

Abstract
Cabazitaxel has been used to treat castration-resistant prostate cancer since its approval by the US Food and Drug Administration in 2010. However, whether cabazitaxel may inhibit the proliferation of other tissue-derived cancer cells, and its underlying mechanism, remains unknown. In the present study, the A549 lung adenocarcinoma cancer cell line was exposed to cabazitaxel, in order to investigate its cytotoxic effect and determine the underlying mechanism. The results demonstrated that cabazitaxel was able to induce autophagy in A549 cells, as evidenced by the formation of autophagosomes, upregulated LC3-II expression and increased LC3 puncta. Cabazitaxel-induced autophagy had a cytotoxic effect on A549 cells, as evidenced by the induction of cell death and cell cycle arrest at G2/M phase, which was independent of the apoptotic pathway. Furthermore, transfection with Beclin1 small interfering RNA and treatment with the autophagy inhibitor 3-methyladenine protected cells from cabazitaxel-induced cell death, thus confirming that cabazitaxel-induced autophagy contributed to A549 cell death. In addition, cabazitaxel targeted the phosphoinositide 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway to induce autophagy, as indicated by reduced phosphorylation of Akt and mTOR. In conclusion, the present study demonstrated that cabazitaxel exerts a cytotoxic effect on A549 cells by acting on the PI3K/Akt/mTOR pathway to promote autophagic cell death. This result supports the potential use of cabazitaxel as a chemotherapeutic agent for the treatment of lung cancer.
Keywords: cabazitaxel, A549 lung adenocarcinoma cancer cell line, autophagic cell death, PI3K/Akt/mTOR signaling pathway
Introduction
Autophagy is a physiological process that is vital to homeostasis, which is responsible for the elimination of damaged or aged organelles from the cell (1). Autophagy can be stimulated by various stressors, including starvation and oxidative stress, or by treatment with some pharmacological agents (2). In addition to its roles in maintaining normal cellular homeostasis, autophagy has an important role in the occurrence of cancer, as well as infectious, inflammatory, neurodegenerative and metabolic diseases (3–7). It has previously been reported that autophagy acts as a 'double-edged sword' in tumor therapy; namely, it enables tumor cell survival in response to stress, thus promoting tumorigenesis; however, it also induces tumor cell death, thus preventing tumor occurrence (8). Autophagy-induced cell death is classified as type II programmed cell death (PCD), which differs from apoptosis, which is classified as type I PCD (9,10). In this setting, autophagy is considered an important underlying mechanism of tumor suppression via the induction of non-apoptotic cell death. During autophagy, the presence of autophagosomes is considered the typical morphological feature, the outer membrane of which fuses with lysosomes to form autolysosomes in which luminal materials, including the internal membrane, are degraded (11). Microtubule-associated protein light chain 3 (LC3)-II, which is involved in autophagosome biogenesis and is localized to the autophagosome membrane, is a marker of autophagosomes. In addition, sequestosome 1 (SQSTM1)/p62 is another protein marker that can be used to monitor autophagic flux (12).
Chemotherapy is currently considered a valuable therapeutic strategy for the treatment of tumors. Chemotherapy-induced apoptosis, which is associated with the release of cytochrome c from the mitochondria via extrinsic or intrinsic pathways, is the predominant mechanism underlying cancer cell growth inhibition by chemotherapeutic drugs such as fluorouracil, cisplatin, cyclophosphamide and paclitaxel (13–15). Furthermore, autophagy-induced cell death has an important role in tumor cell death under certain conditions. Mono-Pt, which is a novel cytoprotective monofunctional platinum (II) complex, inhibits cell growth and proliferation via autophagic cell death in apoptosis-resistant ovarian cancer. Treatment of Caov-3 ovarian carcinoma cells with Mono-Pt resulted in punctate distribution of LC3, an increased ratio of LC3-II to LC3-I, and accumulation of autophagic vacuoles in the cytoplasm (16). In addition, Q6, gefitinib, vitexin 6, and other compounds or plant extracts exert potent antiproliferative effects via the autophagy-dependent degradation pathway (17–19). Autophagic cell death induced by chemical drugs or plant extracts may shed new light on therapeutic strategies used to suppress tumor growth.
Cabazitaxel was approved by the US Food and Drug Administration in 2010 for use in treating paclitaxel resistance or advanced prostate cancer. In addition, it has been reported that cabazitaxel possesses broad-spectrum antitumor activity against the proliferation of colon cancer cells, as well as pancreatic cancer, gastric cancer, head and neck cancer, breast cancer, etc. (20,21). Taxane agents, including paclitaxel, have been demonstrated to induce autophagy in various cancer cells (22). As a broad-spectrum antitumor and novel taxane agent, it remains unclear whether cabazitaxel may induce cytotoxic effects via the autophagic pathway in A549 cells. In order to extend the future clinical application of cabazitaxel, the present study aimed to investigate the cytotoxic effects induced by cabazitaxel and to illustrate its underlying mechanism in A549 cells. The results demonstrated that cabazitaxel may induce autophagic cell death in a phosphoinositide 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway-dependent manner.
Materials and methods
Cell culture
The A549 human lung adenocarcinoma epithelial cell line was purchased from Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). The cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS; Sijiqion, Hangzhou, China) and 100 U of penicillin G with 100 µg of streptomycin per ml, cells were grown in a humidified atmosphere containing 5% CO2 at 37°C.
Chemicals, reagents and antibodies
Cabazitaxel was kindly provided by Professor Shengyong Zhang (Department of Medicinal Chemistry, Fourth Military Medical University, Xi'an, China). Cabazitaxel was dissolved at a concentration of 50 mg/ml in absolute ethyl alcohol as a stock solution, which was stored at 4°C, and was diluted with medium prior to each experiment. z-VAD-FMK and Annexin V/propidium iodide (PI) apoptosis detection kit was obtained from Beyotime Institute of Biotechnology (Hangzhou, China). Anti-LC3 (cat. no. L8918; 1:800), anti-beclin1 (cat. no. SAB5300513-100UL; 1:1,000) and anti-p62 (cat. no. P0067; 1:1,000) antibodies, 5-fluorouracil (5-FU) and 3-methyladenine (3-MA) were purchased from Sigma-Aldrich (Merck Millipore, Darmstadt, Germany). Anti-mTOR (cat no 653401; 1:1,000), anti-phosphorylated (p)-mTOR (Ser2481) (ca. no 651701; 1:500), anti-Akt (cat. no. 603401; 1:500) and anti-p-Akt (Ser473) (cat. no. 649001; 1:500) antibodies were purchased from BioLegend (San Diego, CA, USA). Anti-human β-actin monoclonal antibody was provided by Cwbiotech (Beijing, China; cat. no. CW0096M; 1:2,000).
Apoptosis assay
Apoptosis assay was conducted with Annexin V/PI detection kit according to manufacturer's protocol. Briefly, 1×105 A549 cells/well were treated with 5 µg/ml cabazitaxel and 20 µM 5-FU for 30 h. Next, the treated cells were harvested and washed 3 times with cold PBS. Cells were subsequently stained with 5 µl FITC Annexin V and 10 µl PI for 15 min at room temperature and subjected to flow cytometry for analysis apoptosis.
Cell death assay
Cell death was assessed by trypan blue exclusion assay. Following treatment with various concentration of Cabazitaxel (1, 2, 5, 1 and 20 µg/ml) at different times, suspended and adherent cells were collected and stained with 0.4% trypan blue dye for 3 min at room temperature. Cells were subsequently counted using a hemocytometer under a light microscope. Pharmaceutic inhibitors, z-VAD-FMK and 3-MA, were used to determine the apoptotic and autophagic cell death. Cells were pretreated with 20 µM z-VAD-FMK or 10 mM 3-MA for 1 h at 37°C and then incubated with cabazitaxel. Next, cells were stained with 0.4% trypan blue in order to detect cell death.
Cell cycle analysis
Cell cycle analysis was performed as described previously (23). Briefly, cells were synchronized by culturing in RPMI-1640 medium containing 0.5% FBS overnight. The cells were then incubated in fresh RPMI-1640 medium supplemented with 10% FBS and serially diluted cabazitaxel at 37°C. The treated cells (5 µg/ml cabazitaxel) were harvested after a 24 h incubation and were washed with cold phosphate-buffered saline (PBS) before being fixed with 70% ethanol at 4°C overnight. After thoroughly washing with PBS, the fixed cells were stained in the dark with 50 µg/ml propidium iodile (PI) containing 100 µg/ml RNase A and 1% Triton X-100 for 45 min at room temperature.. The cells were finally subjected to flow cytometric analysis.
Transmission electron microscopy (TEM)
A549 cells were seeded in a 6-well plate and treated with 5 µg/ml cabazitaxel for 24 h. Cells were digested and fixed with 3% glutaraldehyde in PBS (pH 7.8) for 3 h at room temperature, and were then incubated with 0.1% osmium tetroxide for 1 h at 4°C. Cells were subsequently dehydrated in increasing concentrations of ethanol (50, 60, 80 and 100%) and were embedded in Epon resin. Ultrathin sections were loaded onto copper grids and were stained with uranyl acetate and lead citrate, prior to being observed under a transmission electron microscope (JEM-1230; JEOL Ltd., Tokyo, Japan).
Western blotting
A549 cells (seeded at 2×105) were treated with various concentrations (1, 2, 5, 10 and 20 µg/ml) of cabazitaxel for 24 h at 37°C and were lysed in radioimmunoprecipitation assay buffer (Beyotime Institute of Biotechnology). Proteins were quantified using a bicinchoninic acid assay kit (Cwbiotech Co., Ltd., Beijing, China) then 30 µg proteins per well were subjected to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and were transferred to polyvinylidene fluoride membranes. After blocking with 5% bovine serum albumin for 1 h at room temperature, the membranes were sequentially incubated with the corresponding primary antibodies overnight at 4°C, and with horseradish peroxidase-conjugated anti-rabbit immunoglobulin (Ig)G (cat. no. CW0103S; 1:1,000; Cwbiotech) or anti-mouse IgG (cat. no. 0102S, 1:1,000; Cwbiotech) for 1 h at room temperature. Finally, blots were developed and visualized using the SuperSignal West Dura chemiluminescence kit (Pierce; Thermo Fisher Scientific, Inc., Rockford, IL, USA) and an ImageQuant LAS 4000 mini system (GE Healthcare Life Sciences, Pittsburgh, PA, USA). Image J software (version 1.38; National Institutes of Health, Bethesda, MD, USA) was used to quantify protein bands. β-actin was used as a protein loading control.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
TUNEL staining was conducted to assess the levels of apoptosis induced by cabazitaxel or 5-FU (positive control) treatment according to the manufacturer's protocol (Beyotime Institute of Biotechnology). Briefly, A549 cells were fixed in 4% paraformaldehyde for 1 h at room temperature following treatment with cabazitaxel or 5-FU. Subsequently, the cells were permeabilized with 0.1% Triton X-100 and were incubated with 50 µl TUNEL reaction buffer at 37°C for 1 h in the dark. After staining with 4′,6-diamidino-2-phenylindole, slides were visualized by fluorescence microscopy.
Caspase 3 activity assay
Caspase 3 protease activity was measured using a Caspase-3 activity detection kit according to the manufacturer's protocol (Beyotime Institute of Biotechnology). Briefly, A549 cells were lysed at various time points following treatment with cabazitaxel or 5-FU. Cell lysates were centrifuged at 12,000 × g for 15 min at 4°C prior to incubation with Ac-DEVD-pNA for 60 min at 37°C. Absorbance was measured at 405 nm using a Biotek Eon microplate spectrophotometer (Biotek, Winooski, VT, USA) and caspase 3 activity was calculated according to a pNA standard curve.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total mRNA was extracted using TRIzol reagent from the A549 cells treated with various concentrations of cabazitaxel (1, 2, 5, 10 and 20 µg/ml) for 24 h at 37°C. Subsequently, cDNA was synthesized using the RevertAid First Strand cDNA synthesis kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Briefly, the total volume of 20 µl reverse transcription mixer (500 ng RNA, 1 µl oligo(dT)18, 1µl RevertAid RT) was incubated for 60 min at 42°C, then the reaction was terminated by heating at 70°C for 5 min. qPCR was performed on an ABI StepOnePlus™ thermocycler (Applied Biosystems; Thermo Fisher Scientific, Inc., Foster City, CA, USA) using a SYBR® Green PCR kit (Applied Biosystems; Thermo Fisher Scientific, Inc.). Each sample was run in triplicate in a final volume of 25 µl containing 1 µl first-strand cDNA template, 10 pmol of each primer (Table I; obtained from Sangon Biotech Co., Ltd, Shanghai, China), 12.5 µl of SYBR® Green PCR master mix and 11.5 µl distilled water. Cycling conditions of the qPCR were as follows: 1 cycle at 95°C for 30 sec, followed by 40 cycles at 95°C for 20 sec, 60°C for 30 sec and extension at 72°C for 15 sec.. Data analyses were performed according to the 2−ΔΔ Cq method (24).
Table I.
Primer sequences of autophagy-related genes.
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Atg5 | atgacagatgacaaagatg | caacgtcaaataacttactc |
| Atg7 | tgacgatcggatgaatgagcc | gctcatgtcccagattttggaag |
| Atg12 | ctgtgtaattgcgtccccct | gaagctgcaacacagactgc |
| LC3-II | ccgcaccttcgaacaaagag | aagctgcttctcacccttgt |
| β-actin | tgacggggtcacccacactg | aagctgtagccgcgctcggt |
Atg, autophagy related; LC3, microtubule-associated protein light chain 3.
Plasmid and small interfering (si)RNA transfection
Beclin1 siRNA (sense 5′-GGAGCCAUUUAUUGAAACUTT-3′ and antisense 5′-AGUUUCAAUAAAUGGCUCCTT-3′) was synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, China). Transfection of cells with green fluorescent protein (GFP)-LC3 plasmid (provided by Dr. Ye Wei) or Beclin1 siRNA was carried out using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) according the manufacturer's protocol. Briefly, 4 µg GFP-LC3 plasmid, 100 pmol Beclin1 siRNA and 5 µl Lipofectamine 2000 were diluted in 50 µl Opti-MEM I medium (Invitrogen; Thermo Fisher Scientific, Inc.), respectively, and were incubated for 5 min at room temperature. The diluted GFP-LC3 plasmid and Beclin1 siRNA were mixed with Lipofectamine 2000, separately, and were incubated for 20 min at room temperature. The complexes were then added to 6-well plates, 2×105 cells/well for transfection. Cabazitaxel was added to the cells at 5 µg/ml/well 48 h post-transfection, and the cells were incubated for a further 24 h at 37°C. LC3 puncta was visualized by fluorescence microscopy, and the protein expression levels of Beclin1 were determined by western blotting.
Statistical analysis
Data are presented as the mean ± standard deviation. Student's t-test and one-way analysis of variance were used to analyze data. Differences between experimental groups were assessed by non-parametric analysis using SPSS software, version 12 (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Cabazitaxel treatment induces autophagy in A549 cells
An increasing body of evidence has suggested that manipulation of autophagy may provides a useful way to prevent cancer development (25,26); therefore, the present study aimed to investigate whether cabazitaxel could induce autophagy in A549 lung cancer cells. Autophagosomal formation and the expression of autophagy-associated proteins were used to evaluate cabazitaxel-induced autophagy. Following exposure to various concentrations of cabazitaxel for 24 h, RT-qPCR was conducted to determine the mRNA expression of autophagy related (Atg) genes; the results demonstrated that the expression levels of LC3, Atg5, Atg7 and Atg12 were increased (Fig. 1A), with Atg7 exhibiting the highest expression levels. Furthermore, immunoblotting indicated that LC3-II protein expression and the conversion of LC3-I to LC3-II were significantly upregulated in cabazitaxel-treated cells compared with the control group (Fig. 1B). Conversely, SQSTM1/p62 expression was decreased following exposure to cabazitaxel (Fig. 1C).
Figure 1.

Cabazitaxel induces autophagy in A549 cells. (A) A549 cells were treated with various concentrations of cabazitaxel for 24 h; total RNA was extracted by TRIzol, and autophagy related (Atg) mRNA expression levels were analyzed by quantitative polymerase chain reaction using specific primers. (B and C) Proteins were extracted by radioimmunoprecipitation assay buffer following treatment with cabazitaxel. Samples underwent western blotting to determine protein expression levels. (D) A549 cells were transfected with green fluorescent protein-microtubule-associated protein light chain 3 (LC3) plasmids for 48 h, and were then treated with 5 µg/ml cabazitaxel for another 24 h. LC3 puncta were observed by fluorescence microscope. Magnification × 20, close-up image magnification, ×40. (E) Cabazitaxel-treated A549 cells were fixed and dehydrated, and subjected to transmission electron microscopy after staining with uranyl acetate and lead citrate. Bottom panel, magnification ×8,000. Top panel, ×30,000) Data are presented as the mean ± standard deviation of three independent experiments *P<0.05, **P<0.01 vs. control group. SQSTM1, sequestosome 1.
Since LC3 is a specific marker for autophagosomal formation, GFP-tagged LC3 was used to measure autophagic flux. As shown in Fig. 1D, the green puncta were distributed in the cytoplasm post-transfection, and the percentage of cells expressing LC3 was much higher in the treated group compared with in the control group. In addition, the presence of autophagic compartments was detected in cabazitaxel-treated cells by TEM. Following exposure to cabazitaxel for 24 h, accumulation of massive autophagic vacuoles (autophagosomes) was detected in the cytoplasm under TEM, and these autophagosomes consisted of double membrane structural compartments, which contain recognizable cellular organelles and a high electron density substance. Conversely, fewer autophagosomes were observed in the control group (Fig. 1E). Furthermore, typical apoptotic morphology was not observed under TEM, including chromatin condensation and apoptotic bodies; the nuclear membrane integrity and chromatin distribution of cabazitaxel-treated cells were normal. These results indicate that cabazitaxel could not induce apoptosis of A549 cells. Taken together, these findings support the hypothesis that cabazitaxel induces autophagy in A549 cells.
Cabazitaxel promotes cell death by inducing cell cycle arrest at G2/M phase
The present study subsequently assessed whether cabazitaxel-induced autophagy promoted cell survival or cell death. When A549 cells were exposed to various concentrations of cabazitaxel, cells detached from the bottom of the flask and were observed to be round-shaped. Therefore, it was hypothesized that cabazitaxel exhibited cytotoxicity in A549 cells. Cell death was observed by trypan blue assay following treatment with various doses of cabazitaxel (1–20 µg/ml) for 12, 24 or 48 h. As shown in Fig. 2A, cabazitaxel induced a time- and dose-dependent increase in A549 cell death; 1 µg/ml cabazitaxel treatment for 12 h induced slight cell death of A549 cells compared with the control group; subsequently, cell death was significantly increased in a dose- and time-dependent manner.
Figure 2.

Cabazitaxel promotes cell death and induce A549 cells cycle arrest at G2/M phase. (A) A549 cells were incubated with various concentrations of cabazitaxel for various durations, and cell death was determined by trypan blue dye exclusion assay. (B) A549 cells were synchronized overnight, and were then incubated with various concentrations of cabazitaxel for 24 h. The cells were harvested and stained with propidium iodide, and were subjected to cell cycle analysis by flow cytometry. Data are presented as the mean ± standard deviation of three independent experiments *P<0.05, **P<0.01 vs. control group.
To gain insight into the mechanism underlying the antiproliferative effects of cabazitaxel, the present study investigated the effects of cabazitaxel treatment on the cell cycle. The number of A549 cells at G2/M phase was significantly increased to 80–90% following incubation with various doses (5, 10, 20 µg/ml) of cabazitaxel for 24 h (Fig. 2B). Taken together, these results suggest that cabazitaxel exerts a cytotoxic cell death-promoting effect via the induction of G2/M phase arrest.
Cabazitaxel induces non-apoptotic cell death
Apoptosis is one of the major cell death pathways. To determine whether cabazitaxel-mediated cell death was induced via the apoptotic pathway, cabazitaxel-treated cells were stained with Annexin V/PI and TUNEL-fluorescein isothiocyanate (FITC). Following exposure to 5 µg/ml cabazitaxel for 24 h, the percentage of apoptotic cells was not significantly altered compared with in the control group, as determined by Annexin/PI staining and flow cytometry. Conversely, the majority of 5-FU-treated A549 cells were positive for apoptosis staining (Fig. 3A). To further confirm these results, a TUNEL-FITC assay was used to detect the apoptosis of cabazitaxel-treated A549 cells. Only weak fluorescence was detected in a very small number of cabazitaxel-treated and control A549 cells; however, strong fluorescence was detected in 5-FU-treated A549 cells (Fig. 3B).
Figure 3.

Cabazitaxel induces non-apoptotic cell death in A549 cells. (A and B) A549 cells were exposed to 5 µg/ml cabazitaxel or 20 µM 5-fluorouracil (5-FU) for 24 h at 37°C, then stained with Annexin V/PI or terminal deoxynucleotidyl transferase dUTP nick end labeling-fluorescein isothiocyanate (FITC)/propidium iodide and 4′,6-diamidino-2-phenylindole (DAPI), and were then subjected to flow cytometry or fluorescence imaging. Magnification, ×10. (C) Cabazitaxel (5 µg/ml) or 5-FU (20 µM) treated A549 cells were harvested and lysed, and the collected supernatants were subjected to caspase 3 activity assay. (D) A549 cells were pretreated with 20 µM z-VAD-FMK for 1 h, were incubated with cabazitaxel (5 µg/ml) or 5-FU for 24 h, and cell death was subsequently determined by trypan blue dye exclusion assay. Normal non-treated A549 cells were used as the control. Data are presented as the mean ± standard deviation of three independent experiments *P<0.05, **P<0.01.
Caspase 3 is an apoptosis-associated cysteine peptidase, which has a central role in the execution phase of cell apoptosis. Therefore, the present study investigated caspase 3 activity in A549 cells, and demonstrated that 5-FU, rather than cabazitaxel, activated caspase 3 in A549 cells (Fig. 3C). z-VAD-FMK is a pan-caspase inhibitor, which can be used to block all features of apoptosis, including apoptotic cell death. Notably, z-VAD-FMK was not able to block cabazitaxel-induced cell death; however, it could inhibit 5-FU-induced cell death, thus providing evidence for caspase-independent cell death in A549 cells (Fig. 3D). These results indicate that cabazitaxel-induced cytotoxicity is caused by alternative caspase 3-independent non-apoptotic cell death.
Cabazitaxel-induced autophagy contributes to cell death
As aforementioned, cabazitaxel treatment was able to induce autophagy in A549 cells. Subsequently, the present study used 3-MA, a well-known inhibitor of autophagy, to examine whether cell death was triggered by autophagy. A549 cells were pretreated with 10 mM 3-MA for 1 h, and cabazitaxel was then added to the pretreated cells for an additional 24 h. As shown in Fig. 4A, 3-MA resulted in a partial but significant inhibition of cabazitaxel-induced cell death. Subsequently, cells were transfected with siRNA specific for Beclin1, which is required for autophagy, in order to elucidate the association between autophagy and cabazitaxel-induced cell death. Beclin1-siRNA suppressed Beclin1 expression and decreased cabazitaxel-induced cell death post-transfection (Fig. 4B). These findings indicate that autophagy contributes to the cabazitaxel-induced death of A549 cells.
Figure 4.

Autophagy triggers cell death. (A) A549 cells were pretreated with 10 mM 3-methyladenine (3-MA) for 1 h and were incubated with 5 µg/ml cabazitaxel for 48 h. Subsequently, cell death was determined by trypan blue dye exclusion assay at various time-points. (B) A549 cells were transfected with 100 pmol Beclin1 small interfering (si)RNA, and proteins were extracted 48 h post-transfection. Beclin1 protein expression was detected by western blotting. Transfected cells were incubated with 5 µg/ml cabazitaxel for an additional 48 h, and cell death was determined by trypan blue dye exclusion assay at various time-points. Data are presented as the mean ± standard deviation of three independent experiments *P<0.05, **P<0.01.
PI3K/Akt/mTOR signaling pathway inhibition activates cabazitaxel-induced autophagy
The PI3K/Akt/mTOR pathway is an intracellular signaling pathway that is involved in autophagy regulation; inhibition of the PI3K/Akt/mTOR pathway promotes autophagy. To determine the role of this pathway, two critical proteins, Akt and mTOR, were examined. Following treatment with cabazitaxel for 24 h, the levels of p-mTOR and p-Akt were markedly decreased compared with the control (Fig. 5). These results indicate that cabazitaxel targets the PI3K/Akt/mTOR pathway to induce autophagic cell death via the inhibition of Akt and mTOR phosphorylation.
Figure 5.
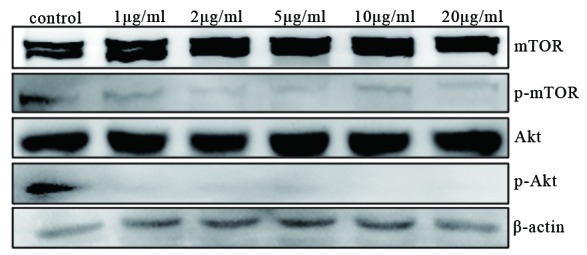
Inhibition of the phosphoinositide 3 -kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling pathway activates cabazitaxel-induced autophagy. Proteins were extracted from cabazitaxel-treated A549 cells and the phosphorylation of mTOR and Akt was detected by western blotting. P-, phosphorylated.
Discussion
Cabazitaxel is a novel agent for the treatment of metastatic hormone-refractory prostate cancer. A previous study reported that cabazitaxel acts as a broad-spectrum chemotherapy agent that may prevent cancer progression (20). In addition, our pilot studies confirmed that cabazitaxel inhibited the proliferation of various types of human cancer cell, including A549 lung cancer cells; SW480 and HT-29 colon cancer cells; LNCaP, DU145 and PC-3 prostate cancer cells; as well as murine M16 melanoma cancer cells. We also demonstrated that morphological alterations of the cabazitaxel-treated A549 cells were more obvious than in the prostate cancer cell lines; the cabazitaxel-treated cells exhibited detachment, a round shape and shrinkage. Furthermore, cell death induced by cabazitaxel was increased in A549 cells compared with in other human tissue-derived cancer cell lines (data not published). Based on these findings, the present study used A549 cells as the target to study the antitumor effects of cabazitaxel.
The present study revealed a novel molecular mechanism underlying the antitumor activity of cabazitaxel in A549 cells. When A549 cells were treated with cabazitaxel, the number of autophagosomes, LC3-II expression and autophagic flux were increased. Concurrently, cabazitaxel was able to promote A549 cell death independent of the apoptotic pathway. Further evidence indicated that cabazitaxel-induced autophagy contributed to A549 cell death via the PI3K/Akt/mTOR pathway. Therefore, it may be concluded that cabazitaxel-induced autophagic cell death is the predominant mechanism underlying A549 cell proliferation inhibition. These data may help to provide the theoretical basis for the clinical application of cabazitaxel.
PCD is a crucial process for the organized destruction of cells, which is essential for the development and maintenance of cellular homeostasis. PCD can be divided into three categories: Apoptotic, autophagic and necrotic cell death, and apoptotic cell death is considered a vital defense mechanism to eliminate cancer cells (27). In the present study, cabazitaxel-treated A549 cells did not exhibit typical apoptotic morphology and increased caspase 3 activity. In addition, z-VAD-FMK was not able to inhibit cell death, thus suggesting that cabazitaxel may induce A549 cell death independent of the apoptotic pathway. Subsequently, the present study investigated whether cabazitaxel was able to induce autophagic cell death. Following exposure to cabazitaxel, typical features of autophagy were observed, including high LC3-II expression, Atg gene upregulation, p62 downregulation and autophagic vacuole formation; however, features associated with apoptosis were not detected in A549 cells. In addition, siRNA Beclin1 gene silencing and treatment with an autophagy inhibitor rescued cabazitaxel-induced cell death. There are three criteria to certify autophagic cell death: i) Cell death occurs without the involvement of apoptosis; ii) there is an increase in autophagic flux, alongside an increase in autophagic markers, in the dying cells; and iii) suppression of autophagy via pharmacological inhibitors or genetic approaches is able to rescue or prevent cell death (28). The results of the present study fully coincided with the aforementioned criteria of autophagic cell death. Therefore, the present study concluded that autophagy contributes to cabazitaxel-induced A549 cell death, which serves as a protective mechanism to facilitate the elimination of cancer cells.
Previous studies have demonstrated that autophagic cell death is triggered by numerous signaling pathways, such as the adenosine monophosphate-activated protein kinase pathway, the PI3K/AKT/mTOR pathway, and the mitogen-activated protein kinases (extracellular signal-regulated kinases, p38 and c-Jun N-terminal kinases) pathway (29–32). The PI3K/AKT/mTOR signaling pathway represents one of the major survival pathways that is dysregulated in various types of human cancer, and contributes to cancer pathogenesis and therapy resistance. It has previously been demonstrated that inhibition of the PI3K/Akt/mTOR pathway induces G2/M arrest, whereas Akt promotes cell cycle progression through the G2/M transition via suppression of the cyclin-dependent kinase cell division control (Cdc)2 and Cdc25c pathway (33). In the present study, A549 cells were arrested at G2/M phase following treatment with cabazitaxel, thus suggesting that cabazitaxel-mediated G2/M arrest may be induced via inhibition of the PI3K/Akt/mTOR pathway. Mounting evidence has indicated that the PI3K/AKT/mTOR signaling cascade regulates autophagy. As a member of the PI3K-related kinase family, mTOR is associated with cell proliferation and cell metabolism. Previous studies have reported that inhibition of Akt and its downstream target mTOR contributes to the initiation of autophagy (34,35). Activation of the PI3K/Akt/mTOR pathway has also been associated with pathogenesis of non-small cell lung cancer (NSCLC), and inhibition of the PI3K/AKT/mTOR signaling pathway has been suggested as a potential therapeutic target in NSCLC (36). The present study demonstrated that Akt and mTOR phosphorylation were significantly decreased in the cabazitaxel-treated A549 cells, thus indicating that cabazitaxel may be considered a candidate agent for the treatment of lung cancer through inhibition of the PI3K/AKT/mTOR signaling pathway.
In conclusion, the results of the present study suggested that cabazitaxel may induce autophagic cell death in human adenocarcinoma lung cancer cells via PI3K/Akt/mTOR signaling inhibition. These findings represent a novel anticancer mechanism of cabazitaxel, and may help expand its application in the chemotherapeutic treatment of various cancers other than those of prostate gland origin.
Acknowledgments
The present study was supported by the Science and Technolgy Innovation Project Plan of Shaanxi Province (grant nos. 2015KTCL03-01 and 2014FWPT-11).
References
- 1.McLeod IX, Jia W, He YW. The contribution of autophagy to lymphocyte survival and homeostasis. Immunol Rev. 2012;249:195–204. doi: 10.1111/j.1600-065X.2012.01143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y, Qin ZH. Coordination of autophagy with other cellular activities. Acta Pharmacol Sin. 2013;34:585–594. doi: 10.1038/aps.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011;8:528–539. doi: 10.1038/nrclinonc.2011.71. [DOI] [PubMed] [Google Scholar]
- 4.Deretic V. Autophagy in infection. Curr Opin Cell Biol. 2010;22:252–262. doi: 10.1016/j.ceb.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang S, Dupont N, Castillo EF, Deretic V. Secretory versus degradative autophagy: Unconventional secretion of inflammatory mediators. J Innate Immun. 2013;5:471–479. doi: 10.1159/000346707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janda E, Isidoro C, Carresi C, Mollace V. Defective autophagy in Parkinson's disease: Role of oxidative stress. Mol Neurobiol. 2012;46:639–661. doi: 10.1007/s12035-012-8318-1. [DOI] [PubMed] [Google Scholar]
- 7.Ren SY, Xu X. Role of autophagy in metabolic syndrome-associated heart disease. Biochim Biophys Acta. 2015;1852:225–231. doi: 10.1016/j.bbadis.2014.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308–5316. doi: 10.1158/1078-0432.CCR-07-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC, Kroemer G. To die or not to die: That is the autophagic question. Curr Mol Med. 2008;8:78–91. doi: 10.2174/156652408783769616. [DOI] [PubMed] [Google Scholar]
- 10.Shen S, Kepp O, Kroemer G. The end of autophagic cell death? Autophagy. 2012;8:1–3. doi: 10.4161/auto.8.1.16618. [DOI] [PubMed] [Google Scholar]
- 11.Kaminskyy V, Abdi A, Zhivotovsky B. A quantitative assay for the monitoring of autophagosome accumulation in different phases of the cell cycle. Autophagy. 2011;7:83–90. doi: 10.4161/auto.7.1.13893. [DOI] [PubMed] [Google Scholar]
- 12.Jiang P, Mizushima N. LC3- and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods. 2015;75:13–18. doi: 10.1016/j.ymeth.2014.11.021. [DOI] [PubMed] [Google Scholar]
- 13.Torrisi R, Balduzzi A, Ghisini R, Rocca A, Bottiglieri L, Giovanardi F, Veronesi P, Luini A, Orlando L, Viale G, et al. Tailored preoperative treatment of locally advanced triple negative (hormone receptor negative and HER2 negative) breast cancer with epirubicin, cisplatin, and infusional fluorouracil followed by weekly paclitaxel. Cancer Chemother Pharmacol. 2008;62:667–672. doi: 10.1007/s00280-007-0652-z. [DOI] [PubMed] [Google Scholar]
- 14.Kaufmann SH, Earnshaw WC. Induction of apoptosis by cancer chemotherapy. Exp Cell Res. 2000;256:42–49. doi: 10.1006/excr.2000.4838. [DOI] [PubMed] [Google Scholar]
- 15.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: Therapeutic implications. Mol Cancer Ther. 2011;10:1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo WJ, Zhang YM, Zhang L, Huang B, Tao FF, Chen W, Guo ZJ, Xu Q, Sun Y. Novel monofunctional platinum (II) complex Mono-Pt induces apoptosis-independent autophagic cell death in human ovarian carcinoma cells, distinct from cisplatin. Autophagy. 2013;9:996–1008. doi: 10.4161/auto.24407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu XW, Cai TY, Zhu H, Cao J, Su Y, Hu YZ, He QJ, Yang B. Q6, a novel hypoxia-targeted drug, regulates hypoxia-inducible factor signaling via an autophagy-dependent mechanism in hepatocellular carcinoma. Autophagy. 2014;10:111–122. doi: 10.4161/auto.26838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dragowska WH, Weppler SA, Wang JC, Wong LY, Kapanen AI, Rawji JS, Warburton C, Qadir MA, Donohue E, Roberge M, et al. Induction of autophagy is an early response to gefitinib and a potential therapeutic target in breast cancer. PLoS One. 2013;8:e76503. doi: 10.1371/journal.pone.0076503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou J, Hu H, Long J, Wan F, Li L, Zhang S, Shi YE, Chen Y. Vitexin 6, a novel lignan, induces autophagy and apoptosis by activating the Jun N-terminal kinase pathway. Anticancer Drugs. 2013;24:928–936. doi: 10.1097/CAD.0b013e328364e8d3. [DOI] [PubMed] [Google Scholar]
- 20.Vrignaud P, Sémiond D, Lejeune P, Bouchard H, Calvet L, Combeau C, Riou JF, Commerçon A, Lavelle F, Bissery MC. Preclinical antitumor activity of cabazitaxel, a semisynthetic taxane active in taxane-resistant tumors. Clin Cancer Res. 2013;19:2973–2983. doi: 10.1158/1078-0432.CCR-12-3146. [DOI] [PubMed] [Google Scholar]
- 21.Azarenko O, Smiyun G, Mah J, Wilson L, Jordan MA. Antiproliferative mechanism of action of the novel taxane cabazitaxel as compared with the parent compound docetaxel in MCF7 breast cancer cells. Mol Cancer Ther. 2014;13:2092–2103. doi: 10.1158/1535-7163.MCT-14-0265. [DOI] [PubMed] [Google Scholar]
- 22.Liu F, Liu D, Yang Y, Zhao S. Effect of autophagy inhibition on chemotherapy-induced apoptosis in A549 lung cancer cells. Oncol Lett. 2013;5:1261–1265. doi: 10.3892/ol.2013.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu D, Makkar G, Dong T, Strickland DK, Sarkar R, Monahan TS. MARCKS signaling differentially regulates vascular smooth muscle and endothelial cell proliferation through a KIS-, p27kip1- dependent mechanism. PLoS One. 2015;10:e0141397. doi: 10.1371/journal.pone.0141397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Sharma K, Le N, Alotaibi M, Gewirtz DA. Cytotoxic autophagy in cancer therapy. Int J Mol Sci. 2014;15:10034–10051. doi: 10.3390/ijms150610034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kung CP, Budina A, Balaburski G, Bergenstock MK, Murphy M. Autophagy in tumor suppression and cancer therapy. Crit Rev Eukaryot Gene Expr. 2011;21:71–100. doi: 10.1615/CritRevEukarGeneExpr.v21.i1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen HM, Codogno P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy. 2011;7:457–465. doi: 10.4161/auto.7.5.14226. [DOI] [PubMed] [Google Scholar]
- 28.Choi KS. Autophagy and cancer. Exp Mol Med. 2012;44:109–120. doi: 10.3858/emm.2012.44.2.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Høyer-Hansen M, Jäättelä M. AMP-activated protein kinase: A universal regulator of autophagy? Autophagy. 2007;3:381–383. doi: 10.4161/auto.4240. [DOI] [PubMed] [Google Scholar]
- 31.Shi WY, Xiao D, Wang L, Dong LH, Yan ZX, Shen ZX, Chen SJ, Chen Y, Zhao WL. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. 2012;3:e275. doi: 10.1038/cddis.2012.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corcelle E, Djerbi N, Mari M, Nebout M, Fiorini C, Fénichel P, Hofman P, Poujeol P, Mograbi B. Control of the autophagy maturation step by the MAPK ERK and p38: Lessons from environmental carcinogens. Autophagy. 2007;3:57–59. doi: 10.4161/auto.3424. [DOI] [PubMed] [Google Scholar]
- 33.Li Y, Zhang P, Qiu F, Chen L, Miao C, Li J, Xiao W, Ma E. Inactivation of PI3K/Akt signaling mediates proliferation inhibition and G2/M phase arrest induced by andrographolide in human glioblastoma cells. Life Sci. 2012;90:962–967. doi: 10.1016/j.lfs.2012.04.044. [DOI] [PubMed] [Google Scholar]
- 34.Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, Kondo S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65:3336–3346. doi: 10.1158/0008-5472.CAN-04-3640. [DOI] [PubMed] [Google Scholar]
- 35.Paglin S, Lee NY, Nakar C, Fitzgerald M, Plotkin J, Deuel B, Hackett N, McMahill M, Sphicas E, Lampen N, Yahalom J. Rapamycin-sensitive pathway regulates mitochondrial membrane potential, autophagy, and survival in irradiated MCF-7 cells. Cancer Res. 2005;65:11061–11070. doi: 10.1158/0008-5472.CAN-05-1083. [DOI] [PubMed] [Google Scholar]
- 36.Fumarola C, Bonelli MA, Petronini PG, Alfieri RR. Targeting PI3K/AKT/mTOR pathway in non small cell lung cancer. Biochem Pharmacol. 2014;90:197–207. doi: 10.1016/j.bcp.2014.05.011. [DOI] [PubMed] [Google Scholar]