2-induced expression of inducible nitric oxide synthase (iNOS) and NADPH oxidase (NOX) in RVSMCs. In addition, TA significantly decreased the production of tumor necrosis factor-α (TNF-α), interleukin 6 (IL-6) and IL-1β. Furthermore, TA pretreatment prevented nuclear factor-κB (NF-κB) subunit p65 phosphorylation and NF-κB inhibitor α (IκBα) degradation induced by H2O2 in RVSMCs. TA is, therefore, suggested to inhibit H2O2-induced oxidative stress and inflammation in RVSMCs via inhibition of the NF-κB signaling pathway. TA may have potential as a pharmacological agent in the prevention or treatment of atherosclerosis.
Keywords: tormentic acid, atherosclerosis, H2O2, rat vascular smooth muscle cells
Introduction
Atherosclerosis is a complex pathology involving several processes, including subendothelial retention of atherogenic lipoproteins, oxidative stress, inflammation and cellular proliferation (1). Vascular smooth muscle cells (VSMCs) may contribute to the development of atherosclerosis through the production of inflammatory cytokines, such as monocyte chemoattractant protein-1, and the synthesis of matrix proteins (2).
Reactive oxygen species (ROS), for example superoxide anion (O2−) and hydrogen peroxide (H2O2), are physiological and pathophysiological signaling molecules that participate in the development of atherosclerosis (3,4). Excessive production of ROS, partially through upregulation of DNA damage pathways, is a central mechanism that mediates pathological activation of VSMCs. In addition, ROS activate multiple pro-inflammatory transcription factors, including nuclear factor erythroid 2-related factor 2, nuclear factor-κB (NF-κB), and activator protein 1, which regulate the expression of adhesion molecules and chemokines in VSMCs (5). Therefore, targeting ROS is an important therapeutic strategy for atherosclerosis.
Tormentic acid (TA) is a triterpene isolated from the stem bark of the plant Vochysia divergens. Previous studies have demonstrated that TA has anticancer, anti-oxidant, anti-inflammatory and hypoglycemic properties (6–9). TA was demonstrated to suppress high-fat diet-induced diabetes and hyperlipidemia via glucose transporter 4 and adenosine monophosphate-activated protein kinase phosphorylation (10). Fogo et al (11) previously reported that TA significantly reduced VSMC proliferation and survival. In addition, TA inhibited lipopolysaccharide-induced inducible nitric oxide synthase (iNOS), cyclooxygenase-2, and tumor necrosis factor-α (TNF-α) expression in RAW264.7 cells (12). However, the impact of TA on H2O2-induced oxidative stress and inflammation in rat VSMCs (RVSMCs) remains unclear. Therefore, the aim of the present study was to investigate whether TA suppressed H2O2-induced oxidative stress and inflammation in RVSMCs, and to determine the molecular mechanisms.
Materials and methods
Animal and RVSMC preparation
Female Sprague Dawley (SD) rats (age, 6 weeks; weight, 180–200 g) were obtained from the Animal Breeding Center of the People's Hospital of Tianjin City (Tianjin, China). They were housed in barrier facilities with 12 h light/dark cycles at 22±2°C, and had access to laboratory chow (Jiangsu Xietong Medicine Biological Engineering Co., Ltd., Jiangsu, China) and tap water ad libitum. After 1 week of feeding, the animals were anesthetized by subcutaneous injection of sodium pentobarbital (40 mg/kg body weight). All experiments were performed in accordance with the institutional guidelines for animal care. This study was approved by the ethics committee of the People's Hospital of Tianjin City (Tianjin, China).
RVSMCs were enzymatically isolated from the aortas of female SD rats according to the methods described in a previous study (13), and were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine serum (FBS; Sigma-Aldrich; Merck Millipore, Darmstadt, Germany), 100 U/ml penicillin, 100 µg/ml streptomycin and 200 mM L-glutamine in a humidified 5% CO2 atmosphere at 37°C. For all experiments, RVSMCs (used at passages 5–8) were cultured to 70–80% confluence and serum-starved in DMEM without FBS for 24 h.
H2O2-induced oxidant stress
Cells were pretreated with various concentrations of TA (12.5, 25 and 50 µM; Shaanxi Institute for Food and Drug Control, Shaanxi, China) for 2 h, followed by the addition of H2O2 (100 µM final concentration) for a further 24 h. Controls performed were 2 h TA pretreatment without H2O2 stimulation, and 24 h H2O2 treatment without 2 h TA pretreatment.
Cell viability assay
Cell viability was assessed by Cell Counting Kit-8 (CCK-8) assay (Beyotime Institute of Biotechnology, Shanghai, China). In brief, RVSMCs were seeded into 96-well plates at 1×104 cells per well and cultured for 24 h to adhere. Following the described H2O2 and TA treatments, 10 µl CCK-8 reagent was added to each well and the cells incubated for a further 2 h. Finally, the absorbance was read at 570 nm (A570) using a Bio-Rad enzyme-linked immunosorbent assay (ELISA) microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The viability of cells was calculated as (A570 of treated groups/A570 of control group) ×100%.
Measurement of cellular ROS levels
RVSMCs were stained with 5 µM dihydroethidium (DHE; Molecular Probes; Thermo Fisher Scientific, Inc.) for 30 min at 37°C. Fluorescence of DHE was measured with a fluorescence microscope (excitation wavelength 488 nm and emission wavelength 585 nm), quantified using ImageJ software (version, 1.46; National Institutes of Health, Bethesda, MD, USA).
ELISA assay
Following RVSMC incubation with TA (12.5, 25 and 50 µM) for 24 h, the cells were exposed to H2O2 for a further 2 h. Samples of the supernatant were collected from each well to measure TNF-α (cat. no. RAB0479; Sigma-Aldrich), interleukin (IL)-6 (cat. no. 10406; Sigma-Aldrich) and IL-1β (cat. no. RAB0278; Sigma-Aldrich) levels by ELISA.
Western blot analysis
Cell lysate was prepared from RVSMCs using lysis buffer (Cell Signaling Technology, Inc., Danvers, MA, USA). Equal amounts of protein samples (30 µg total protein per lane) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis on 10% gels and transferred onto polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). After blocking with 2% non-fat dry milk in Tris-buffered saline (TBS) for 1 h at room temperature, the membranes were probed with the following primary antibodies in TBS plus 0.1% Tween-20 (TBST) containing 5% BSA (Sigma-Aldrich; Merck Millipore) at 4°C overnight: Mouse anti-rabbit nitric oxide synthase (NOS; cat. no. N7782; dilution, 1:1,000; Sigma-Aldrich; Merck Millipore); mouse anti-rabbit nicotinamide adenine dinucleotide phosphate (NADPH) -oxidase 1 (NOX1; cat. no. SAB2108601; dilution, 1:2,000; Sigma-Aldrich; Merck Millipore); mouse anti-rabbit neutrophil cytosolic factor 1 (p47phox; cat. no. sc-14015; dilution, 1:1,500; Santa Cruz Biotechnology, Inc., Dallas, TX, USA); and mouse anti-rabbit glyceraldehyde 3-phosphate dehydrogenase (GAPDH; cat. no. sc-25778; dilution, 1:1,500; Santa Cruz Biotechnology, Inc.). Membranes were then washed three times in TBST for 5 min per wash before they were incubated with goat anti-rabbit horseradish peroxidase-conjugated anti-IgG secondary antibody (cat. no. sc-2054; dilution, 1:3,000; Santa Cruz Biotechnology, Inc.) for 1 h at room temperature. Immune complexes were visualized using enhanced chemiluminescence reagent (Gibco; Thermo Fisher Scientific, Inc.). Developed films were scanned, and the optical densities were analyzed using ImageJ software (version, 1.37; National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data are expressed as the mean ± standard deviation. Statistical differences were analyzed using one-way analysis of variance, followed by Dunnett's multiple comparison post-hoc test. Differences in the cumulative clinical score were analyzed using the non-parametric Mann-Whitney test. P<0.05 was considered to indicate a statistically significant difference.
Results
Effects of TA on RVSMC viability
The effect of TA on cell viability in H2O2-induced RVSMCs was examined by CCK-8 assay. The significant decrease in cell viability resulting from H2O2 treatment (P=0.017) compared with untreated control; Fig. 1A), was significantly attenuated by TA i n a dose- dependent manner (P<0.05; Fig. 1A). To exclude any proliferative effect of TA from analysis, cell viability was assessed following TA treatment alone, and was demonstrated to be unaffected by treatment with TA at any of the concentrations tested (12.5, 25, and 50 µM) compared with untreated control (Fig. 1B). Thus, the concentrations 12.5, 25 and 50 µM were used in subsequent experiments.
Figure 1.

Effect of TA on RVSMC viability. (A) RVSMC viability was determined by CCK-8 assay following pretreatment with various concentrations of TA (0, 12.5, 25 and 50 µM) for 2 h and H2O2 treatment (final concentration 100 µM) for a further 24 h. (B) RVSMC viability was determined by CCK-8 assay following pretreatment with various concentrations of TA (0, 10, 50 and 100 µM) for 24 h. Data are presented as the mean ± standard deviation obtained from five individual experiments performed in triplicate. *P<0.05 vs. untreated control; #P<0.05 vs. H2O2-treated control. TA, tormentic acid; RVSMC, rat vascular smooth muscle cell; CCK, cell counting kit; H2O2, hydrogen peroxide.
Effect of TA on ROS generation in RVSMCs exposed to H2O2
As increased ROS levels, resulting in oxidative stress, are considered to be important in the pathogenesis of atherosclerosis, the effect of TA on ROS generation in RVSMCs exposed to H2O2 was investigated. Treatment with H2O2 for 2 h significantly increased the production of ROS compared with untreated control cells (P=0.019; Fig. 2). However, pretreatment with 25 µM (P=0.041) and 50 µM TA (P=0.024) significantly inhibited ROS generation compared with cells treated with H2O2 only (Fig. 2).
Figure 2.
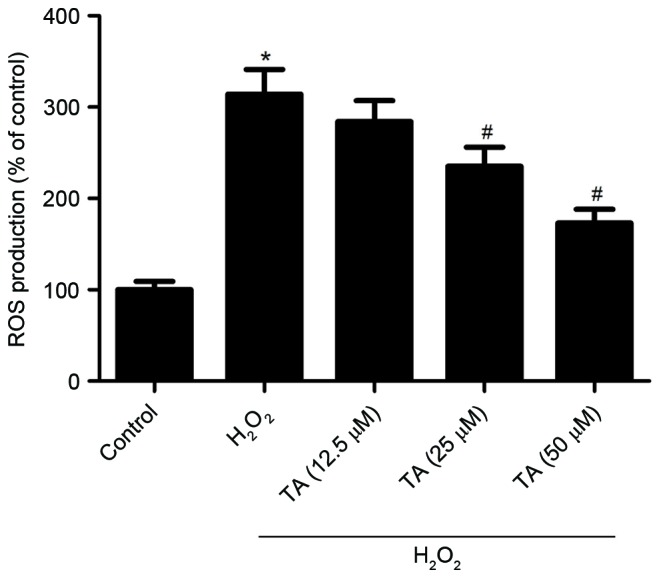
Effect of TA on ROS generation in RVSMCs exposed to H2O2. The level of intracellular ROS was measured following RVSMC 2 h pretreatment with various concentrations of TA (12.5, 25 and 50 µM) and H2O2 treatment (final concentration 100 µM) for a further 24 h. Data are presented as the mean ± standard deviation obtained from five individual experiments performed in triplicate. *P<0.05 vs. untreated control; #P<0.05 vs. H2O2-treated control. ROS, reactive oxygen species; TA, tormentic acid; H2O2, hydrogen peroxide; RVSMC, rat vascular smooth muscle cell.
Effect of TA on iNOS, NOX1 and p47phox protein expression in RVSMCs exposed to H2O2
The effect of TA on iNOS, NOX1 and p47phox protein expression levels were evaluated by western blot analysis in RVSMCsexposed to H2O2. Treatment with H2O2 for 2 h significantly increased the protein expression levels of iNOS (P=0.017), NOX1 (P=0.026) and p47phox (P=0.031) compared with untreated control cells (F ig. 3A and B). However, pretreatment with TA significantly inhibited the H2O2-induced expression of all three proteins in RVSMCs in a dose-dependent manner, compared with cells treated with H2O2 only (P<0.05; Fig. 3A and B).
Figure 3.
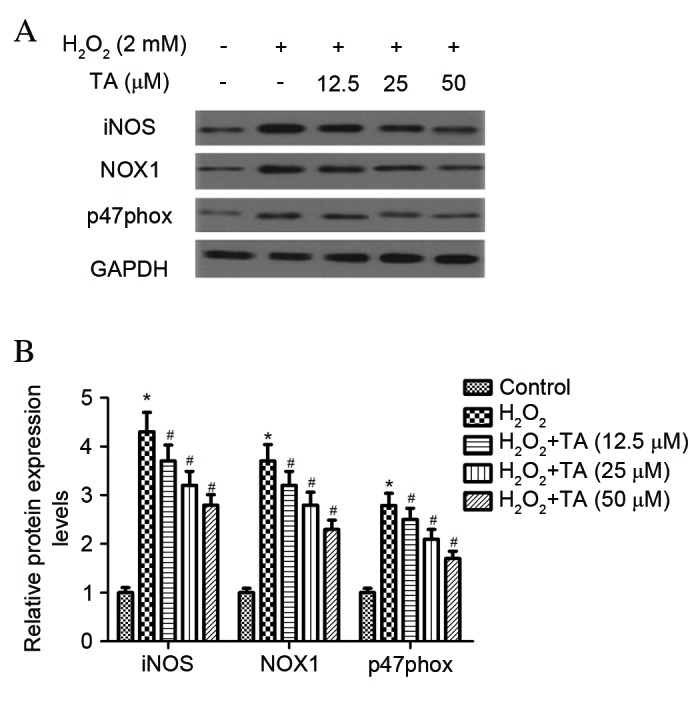
Effect of TA on iNOS and NADPH oxidase expression in RVSMCs exposed to H2O2. RVSMCs were pretreated with various concentrations of TA (0, 12.5, 25 and 50 µM) for 2 h, followed by the addition of H2O2 (100 µM as final concentration) for another 24 h. (A) Proteins were subjected to western blot analysis and (B) the relative protein levels of iNOS, p47phox and NADPH oxidase 1 were analyzed, using GAPDH as the loading control. Data are presented as the mean ± standard deviation obtained from five individual experiments performed in triplicate. *P<0.05 vs. untreated control; #P<0.05 vs. H2O2-treated control. RVSMC, rat vascular smooth muscle cell; TA, tormentic acid; iNOS, inducible nitric oxide synthase; NADPH, nicotinamide adenine dinucleotide phosphate; H2O2, hydrogen peroxide; NOX1, NADPH oxidase 1; p47phox, neutrophil cytosolic factor 1; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Effect of TA on TNF-α, IL-6 and IL-1β production in RVSMCs induced with H2O2
To investigate the anti-inflammatory effects of TA on RVSMCs, TNF-α, IL-6 and IL-1β production was evaluated using ELISA, revealing that H2O2 significantly increased the production of TNF-α (P= 0.021; Fig. 4A), IL-6 (P=0.016; Fig. 4B) and IL-1β (P=0.031; Fig. 4C) in the RVSMCs, compared with untreated cells. Compared with cells treated with H2O2 only, TA significantly decreased the production of TNF-α (P=0.028; Fig. 4A), IL-6 (P=0.023; Fig. 4B) and IL-1β (P= 0.016; Fig. 4C) in a dose-dependent manner.
Figure 4.
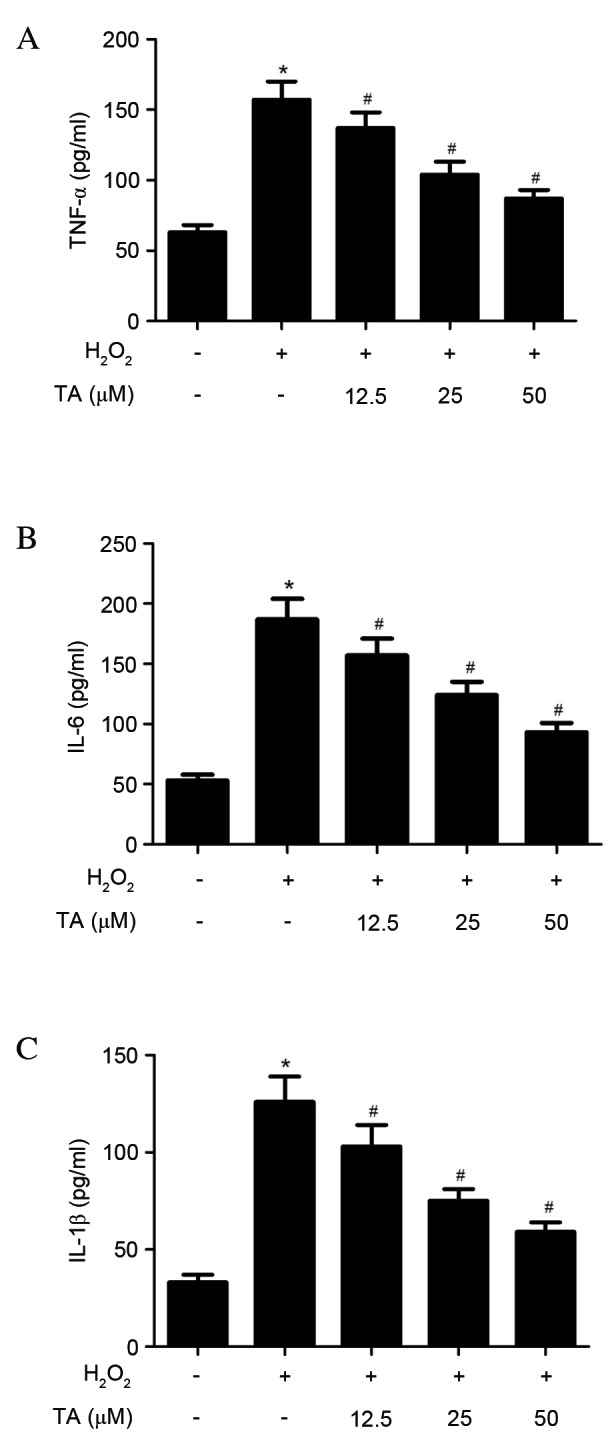
Effects of TA on TNF-α, IL-6 and IL-1β production in RVSMCs induced with H2O2. RVSMCs were pretreated with various concentrations of TA (0, 12.5, 25 and 50 µM) for 2 h, followed by treatment with H2O2 (final concentration 100 µM) for a further 24 h. Enzyme-linked immunosorbent assay was performed to quantify (A) TNF-α, (B) IL-6 and (C) IL-1β levels. Data are presented as the mean ± standard deviation obtained from five individual experiments performed in triplicate. *P<0.05 vs. untreated control; #P<0.05 vs. H2O2-treated control. RVSMC, rat vascular smooth muscle cell; TNF-α, tumor necrosis factor-α; H2O2, hydrogen peroxide; TA, tormentic acid; IL, interleukin.
Effects of TA on NF-ĸB signaling pathway in H2O2-induced RVSMCs
As NF-ĸB has been previously reported to be important in the regulation of cytokine production, the effects of TA on H2O2-induced NF-ĸB activation were investigated. As demonstrated in Fig. 5, H2O2 significantly increased NF-ĸB p65 phosphorylation (P=0.018; Fig. 5A and B) and IĸBα degradation (P=0.027; Fig. 5A and C) compared with untreated cells. However, TA pretreatment prevented NF-ĸB p65 phosphorylation (P=0.013; Fig. 5A and B) and IĸBα degradation (P=0.019; Fig. 5A and C) induced by H2O2 in RVSMCs in a dose-dependent manner, compared with cells treated with H2O2 only. No differences in the levels of NF-κB p65 were observed in H2O2-induced RVSMC (Fig. 5A; quantitative data not shown).
Figure 5.
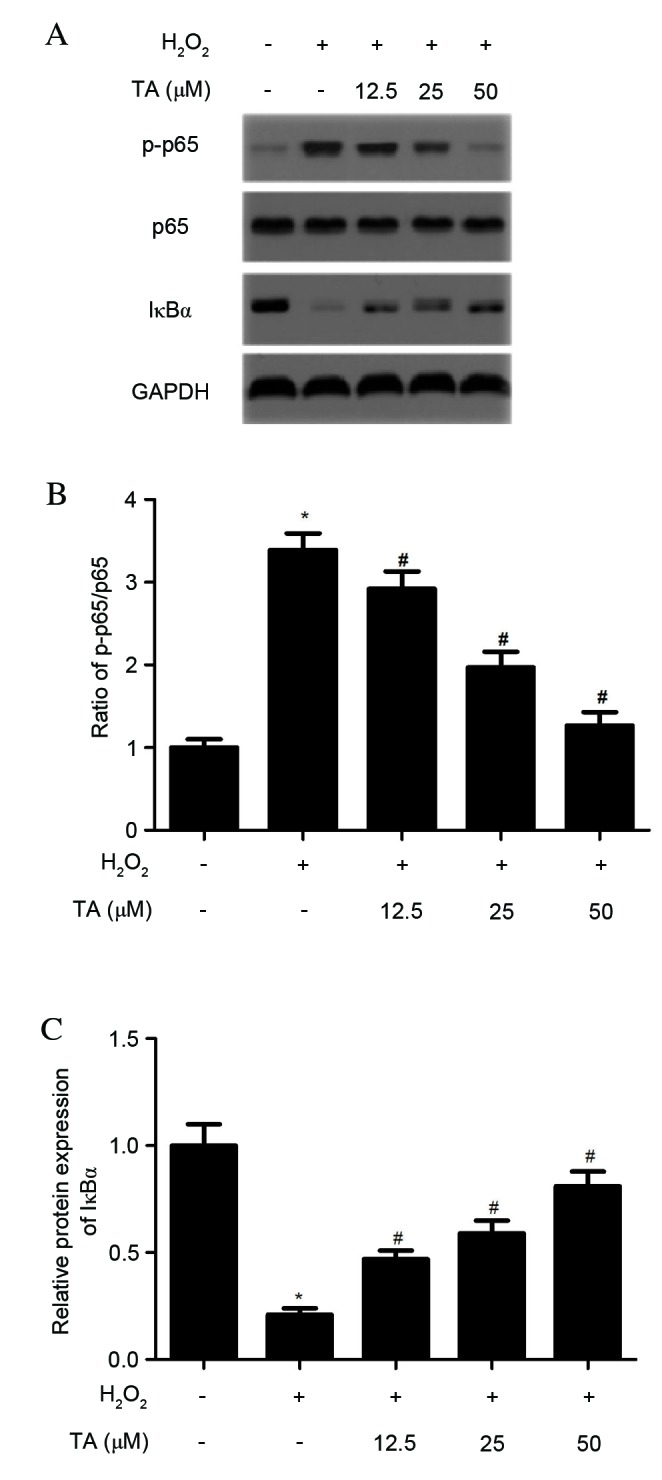
Effects of TA on the NF-ĸB signaling pathway in H2O2-induced RVSMCs. RVSMCs were pretreated with various concentrations of TA (0, 12.5, 25 and 50 µM) for 2 h, followed by the addition of H2O2 (final concentration 100 µM) for a further 24 h. (A) Proteins were subjected to western blot analysis to detect relative protein levels of NF-ĸB p65, p-NF-ĸB p65 and IĸBα, using GAPDH as the loading control. For each treatment, (B) the ratio of phosphorylated to non-phosphorylated NF-ĸB p65, and (C) the relative protein levels of IĸBα were analyzed. Data are presented as the mean ± standard deviation obtained from five individual experiments performed in triplicate. *P<0.05 vs. untreated control; #P<0.05 vs. H2O2-treated control. RVSMC, rat vascular smooth muscle cell; NF-ĸB, nuclear factor-ĸB; H2O2, hydrogen peroxide; TA, tormentic acid; p-, phosphorylated; IĸBα, NF-κB inhibitor-α.
Discussion
The present study demonstrated that TA inhibits H2O2-induced ROS generation in RVSMCs, and H2O2-induced expression of iNOS and NOX1 in RVSMCs. In addition, TA was demonstrated to significantly decrease the production of TNF-α, IL-6 and IL-1β. Furthermore, TA pretreatment reduced NF-ĸB p65 phosphorylation and IĸBα degradation induced by H2O2 in RVSMCs.
Oxidative stress is frequently involved in cardiovascular disease and is a common feature of early stage-atherosclerosis as a response to vascular injury (14,15). H2O2 has previously been demonstrated to activate signaling pathways to stimulate ROS production in vascular cells (16–18). The present study demonstrated that treatment with TA significantly inhibits the generation of ROS induced by H2O2 i n RVSMCs.
Previous studies have reported that iNOS may exacerbate atherosclerosis, as ApoE−/− mice lacking the iNOS gene exhibit decreased atherosclerotic lesion formation compared with ApoE−/− mice (19). NOXs are transmembrane enzymes that transport electrons from cytoplasmic NADPH to molecular oxygen, leading to superoxide generation, and are therefore an important source of vascular ROS (20). Several studies have demonstrated that NOX1 expression is increased in atherosclerosis (21,22), and that p47phox is an essential component of NOX (23). In smooth muscle cells, H2O2 activates NOX, resulting in the production of O2−, and, consequently, oxidant-induced injury (24). Similarly, the present study observed that H2O2 significantly increases the expression of iNOS, NOX1 and p47phox in RVSMCs, whereas pretreatment with TA significantly abrogates this effect.
Inflammatory cytokines are involved in the early stages of atherosclerosis (25). TNF-α is the earliest and primary endogenous mediator in the process of inflammation, and is involved in promoting inflammatory cell infiltration, injuring vascular endothelial cells and stimulating the generation of ROS (26). IL-1β is one of the most potent pro-inflammatory cytokines and endogenous pyrogens, and stimulates the acute phase response (27). In response to oxidative stimuli, VSMCs undergo a phenotypic change to a 'proliferative, migrating and synthetic' state, characterized by excess extracellular matrix and inflammatory cytokine production (28). In accordance with these results, the present study demonstrated that H2O2 significantly increases the production of TNF-α, IL-6 and IL-1β in RVSMCs. However, TA significantly attenuated H2O2-induced production of TNF-α, IL-6 and IL-1β i n RVSMCs.
NF-ĸB represents a family of transcription factors, including p50 and p65 that are important in the regulation of inflammatory responses (29). In response to oxidative stress, Activated IκB kinase phosphorylates the NF-κB inhibitor, IκB, resulting in its polyubiquitination and proteasomal degradation (30). IκB degradation leads to the translocation of NF-κB p50 and p65 to the nucleus, which results in the transcription of a variety of genes participating in diverse cellular processes, including inflammation, proliferation, apoptosis, and cellular senescence (31). Pierce et al (32) observed that the NF-κB inhibitor, salsalate, increases IκB expression levels, and decreases the levels of NF-κB and p47phox NADPH oxidase subunit in endothelial cells. The activation of NF-κB by ROS has also previously been demonstrated to induce TNF-α, IL-6 and IL-1β release in VSMCs (33). The present study demonstrated that pretreatment of RVSMCs with TA prevents H2O2-induced NF-ĸB p65 phosphorylation and IĸBα degradation in a dose-dependent manner. This suggests that TA may reduce H2O2-induced ROS generation through the action of NOX, and reduces TNF-α, IL-6 and IL-1β protein expression levels and induction of iNOS through inhibition of NF-ĸB signaling activation.
In conclusion, the present study demonstrated that TA inhibits H2O2-induced oxidative stress and inflammation in RVSMCs via inhibition of the NF-ĸB signaling pathway. TA may, therefore, have potential as a pharmacological agent in the prevention or treatment of atherosclerosis.
References
- 1.Insull W., Jr The pathology of atherosclerosis: Plaque development and plaque responses to medical treatment. Am J Med. 2009;122(Suppl1):S3–S14. doi: 10.1016/j.amjmed.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 2.Rudijanto A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Med Indones. 2007;39:86–93. [PubMed] [Google Scholar]
- 3.Al Ghouleh I, Khoo NK, Knaus UG, Griendling KK, Touyz RM, Thannickal VJ, Barchowsky A, Nauseef WM, Kelley EE, Bauer PM, et al. Oxidases and peroxidases in cardiovascular and lung disease: New concepts in reactive oxygen species signaling. Free Radic Biol Med. 2011;51:1271–1288. doi: 10.1016/j.freeradbiomed.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Csányi G, Taylor WR, Pagano PJ. NOX and inflammation in the vascular adventitia. Free Radic Biol Med. 2009;47:1254–1266. doi: 10.1016/j.freeradbiomed.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clempus RE, Griendling KK. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc Res. 2006;71:216–225. doi: 10.1016/j.cardiores.2006.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banno N, Akihisa T, Tokuda H, Yasukawa K, Higashihara H, Ukiya M, Watanabe K, Kimura Y, Hasegawa J, Nishino H. Triterpene acids from the leaves of Perilla frutescens and their anti-inflammatory and antitumor-promoting effects. Biosci Biotechnol Biochem. 2004;68:85–90. doi: 10.1271/bbb.68.85. [DOI] [PubMed] [Google Scholar]
- 7.Park GH, Lee JY, Kim DH, Cho YJ, An BJ. Anti-oxidant and antiinflammatory effects of Rosa multiform root. J Life Sci. 2011;21:1120–1126. doi: 10.5352/JLS.2011.21.8.1120. [DOI] [Google Scholar]
- 8.Bortalanza LB, Ferreira J, Hess SC, Delle Monache F, Yunes RA, Calixto JB. Anti-allodynic action of the tormentic acid, a triterpene isolated from plant, against neuropathic and inflammatory persistent pain in mice. Eur J Pharmacol. 2002;453:203–208. doi: 10.1016/S0014-2999(02)02428-7. [DOI] [PubMed] [Google Scholar]
- 9.Ivorra M, Paya M, Villar A. Hypoglycemic and insulin release effects of tormentic acid: A new hypoglycemic natural product. Planta Med. 1988;54:282–285. doi: 10.1055/s-2006-962433. [DOI] [PubMed] [Google Scholar]
- 10.Wu JB, Kuo YH, Lin CH, Ho HY, Shih CC. Tormentic acid, a major component of suspension cells of Eriobotrya japonica, suppresses high-fat diet-induced diabetes and hyperlipidemia by glucose transporter 4 and AMP-activated protein kinase phosphorylation. J Agric Food Chem. 2014;62:10717–10726. doi: 10.1021/jf503334d. [DOI] [PubMed] [Google Scholar]
- 11.Fogo AS, Antonioli E, Calixto JB, Campos AH. Tormentic acid reduces vascular smooth muscle cell proliferation and survival. Eur J Pharmacol. 2009;615:50–54. doi: 10.1016/j.ejphar.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 12.An HJ, Kim IT, Park HJ, Kim HM, Choi JH, Lee KT. Tormentic acid, a triterpenoid saponin, isolated from Rosa rugosa, inhibited LPS-induced iNOS, COX-2, and TNF-α expression through inactivation of the nuclear factor-κb pathway in RAW 264.7 macrophages. Int Immunopharmacol. 2011;11:504–510. doi: 10.1016/j.intimp.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 13.Chamley-Campbell J, Campbell GR, Ross R. The smooth muscle cell in culture. Physiol Rev. 1979;59:1–61. doi: 10.1152/physrev.1979.59.1.1. [DOI] [PubMed] [Google Scholar]
- 14.Parthasarathy S, Khan-Merchant N, Penumetcha M, Santanam N. Oxidative stress in cardiovascular disease. J Nucl Cardiol. 2001;8:379–389. doi: 10.1067/mnc.2001.114150. [DOI] [PubMed] [Google Scholar]
- 15.Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol. 2003;91:7A–11A. doi: 10.1016/S0002-9149(02)03144-2. [DOI] [PubMed] [Google Scholar]
- 16.Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. Biofactors. 2003;17:287–296. doi: 10.1002/biof.5520170128. [DOI] [PubMed] [Google Scholar]
- 17.Griendling KK, Sorescu D, Lassègue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–2183. doi: 10.1161/01.ATV.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 18.Taniyama Y, Griendling KK. Reactive oxygen species in the vasculature: Molecular and cellular mechanisms. Hypertension. 2003;42:1075–1081. doi: 10.1161/01.HYP.0000100443.09293.4F. [DOI] [PubMed] [Google Scholar]
- 19.Detmers PA, Hernandez M, Mudgett J, Hassing H, Burton C, Mundt S, Chun S, Fletcher D, Card DJ, Lisnock J, et al. Deficiency in inducible nitric oxide synthase results in reduced atherosclerosis in apolipoprotein E-deficient mice. J Immunol. 2000;165:3430–3435. doi: 10.4049/jimmunol.165.6.3430. [DOI] [PubMed] [Google Scholar]
- 20.Bedard K, Krause KH. The NOX family of ROS-generating NAPDH oxidase: Physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 21.Sheehan AL, Carrell S, Johnson B, Stanic B, Banfi B, Miller FJ., Jr Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis. 2011;216:321–326. doi: 10.1016/j.atherosclerosis.2011.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dikalova AE, Góngora MC, Harrison DG, Lambeth JD, Dikalov S, Griendling KK. Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via eNOS uncoupling. Am J Physiol Heart Circ Physiol. 2010;299:H673–H679. doi: 10.1152/ajpheart.00242.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barry-Lane PA, Patterson C, van der Merwe M, Hu Z, Holland SM, Yeh ET, Runge MS. p47phox is required for atherosclerotic lesion progression in ApoE(−/−) mice. J Clin Invest. 2001;108:1513–1522. doi: 10.1172/JCI200111927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li WG, Miller FJ, Jr, Zhang HJ, Spitz DR, Oberley LW, Weintraub NL. H(2)O(2)-induced O(2) production by a non-phagocytic NAD(P)H oxidase causes oxidant injury. J Biol Chem. 2001;276:29251–29256. doi: 10.1074/jbc.M102124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 26.Li D, Fu Y, Zhang W, Su G, Liu B, Guo M, Li F, Liang D, Liu Z, Zhang X, et al. Salidroside attenuates inflammatory responses by suppressing nuclear factor-κB and mitogen activated protein kinases activation in lipopolysaccharide-induced mastitis in mice. Inflamm Res. 2013;62:9–15. doi: 10.1007/s00011-012-0545-4. [DOI] [PubMed] [Google Scholar]
- 27.Dinarello C. The IL-1 family and inflammatory diseases. Clin Exp Rheumatol. 2002;20(5 Suppl 27):S1–S13. [PubMed] [Google Scholar]
- 28.O'Brien KD, McDonald TO, Chait A, Allen MD, Alpers CE. Neovascular expression of E-selectin, intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in human atherosclerosis and their relation to intimal leukocyte content. Circulation. 1996;93:672–682. doi: 10.1161/01.CIR.93.4.672. [DOI] [PubMed] [Google Scholar]
- 29.Barnes PJ, Karin M. Nuclear factor-kappaB: A pivotal transcription factor in chronic inflammatory disease. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 30.Tumur Z, Shimizu H, Enomoto A, Miyazaki H, Niwa T. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-kappaB activation. Am J Nephrol. 2010;31:435–441. doi: 10.1159/000299798. [DOI] [PubMed] [Google Scholar]
- 31.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 32.Pierce GL, Lesniewski LA, Lawson BR, Beske SD, Seals DR. Nuclear factor-{kappa}B activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans. Circulation. 2009;119:1284–1292. doi: 10.1161/CIRCULATIONAHA.108.804294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. 2009;78:539–552. doi: 10.1016/j.bcp.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]