

Could a greater miracle take place than for us to look through each other's eyes for an instant?Henry David Thoreau
Introduction
As seen from both patients’ and physicians’ points of view, there is wide agreement that systemic sclerosis (SSc) is one of the autoimmune disorders with the highest morbidity and mortality rates. In this review, we will follow the evolution of a new approach to its treatment. Support for using hematopoietic stem cell transplantation (HSCT) for SSc arose from seminal studies of genetic and antigen‐induced experimental models of autoimmune disease that demonstrated that high‐dose immunosuppression followed by either allogeneic (same species) or autologous (self) bone marrow transplantation (BMT) could prevent and even reverse damage from autoimmune diseases. Three decades after these initial preclinical observations, our understanding of the therapeutic potential of immune restoration following autologous HSCT has deepened, and the clinical evidence for its application in scleroderma has broadened.
In this review we will examine the outcome of conventional therapy for scleroderma lung disease; detail the techniques, toxicities, and results of HSCT for SSc; and explore the biology of immune restoration following autologous transplantation. We will compare the design and outcomes of randomized trials comparing HSCT with cyclophosphamide (CYC) treatment and formulate criteria for the timely referral of patients with scleroderma lung disease for HSCT.
Pathogenesis of scleroderma
The heterogeneous manifestations of SSc are the result of 3 primary pathogenic processes: vasculopathy, inflammation, and fibrosis. As with the other connective tissue diseases, there is no single genetic abnormality conferring susceptibility to the disease; however, a permissive genetic environment is likely necessary for the initiation of disease. For example, there is an increased relative risk of SSc in first‐degree relatives of patients with the condition, and a number of HLAs are associated with SSc, several of which correlate with specific ethnic populations or clinical features 1. Genomic analyses can differentiate normal from scleroderma skin and disclose associated fibroinflammatory and interferon (IFN)–associated gene signatures 2, 3. Among the chemokines, CXCL4 has been shown to predict the risk and progression of SSc 4. Epigenetic modification, including DNA methylation, histone modification, and microRNA expression, also appear to play roles in the pathophysiology of SSc.
Phenotypic changes in SSc are driven by vascular damage, immune dysregulation, fibroblast activation, and collagen deposition 1. In a permissible genetic environment, increased circulating factors such as endothelin 1 (ET‐1) promote vasoconstriction and injury to the endothelium. Vascular injury leads to a cascade of inflammatory cytokines, growth factors, and reactive oxygen species that propagate tissue damage. Platelet‐derived growth factor can promote complement‐directed endothelial cell injury, and aberrant transforming growth factor β (TGFβ) and ET‐1 signaling appear essential for myofibroblast activation and profibrotic signaling.
Mortality in scleroderma
While SSc is a rare disease with an incidence of only 20 patients per million adults per year, it disproportionately impacts women in their 30s to 50s and is both debilitating and deadly. Independent risk factors for mortality in SSc include the presence of anti–topoisomerase I antibodies as well as pulmonary, renal, or cardiac organ involvement. Rapidity of progression of skin thickness has been shown to predict internal organ involvement and mortality 5. Meta‐analysis of 40 years of publications comprising 2,691 patients with SSc has provided standardized mortality ratios (SMRs), the ratio of deaths compared to the expected deaths in the general population matched for age and sex, for the disease. Analysis showed that the pooled SMR for SSc was 3.5 (95% confidence interval [95% CI] 3.0–4.1) with little apparent change over the past 4 decades 6. Similarly, a 15‐year follow‐up of 398 patients with SSc enrolled between 1995 and 1999 at the Royal Free Hospital (London, UK) showed an overall SMR of 3.8 (i.e., 280% higher mortality than in the general population) 7. Patients with diffuse cutaneous SSc had a worse outcome than those with limited cutaneous disease, and women with diffuse cutaneous SSc had a remarkable SMR of 7.1. Organ (often lung) involvement denoted a much poorer prognosis compared to patients without organ complications 7. Scleroderma (either limited or diffuse) with organ involvement remains a progressively fatal disease without evidence of a survival plateau (Figure 1).
Figure 1.
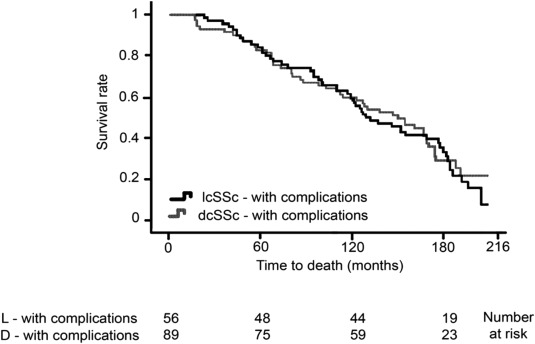
Survival of patients with limited cutaneous systemic sclerosis (lcSSc) or diffuse cutaneous SSc (dcSSc) with internal organ complications enrolled at the Royal Free Hospital (London, UK). Adapted, with permission, from ref. 7.
Scleroderma lung disease
While multiple target tissues may be damaged, pulmonary involvement is the leading cause of death in SSc 8. Detection of scleroderma‐associated interstitial lung disease (ILD) requires both pulmonary function testing (PFT) and high‐resolution computed tomographic (CT) imaging of the lung 9. In a systematic review of 20 publications comprising 1,524 patients, diffusing capacity of the lung for carbon monoxide (DLCO) was the most consistent predictor of mortality in scleroderma lung disease, while the extent of disease on high‐resolution CT imaging was an independent predictor for overall mortality and ILD progression 10. Pulmonary fibrosis on high‐resolution CT and either a forced vital capacity (FVC) of <80% predicted or a decline of >10% in the FVC or a decline of >15% in the DLCO on serial measures constitute accepted indications for treatment 11. However, lung fibrosis is rarely reversible with conventional therapies.
While uncontrolled case series have shown varying responses to several disease‐modifying antirheumatic drugs (DMARDs) used alone or in combination, only CYC given for 1 year has been shown to be of benefit when compared to placebo administration 12. In the Scleroderma Lung Study (SLS) the degree of FVC benefit first conferred by CYC treatment was modest (2.5% mean difference in the adjusted 12‐month FVC between the arms), peaked at 6 months off therapy, and was lost by 12 months off treatment 13. In a recently completed second SLS, there was comparable stabilization and improvement in FVC with a 2‐year course of mycophenolate mofetil (MMF) when compared to a 1‐year course of CYC 14. Moreover, MMF appeared better tolerated than CYC. Other, ongoing studies are exploring agents that inhibit fibrosis, TGFβ or Wnt signaling, and B cell activity.
Several issues emerge in a review of the published literature on the treatment of scleroderma lung disease. Other than the aforementioned SLS, most trials are comprised of fewer than 30 participants and can suffer from selection and publication bias, while studies demonstrating ineffectiveness may be underreported. Most studies measure the change in percent predicted FVC, DLCO, and/or changes on high‐resolution CT scans while others also include disease activity measures, such as the modified Rodnan skin thickness score (MRSS) and Health Assessment Questionnaire (HAQ) score. Few studies compare the treatment group with a control. Outside of CYC, MMF has been the next most commonly studied drug, and dosages of 2–3 gm daily were generally associated with the stability of PFT findings. However, many studies lasted only 6–12 months and a longer treatment course and follow‐up may be necessary.
Animal models of autoimmune diseases treated with myeloablation and HSCT
Preclinical models of autoimmune disease published in the 1980s demonstrated a remarkable ability to prevent or reverse autoimmune disease following lymphomyeloablative conditioning and either allogeneic or autologous BMT. It was postulated that this efficacy was due to the effects of total body irradiation (TBI) ablating the host autoreactive immune repertoire, which was then replaced with normal allogeneic or autologous stem cells in the absence of the original antigenic trigger of the autoimmune disease. Genetic murine models of diabetes mellitus showed that TBI (which ablates both resting and dividing lymphoid immune cells) and allogeneic BMT could prevent subsequent development of the autoimmune disease 15. Importantly, established organ involvement in the genetic diabetes model (loss of islet cells in the pancreas) or in the lupus model (presence of immune complexes in the kidney) could be reversed after TBI and allogeneic BMT. Similar effects in a rat model of antigen‐induced arthritis were observed after TBI and autologous BMT 16. In these animal experiments, myeloablative doses of TBI and CYC were significantly more effective than nonablative CYC alone in producing sustained elimination of the autoimmune disease following autologous BMT 17.
Initial clinical experience of HSCT for autoimmune diseases
By 1997, several teams had published clinical designs for HSCT for autoimmune disorders 18, 19. In the rare situation of an autoimmune disease co‐existing with a hematologic malignancy, myeloablative allogeneic BMT appeared to offer long‐term remission of both diseases 20. The first report of an individual undergoing successful autologous HSCT for SSc was published in 1997 21. Shortly thereafter, European registry experience showed dramatic improvement in dermal fibrosis after autologous HSCT in SSc 22. By 2009, the number of reported patients with SSc treated with autologous HSCT had reached 224 individuals in Europe 23 and 97 in North and South America 24. In these registry reports of transplants for autoimmune diseases, scleroderma was second only to multiple sclerosis as an indication for autologous HSCT.
The process of autologous HSCT
Spurred by research involving ionizing radiation following World War II, the first successful human autologous HSCT was performed in 1957. Once comprehensive worldwide registries of HSCT became available, records showed that more than 1 million individuals with malignant and nonmalignant diseases had received autologous (58%) or allogeneic (42%) hematopoietic cell transplants between January 2006 and December 2014 in 1,516 centers in 75 countries 25. During this 9‐year period, there were 1,162 transplants for autoimmune diseases: 1,058 autologous and 104 allogeneic.
HSCT is a multistep process involving: 1) hematopoietic cell mobilization and cell harvesting, selection, and cryopreservation, 2) preparative conditioning with chemotherapy with or without irradiation, 3) infusion of the stem cell graft, and 4) supportive care after transplantation 26, 27, 28. Mobilization of CD34+ hematopoietic stem cells from the marrow to the bloodstream is promoted by administration of hematopoietic growth factors, such as granulocyte colony‐stimulating factor (G‐CSF), with or without CYC priming. Circulating blood stem cells are then collected by leukapheresis, often in a 1‐day collection. To further purify stem cells and remove differentiated lymphoid or other contaminating cells, positive stem cell selection by CD34+ isolation techniques (fluorescence or magnetic‐activated cell sorting) can be used before cryopreservation of the autologous cells 28. Once thawed, cells are infused intravenously and home to the marrow. These hematopoietic stem cells are able to repopulate and differentiate into erythrocytes, megakaryocytes, and immune cells. Mediators of innate immunity, such as macrophages, granulocytes, and natural killer cells, repopulate and function early after HSCT. Adaptive immune cells, including effector and regulatory B and T cells, also recover early, while full function and immune memory return more gradually 29.
Multiple regimens have been used to prepare patients to receive a transplant, ranging from nonmyeloablative conditioning (CYC 200 mg/kg) to fully lymphomyeloablative regimens (high‐dose chemotherapy with or without TBI). For the less intensive regimens, most patients can be cared for in outpatient “day hospitals” while residing in apartments near the transplant center. Others remain inpatient until blood counts have engrafted. Sustained granulocyte recovery occurs 10–12 days after transplantation, and most patients return home shortly thereafter. While at the center, family or friends assist as accompanying caregivers, a vital role in the recovery of the patient.
Complications associated with autologous HSCT include preparative regimen–related organ toxicity, opportunistic infections, infertility, recurrence of disease, and secondary malignancies 30. Autologous transplantation–related mortality is ∼3–5% for patients with myeloma and lymphoma and ∼5–10% for those with scleroderma 23, 28, 31, 32.
Transplantation is an intensive procedure with significant side effects including nausea, vomiting, diarrhea, and oral mucositis, which usually resolve with blood count recovery. Organ toxicity may be due to the preparative regimen and any underlying organ damage from SSc. Reports of acute kidney injury led to the use of several scleroderma‐specific renal protective regimens 33, 34. Gastrointestinal endoscopy has revealed gastric antral vascular ectasia (GAVE) in 22% of scleroderma patients screened before HSCT 35. Coagulation therapy of the GAVE lesions has been important in preventing hemorrhage during thrombocytopenia. All referred patients with scleroderma ILD will have diminished PFT findings. For safety, most centers exclude individuals with an FVC or DLCO of <45% predicted, although the lower limits are difficult to assess given other comorbidities 36. Conditioning regimens include varying doses of CYC, which can be associated with acute cardiotoxicity that appears to be dose‐ and schedule‐dependent. At a normally tolerated CYC dose of 200 mg/kg given before transplantation, cardiac deaths have been reported, which raises the question of additional comorbidity from existing SSc‐related heart disease 31.
Opportunistic infections are the leading cause of mortality after HSCT for autoimmune diseases 23. While neutrophils recover within 2 weeks of transplantation, other components of immune recovery may take up to 6–24 months 29. That said, total infections and serious infections (i.e., those treated as an inpatient) cluster within the first month after autologous HSCT. In 4 time intervals (days 0–30, days 31–180, days 181–365, and days 366–730), the rates of total/serious infections were 5.75/4.90, 0.94/0.54, 0.29/0.05, and 0.16/0.08, respectively, among 56 individuals who were treated with CYC, TBI, and antithymocyte globulin (ATG) before autologous HSCT for autoimmune disease 29. Prevention of infection after transplantation is becoming increasingly refined since the introduction of sensitive polymerase chain reaction assays for a number of viral pathogens 37.
Phase I and II trials of autologous HSCT for patients with SSc
While allogeneic HSCT for autoimmune disease has been reported, most centers have focused on autologous transplantation, where fewer complications are observed. Consensus conferences were held in Basel and Seattle to establish frameworks for clinical trials and reporting 18, 19. Follow‐up studies refined the pretransplant conditioning regimens. Protocols enrolled high‐risk scleroderma patients with internal organ involvement but for safety reasons excluded individuals with severely compromised organ function 38. Results of initial pilot trials demonstrated dramatic improvement in skin scores and quality‐of‐life measures with stabilization of pulmonary function 22, 23, 39. Figure 2 depicts outcomes in 34 patients with SSc with up to 8 years of follow‐up after a phase II trial of myeloablative autologous HSCT 39. Before transplantation, mean measures for the cohort included an MRSS of 30.1, a modified HAQ score of 1.85, FVC of 71% predicted, and DLCO of 60% predicted. Over time, there were significant and durable improvements in the MRSS and HAQ, with stabilization of or modest improvement in FVC and DLCO.
Figure 2.
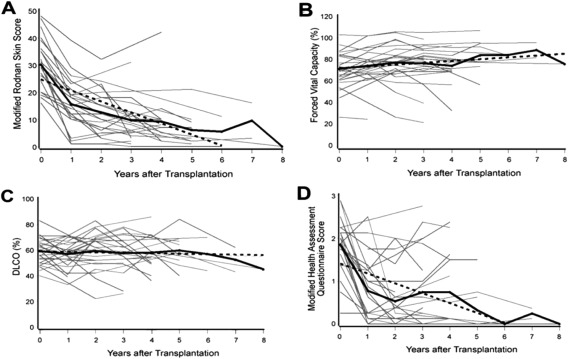
Results after total body irradiation, cyclophosphamide and antithymocyte globulin preparative conditioning, and CD34+ selected autologous hematopoietic stem cell transplantation in 34 patients with severe scleroderma 39. A, Modified Rodnan skin thickness score. B, Forced vital capacity. C, Diffusing capacity for carbon monoxide (DLCO). D, Quality of life measures (modified Health Assessment Questionnaire). Solid black lines show the mean; broken lines represent the generalized estimating equation; gray lines represent individual patient values.
Phase III trials of autologous HSCT for patients with SSc
While encouraging, pilot studies underscored the need to control for potential selection or reporting biases by conducting prospective, randomized clinical trials comparing HSCT to CYC (the only DMARD shown to be of benefit in a controlled trial of scleroderma lung disease). Results from the randomized clinical trials the American Scleroderma Stem Cell versus Immune Suppression Trial (ASSIST) 40 and the Autologous Stem Cell Transplantation International Scleroderma (ASTIS) 32 trial have been published, while the Scleroderma: Cyclophosphamide or Transplantation (SCOT) randomized trial results will be available in late 2016 after the last enrolled subject is evaluated at the 54th month primary end point. Table 1 details these 3 controlled studies. As shown, there are both similarities (inclusion and exclusion criteria) and differences (primary end points, preparative regimens, stem cell mobilization and selection techniques, and length of follow‐up) across the trials. Most randomized subjects had scleroderma with internal organ involvement (predominantly lung), although the ASTIS trial enrolled high‐risk subjects with only skin disease late in the study 5, 32. High‐dose CYC was used for both stem cell mobilization and preparative conditioning in the nonmyeloablative ASSIST and ASTIS trials. In contrast, the SCOT trial mobilizes with G‐CSF alone, uses a lower dose of pretransplant CYC, and uses TBI conditioning, which is fully lymphomyeloablative. A significantly higher dose of CYC before transplantation may account for reported cardiotoxicity in the nonmyeloablative scleroderma trials 31. In the single‐center ASSIST trial, all patients in the CYC arm failed to respond to treatment, and all patients who received HSCT improved. Criticism has been voiced concerning the small number of patients, short duration of follow‐up, and crossover design 41.
Table 1.
Randomized trials of HSCT in SSca
| ASSIST | ASTIS | SCOT | |
|---|---|---|---|
| No. of centers | 1 | 29 | 25b |
| Location of centers | US | Europe | US |
| Year of first randomization | 2006 | 2001 | 2006 |
| Year of last randomization | 2009 | 2009 | 2011 |
| No. of patients enrolled or screened | 45 | NA | 205c |
| No. of patients randomized | 19 | 156 | 75 |
| No. of patients randomized per year | 6 | 19 | 15 |
| Age range, years | <60 | 18–65 | 18–65 |
| Diffuse cutaneous SSc | Yes | Yes | Yes |
| Primary end point | Improvement at 12 months | Organ failure–free survival at 24 months | Organ failure–free survival at 54 months |
| Inclusion criteria | |||
| SSc duration, years | ≤4 | <2 or ≤4 | ≤5 |
| MRSS | ≥15 | ≥20 or ≥15 | ≥16 |
| SSc internal disease | Yes | Yes or no | Yes |
| FVC, % predicted | <80 or 10% decrease | <80 | 70–45 |
| DLCO, % predicted | <80 | 80–40 | 70–45 |
| ILDd | Yes | Yes or no | Yes |
| Exclusion criteria | |||
| TLC, % predicted | <45 | – | – |
| LVEF, % | <40 | <45 | <50 |
| PAP, mean mm Hg | >25 | >50e | >30 |
| Duration of prior CYC treatment, months | >6 | >5 | >6 |
| Treatment regimen | |||
| CYC arm | CYC 1,000 mg/m2/month IV × 6 (6 gm/m2 over 6 months) | CYC 750 mg/m2/month IV × 12 (9 gm/m2 over 12 months) | CYC 750 mg/m2/month IV × 12 (9 gm/m2 over 12 months) |
| HSCT arm | CYC 200 mg/kg, ATG (rabbit) 6.5 mg/kg, and methylprednisolone 5,000 mg | CYC 200 mg/kg and ATG (rabbit) 7.5 mg/kg | CYC 120 mg/kg, ATG (horse) 90 mg/kg, and TBI 800 cGyf |
| Autologous cells | Unselected | CD34 selected | CD34 selected |
| Stem cell mobilization | CYC 2 gm/m2 and G‐CSF | CYC 4 gm/m2 and G‐CSF | G‐CSF only |
| Crossover treatment | Yes | No | No |
| Follow‐up, years | 2.6 (mean) | 5.8 (median) | Ongoing (minimum 4.5) |
| No. with progression of SSc or no response/no. assessed | |||
| CYC | 9/9 | 19/77 | NA |
| HSCT | 0/10 | 9/79 | NA |
| Overall mortality, no. of deaths/no. assessed | |||
| CYC | 0/9 | 30/77 | NA |
| HSCT | 0/10 | 19/79 | NA |
HSCT = hematopoietic stem cell transplantation; SSc = systemic sclerosis; ASSIST = American Scleroderma Stem Cell versus Immune Suppression Trial; ASTIS = Autologous Stem Cell Transplantation International Scleroderma trial; SCOT = Scleroderma: Cyclophosphamide or Transplantation trial; NA = not available; MRSS = modified Rodnan skin thickness score; FVC = forced vital capacity; DLCO = diffusing capacity for carbon monoxide; ILD = interstitial lung disease; TLC = total lung capacity; LVEF = left ventricular ejection fraction; PAP = pulmonary artery pressure; CYC = cyclophosphamide; IV = intravenous; ATG = antithymocyte globulin; TBI = total body irradiation; G‐CSF = granulocyte colony‐stimulating factor.
Seventeen rheumatology and 8 transplant centers.
Insurance denials for clinical trial coverage often precluded randomization.
Determined by bronchoalveolar lavage or high‐resolution computed tomography.
Determined by cardiac echo; other values were determined by right heart catheterization.
Lungs and kidneys were shielded from transmission of >200 cGy 34.
Figure 3 depicts overall survival and event‐free survival (survival free of organ failure) in the multicenter ASTIS trial. As expected, mortality was higher in the first year after transplantation. The curves crossed at 2 years and thereafter HSCT was statistically superior to CYC for both event‐free survival and overall survival 32. Table 2 presents morbidity outcomes in the ASTIS study. Rodnan scores, FVC, total lung capacity (TLC), disability ratings, and physical functioning were all significantly improved after HSCT compared to pretreatment assessments, and these improvements were superior to similar serial assessments after CYC. Compared to their baseline tests, HSCT recipients had improvement over time in FVC and TLC; conversely, CYC recipients had worsening in pulmonary function over time. The differences in pretransplantation and posttransplantation DLCO values for the 2 groups were not significant. As expected, there were more grade 3 adverse events with HSCT, but there were no significant differences in grade 4 adverse events.
Figure 3.
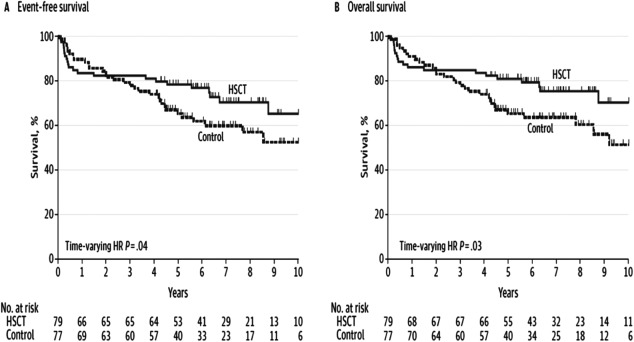
Results from the Autologous Stem Cell Transplantation International Scleroderma trial comparing CD34+ selected autologous hematopoietic stem cell transplantation (HSCT) after conditioning with cyclophosphamide (CYC) and antithymocyte globulin to 12 months of CYC treatment (control) 32. A, Survival free of organ failure. B, Overall survival. HR = hazard ratio.
Table 2.
Change in the area under the time response curve from baseline to year 2 follow‐up by treatment arm in the ASTIS studya
| HSCT | CYC | Difference | P | |
|---|---|---|---|---|
| MRSS | −19.9 | −8.8 | 11.1 | <0.001 |
| FVC, % predicted | 6.3 | −2.8 | −9.1 | 0.004 |
| TLC, % predicted | 5.1 | −1.3 | −6.4 | 0.02 |
| DLCO, % predicted | −4.7 | −4.1 | 0.6 | NS |
| HAQ DI | −0.58 | −0.19 | 0.39 | 0.02 |
| SF‐36 (physical domain) | 10.1 | 4.0 | −6.1 | 0.01 |
| Grade 3 AE | 38 | 20 | – | 0.005 |
| Grade 4 AE | 29 | 21 | – | NS |
Values are the mean area under the curve for the treatment groups in the Autologous Stem Cell Transplantation International Scleroderma (ASTIS) study 32. HSCT = hematopoietic stem cell transplantation; CYC = cyclophosphamide; MRSS = modified Rodnan skin thickness score; FVC = forced vital capacity; TLC= total lung capacity; DLCO = diffusing capacity for carbon monoxide; NS = not significant; HAQ DI = Health Assessment Questionnaire disability index; SF‐36 = Short Form 36; AE = adverse event.
The immunomodulatory effects of autologous HSCT
Since SSc is an aberrant activation of the adaptive and innate immune systems, several immunologic factors have been cited in examining these improved outcomes with HSCT. In both allogeneic and autologous HSCT, stem cell rescue allows delivery of very high doses of potentially disease‐attenuating immunosuppression to patients with autoimmune disease. Allogeneic transplants provide a new immune system from a normal stem cell donor 15, 20. However, clinical experience with allogeneic transplants for autoimmune disease is too limited to afford comparisons with autologous HSCT with regard to long‐term control of the autoreactive disease.
Autologous HSCT could “reset” the host immune system to a point in time when the antigenic triggers of autoimmunity were not present 42. Illustrative of this point is the fact that pre‐HSCT immunity wanes and often disappears after autologous HSCT. In recipients of TBI conditioning and autologous HSCT, the T cell receptor (TCR) repertoire diversity was shown to normalize after lymphoablation and autologous transplantation 43. Thymopoiesis renewal in adult recipients of autologous HSCT is also evidenced by thymic regrowth on imaging, a broadened diversity of the TCR Vβ repertoire among memory cells, and recovery of normal CD4+ T cells. Conversely, individuals who did not respond to autologous transplantation had less T cell diversity early after immune recovery 44. Patients with SSc have diminished Treg cell proportions. Restoration of human Treg cells has also been observed after autologous HSCT 45. In a congenic murine BMT model of proteoglycan‐induced arthritis, the genetic background of the transplanted bone marrow was identical between host and donor except for the congenic T cell marker. Treg cell pools of donor origin were shown to reconstitute a stable, tolerant immune repertoire 46. After human HSCT for juvenile idiopathic arthritis and dermatomyositis, renewal of Treg cells was noted after autologous HSCT with evidence of a more diverse Treg TCR compartment 46, 47. Figures 4A and B illustrate this immune restoration after autologous HSCT.
Figure 4.
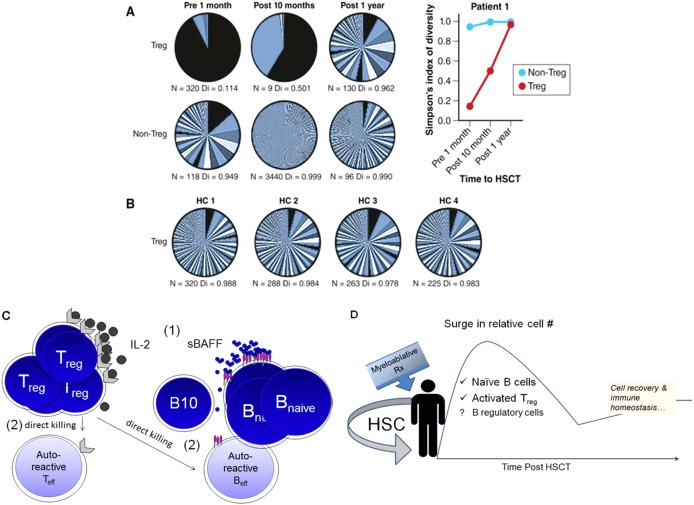
Immune homeostasis following autologous hematopoietic stem cell transplantation (HSCT). A and B, T cell receptor (TCR) diversity in Treg cells restored after autologous HSCT in a patient with autoimmune disease (A) and in 4 healthy controls (HC) (B) 46. The number of different TCR sequences per sample and diversity (Di), where 0 = none and 1 = maximum, before and after transplantation are shown. Adapted, with permission, from ref. 46. C and D, Model of how autologous HSCT results in immune tolerance and immune homeostasis. C, Outcompetition of nonautoreactive lymphocytes results in the death of autoreactive clones. Interleukin‐2 (IL‐2) and serum BAFF (sBAFF) levels increase after lymphomyeloablation 1. The proportions of regulatory T and B cells, both known to suppress or kill effector T and B cells, are increased 2. D, Supranormal numbers of B cells and activated regulatory cells eliminate autoreactive clones during immune recovery. The surge in cell numbers abates as immune homeostasis is achieved.
Future directions
Further explorations of genomic, immunologic, and cell signaling pathways in SSc are currently underway through mechanistic studies associated with the SCOT trial. For example, compared to age‐ and sex‐matched healthy volunteers, prerandomization peripheral blood samples from SCOT patients showed differentially expressed genes, mostly of the IFN signaling pathways. When compared to their baseline samples, HSCT recipients exhibited significant declines in the IFN transcript score at month 26. In contrast, CYC recipients had no change over time 48. These genomic findings could further support the notion that autologous HCT can be associated with restoration of immune homeostasis.
Altered B cell homeostasis and high levels of BAFF have been noted in patients with SSc 49. Patients treated with autologous HSCT have robust B cell recovery associated with a rapid decline in BAFF levels. In addition to a diverse peripheral B cell pool that can potentially outcompete for BAFF, regulatory B cells likely arise after autologous HSCT. Interleukin‐10 (IL‐10)–producing B10 cells have been found to be critical for SSc attenuation in murine models. Taken together, these data suggest that immune tolerance can be induced with HSCT. Figures 4C and D present a working model of how autologous HSCT results in immune tolerance and immune homeostasis.
Indications for autologous HSCT in scleroderma lung disease
Based on the above data, the following abridged criteria broadly serve as indications for referral for HSCT in patients with SSc. Inclusion criteria include diffuse cutaneous SSc with internal organ involvement, age <65 years, disease duration <5 years, MRSS >15, early pulmonary involvement by HRCT and PFTs showing FVC or hemoglobin‐adjusted DLCO between 80% and 45% predicted or a decline in FVC of >10% or DLCO of >15% on serial testing or failure to respond to initial therapy on serial PFT monitoring.
Exclusion criteria include FVC or hemoglobin‐adjusted DLCO of <45% predicted, pulmonary arterial hypertension, cardiac insufficiency/involvement with SSc, renal insufficiency, and prior CYC treatment of >6 months’ duration.
Patient and physician choice of treatment
Barriers to the application of HSCT for autoimmune disease include restrictions in insurance support and physician referral 42. Patients seeking transplants for scleroderma currently face fewer restrictions to health insurer support because more individuals now have health insurance coverage, and two published randomized trials have shown significant clinical benefit with HSCT 32, 40. Physician referral preferences can also be a barrier, as demonstrated by patients’ self‐referral for transplantation consults. Patients wish to learn of the risks as well as the potential long‐term gains with HSCT. However, rheumatologists may be unfamiliar with the details of a treatment outside their specialty or be concerned about the associated risk.
Concern for premature mortality in SSc is the major reason individuals seek consultation for stem cell transplantation. A new sense of urgency in scleroderma lung disease can be likened to the evolution of thinking about rheumatoid arthritis (RA). Once premature mortality in RA was demonstrated (SMRs of 1.2–1.7), a chronic autoimmune disorder became an urgent disease for the rheumatologist 50. If such urgency has been true in RA (with mortality rates 20–70% greater than in the general population), then it is even more pressing in scleroderma lung disease, which has mortality rates of 250–280% greater than expected 6, 7.
While DMARDs and antifibrotic agents may demonstrate initial improvement on PFTs, the key consideration for both patients and physicians is the effect on the premature mortality associated with scleroderma lung disease. Since the ASTIS trial has demonstrated improved long‐term survival and event‐free survival after HSCT, consideration for stem cell transplantation referral is a reasonable action but requires close PFT monitoring before pulmonary compromise becomes too severe for safe transplantation. In such an urgent and evolving situation, it is critical that the practitioner offers timely referral to a transplant center for evaluation and discussion of the techniques, toxicities, and outcomes of HSCT. Without referral, patients will be without the knowledge and context to make informed decisions. Ultimately, this shared decision‐making is a partnership of physician and patient to make the right choice of treatment in light of the individual's situation and values 51.
Conclusions
While there are often multiple disabling manifestations of SSc, lung disease is the major cause of death. Although conventional DMARDs show modest short‐term benefit, the published literature on SSc shows a pooled SMR of 3.5 (250% increase in mortality) with little change over decades of reporting. Hence, premature mortality remains of urgent concern to patients and providers.
The basis for recommending stem cell transplantation in SSc arose from insightful animal experiments showing that HSCT could prevent and reverse organ damage in genetic and antigen‐induced models of autoimmune disease. Subsequent laboratory studies have revealed that benefit arises, in part, from the reinfused autologous stem cells restoring thymopoiesis, broadening diversity of the TCR repertoire, renewing Treg cells, and altering disease signatures of differentially expressed genes. In the absence of the original antigenic triggers, immune homeostasis is restored and self‐tolerance can return.
But do the benefits of autologous HSCT outweigh the risks? Likely not in autoimmune diseases with low associated mortality, but for scleroderma lung disease the benefits appear compelling. Replicated in multiple phase II reports from Europe and the US, dramatic and durable improvements in skin fibrosis and quality‐of‐life measures have been observed along with stabilization of PFTs. Three prospective, randomized clinical trials in patients with SSc and internal organ involvement have compared autologous HSCT treatment to high‐dose intravenous pulse CYC given for up to 12 months. The SCOT trial is still following up all subjects through the 54‐month primary end point, but as detailed above, the ASSIST and ASTIS randomized trials have been completed and both showed statistically significant clinical benefits after stem cell transplantation.
Given the morbidity and inexorable mortality in SSc patients with organ involvement, it appears reasonable to consider consultation for HSCT treatment, and listings of experienced scleroderma research and transplant centers are available online (www.sclerodermatrial.org/announce/sitelist.html and www.scleroderma.org/site/PageServer?pagename=patient_centers_list#.Vry-ObQrJeg).
By choosing to undergo transplantation, patients will be accepting more upfront mortality risk for long‐term gain; hence, they must be carefully informed of risks and fully participate in the decision. Criteria for referral to the HSCT center are given above, but close monitoring of pulmonary function is required for timely referral. During the transplantation consultation, the procedures and toxicities are detailed along with the evidence supporting HSCT as treatment for scleroderma lung disease. Some patients will accept transplantation and others will decline. Not surprisingly, such critical decisions about treatment, as Thoreau observed, can best be reached as patients and practitioners look through each other's eyes, if only for an instant.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article and revising it critically for important intellectual content, and all authors approved the final version to be published.
ACKNOWLEDGMENT
We thank Heidi Oehme for secretarial assistance.
Supported in part by the NIH (National Institute of Allergy and Infectious Diseases awards N01‐AI‐05419 and HHSN‐272‐2011‐00025‐C).
REFERENCES
- 1. Gabrielli A, Avvedimento E, Krieg T. Mechanisms of disease: scleroderma. N Engl J Med 2009;360:1989–2003. [DOI] [PubMed] [Google Scholar]
- 2. Whitfield ML, Finlay DR, Murray JI, Troyanskaya OG, Chi JT, Pergamenschikov A, et al. Systemic and cell type‐specific gene expression patterns in scleroderma skin. Proc Natl Acad Sci U S A 2003;100:12319–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Assassi S, Swindell WR, Wu M, Tan FD, Khanna D, Furst DE, et al. Dissecting the heterogeneity of skin gene expression patterns in systemic sclerosis. Arthritis Rheumatol 2015;67:3016–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van Bon L, Affandi AJ, Broen J, Christmann RB, Marijnissen RJ, Stawski L, et al. Proteome‐wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med 2014;370:433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Domsic RT, Rodriguez‐Reyna T, Lucas M, Fertig N, Medsger TA Jr. Skin thickness progression rate: a predictor of mortality and early internal organ involvement in diffuse scleroderma. Ann Rheum Dis 2011;70:104–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Elhai M, Meune C, Avouac J, Kahan A, Allanore Y. Trends in mortality in patients with systemic sclerosis over 40 years: a systematic review and meta‐analysis of cohort studies. Rheumatology (Oxford) 2012;51:1017–26. [DOI] [PubMed] [Google Scholar]
- 7. Nihtyanova SI, Schreiber BE, Ong VH, Rosenberg D, Moinzadeh P, Coghlan JG, et al. Prediction of pulmonary complications and long‐term survival in systemic sclerosis. Arthritis Rheumatol 2014;66:1625–35. [DOI] [PubMed] [Google Scholar]
- 8. Mayes MD, Lacey JV Jr, Beebe‐Dimmer J, Gillespie BW, Cooper B, Laing TJ, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum 2003;48:2246–55. [DOI] [PubMed] [Google Scholar]
- 9. Suliman YA, Dobrota R, Huscher D, Nguyen‐Kim TD, Maurer B, Jordan S, et al. Pulmonary function tests: high rate of false‐negative results in the early detection and screening of scleroderma‐related interstitial lung disease. Arthritis Rheumatol 2015;67:3256–61. [DOI] [PubMed] [Google Scholar]
- 10. Winstone T, Assayag D, Wilcox P, Dunne J, Hague C, Leipsic J, et al. Predictors of mortality and progression in scleroderma‐associated interstitial lung disease: asystematic review. Chest 2014;146:422–36. [DOI] [PubMed] [Google Scholar]
- 11. Iudici M, Moroncini G, Cipriani P, Giacomelli R, Gabrielli A, Valentini G. Where are we going in the management of interstitial lung disease in patients with systemic sclerosis? Autoimmun Rev 2015;14:575–8. [DOI] [PubMed] [Google Scholar]
- 12. Tashkin DP, Elashoff R, Clements PJ, Goldin J, Roth MD, Furst DE, et al, for the Scleroderma Lung Study Research Group . Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med 2006;22;354:2655–66. [DOI] [PubMed] [Google Scholar]
- 13. Tashkin DP, Elashoff R, Clements PJ, Roth MD, Furst DE, Silver RM, et al, and for the Scleroderma Lung Study Research Group . Effects of 1‐year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med 2007;176:1026–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clements PJ, Tashkin D, Roth M, Khanna D, Furst DE, Tseng C, et al. The Scleroderma Lung Study II (SLS II) shows that both oral cyclophosphamide (CYC) and mycophenolate mofitil (MMF) are efficacious in treating progressive interstitial lung disease (ILD) in patients with systemic sclerosis (SSc) [abstract]. Arthritis Rheumatol 2015;67 Suppl 10 URL: http://acrabstracts.org/abstract/the-scleroderma-lung-study-ii-sls-ii-shows-that-both-oral-cyclophosphamide-cyc-and-mycophenolate-mofitil-mmf-are-efficacious-in-treating-progressive-interstitial-lung-disease-ild-in-patients-w/. [Google Scholar]
- 15. Ikehara S, Ohtsuki H, Good R, Asamoto H, Nakamura T, Sekita K, et al. Prevention of type I diabetes in nonobese diabetic mice by allogenic bone marrow transplantation. Proc Natl Acad Sci U S A 1985;82:7743–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Van Bekkum D, Bohre E, Houben P, Knaan‐Shanzer S. Regression of adjuvant‐induced arthritis in rats following bone marrow transplantation. Proc Natl Acad Sci U S A 1989;86:10090–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van Bekkum D. Conditioning regimens for the treatment of experimental arthritis with autologous bone marrow transplantation. Bone Marrow Transplant 2000;25:357–64. [DOI] [PubMed] [Google Scholar]
- 18. Tyndall A, Gratwohl A. Blood and marrow stem cell transplants in auto‐immune disease: a consensus report written on behalf of the European League against Rheumatism (EULAR) and the European Group for Blood and Marrow Transplantation (EBMT). Bone Marrow Transplant 1997;19:643–5. [DOI] [PubMed] [Google Scholar]
- 19. Sullivan KM, Furst DE. The evolving role of blood and marrow transplantation for the treatment of autoimmune diseases. J Rheumatol Suppl 1997;48:1–4. [PubMed] [Google Scholar]
- 20. Nelson JL, Torrez R, Louie F, Choe O, Storb R, Sullivan KM. Pre‐existing autoimmune disease in patients with long‐term survival after allogeneic bone marrow transplantation. J Rheumatol Suppl 1997;48:23–9. [PubMed] [Google Scholar]
- 21. Tyndall A, Black C, Finke J, Winkler J, Mertlesmann R, Peter HH, et al. Treatment of systemic sclerosis with autologous haemopoietic stem cell transplantation. Lancet 1997;349:254. [DOI] [PubMed] [Google Scholar]
- 22. Binks M, Passweg J, Furst D, McSweeney P, Sullivan K, Besenthal C, et al. Phase I/II trial of autologous stem cell transplantation in systemic sclerosis: procedure related mortality and impact on skin disease. Ann Rheum Dis 2001;60:577–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Farge D, Labopin M, Tyndall A, Fassas A, Mancardi GL, Van Laar J, et al. Autologous hematopoietic stem cell transplantation for autoimmune diseases: an observational study on 12 years’ experience from the European Group for Blood and Marrow Transplantation Working Party on Autoimmune Diseases. Haematologica 2010;95:284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pasquini MC, Voltarelli J, Atkins HL, Hamerschlak N, Zhong X, Ahn KW, et al. Transplantation for autoimmune diseases in North and South America: a report of the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant 2012;18:1471–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gratwohl A, Pasquini MC, Aljurf M, Atsuta Y, Baldomero H, Foeken L, for the Worldwide Network for Blood and Marrow Transplantation (WBMT) . One million haemopoietic stem‐cell transplants: a retrospective observational study [published erratum appears in Lancet Haematol 2015;2:e184]. Lancet Haematol 2015;2:e91–100. [DOI] [PubMed] [Google Scholar]
- 26. Thomas E, Storb R, Clift R, Fefer A, Johnson FL, Neiman PE, et al. Bone‐marrow transplantation (first of two parts). N Engl J Med 1975;292:832–43. [DOI] [PubMed] [Google Scholar]
- 27. Thomas ED, Storb R, Clift RA, Fefer A, Johnson L, Neiman PE, et al. Bone‐marrow transplantation (second of two parts). N Engl J Med 1975;292:895–902. [DOI] [PubMed] [Google Scholar]
- 28. Blume KG, Thomas ED. A review of autologous hematopoietic cell transplantation. Biol Blood Marrow Transplant 2000;6:1–12. [DOI] [PubMed] [Google Scholar]
- 29. Storek J, Zhao Z, Lin E, Berger T, McSweeney P, Nash R, et al. Recovery from and consequences of severe iatrogenic lymphopenia (induced to treat autoimmune diseases). Clin Immunol 2004;113:285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Daikeler T, Tichelli A, Passweg J. Complications of autologous hematopoietic stem cell transplantation for patients with autoimmune diseases. Pediatr Res 2012;71:439–44. [DOI] [PubMed] [Google Scholar]
- 31. Burt R, Oliveira M, Shah S, Moraes D, Simoes B, Gheorghiade M, et al. Cardiac involvement and treatment‐related mortality after non‐myeloablative haemopoietic stem‐cell transplantation with unselected autologous peripheral blood for patients with systemic sclerosis: a retrospective analysis. Lancet 2013;381:1116–24. [DOI] [PubMed] [Google Scholar]
- 32. Van Laar JM, Farge D, Sont JK, Naraghi K, Marjanovic Z, Larghero J, et al, for the EBMT/EULAR Scleroderma Study Group . Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA 2014;311:2490–8. [DOI] [PubMed] [Google Scholar]
- 33. Hosing C, Nash R, McSweeney P, Mineishi S, Seibold J, Griffith LM, et al. Acute kidney injury in patients with systemic sclerosis participating in hematopoietic cell transplantation trials in the United States. Biol Blood Marrow Transplant 2011;17:674–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Craciunescu O, Steffey BA, Kelsey CR, Larrier NA, Paarz‐Largay CJ, Prosnitz RG, et al. Renal shielding and dosimetry for patients with severe systemic sclerosis receiving immunoablation with total body irradiation in the Scleroderma: Clophosphamide or Transplantation trial. Int J Radiat Oncol Biol Phys 2011;79:1248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hung EW, Mayes MD, Sharif R, Assassi S, Machicao VI, Hosing C, et al. Gastric antral vascular ectasia and its clinical correlates in patients with early diffuse systemic sclerosis in the SCOT trial. J Rheumatol 2013;40:455–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chien JW, Sullivan KM. Carbon monoxide diffusion capacity: how low can you go for hematopoietic cell transplantation eligibility? Biol Blood Marrow Transplant 2009;15:447–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tomblyn M, Chiller T, Einsele H, Gress R, Sepkowitz K, Storek J, et al. Guidelines for preventing infectious complications among hematopoietic cell transplantation recipients: a global perspective. Biol Blood Marrow Transplant 2009;15:1143–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McSweeney PA, Nash R, Storb R, Furst D, Gauthier J, Sullivan KM. Autologous stem cell transplantation for autoimmune diseases: issues in protocol development. J Rheumatol Suppl 1997;48:79–84. [PubMed] [Google Scholar]
- 39. Nash RA, McSweeney PA, Crofford LJ, Abidi M, Chen CS, Godwin JD, et al. High‐dose immunosuppressive therapy and autologous hematopoietic cell transplantation for severe systemic sclerosis: long‐term follow‐up of the US multicenter pilot study. Blood 2007;110:1388–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Burt RK, Shah SJ, Dill K, Grant T, Gheorghiade M, Schroeder J, et al. Autologous non‐myeloablative haemopoietic stem‐cell transplantation compared with pulse Cyclophosphamide once per month for systemic sclerosis (ASSIST): an open‐label, randomised phase 2 trial. Lancet 2011;378:498–506. [DOI] [PubMed] [Google Scholar]
- 41. Sullivan KM, Wigley F, Denton C, van Laar J, Furst DE. Haemopoietic stem‐cell transplantation for systemic sclerosis. Lancet 2012;379:219–20. [DOI] [PubMed] [Google Scholar]
- 42. Sullivan KM, Muraro P, Tyndall A. Hematopoietic cell transplantation for autoimmune disease: updates from Europe and the United States. Biol Blood Marrow Transplant 2010;16 Suppl:S48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Muraro P, Douek D, Packer A, Chung K, Guenaga F, Cassiani‐Ingoni R, et al. Thymic output generates a new and diverse TCR repertoire after autologous stem cell transplantation in multiple sclerosis patients. J Exp Med 2005;201:805–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Muraro PA, Robins H, Malhotra S, Howell M, Phippard D, Desmarais C, et al. T cell repertoire following autologous stem cell transplantation for multiple sclerosis. J Clin Invest 2014;124:1168–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. De Kleer I, Vastert B, Klein M, Teklenburg G, Arkesteijn G, Yung GP. Autologous stem cell transplantation for autoimmunity induces immunologic self‐tolerance by reprogramming autoreactive T cells and restoring the CD4+CD25+ immune regulatory network. Blood 2006;107:1696–702. [DOI] [PubMed] [Google Scholar]
- 46. Delemarre EM, van den Broek T, Mijnheer G, Meerding J, Wehrens EJ, Olek S. Autologous stem cell transplantation aids autoimmune patients by functional renewal and TCR diversification of regulatory T cells. Blood 2016;127:91–101. [DOI] [PubMed] [Google Scholar]
- 47. Snowden JA. Rebooting autoimmunity with autologous HSCT. Blood 2016;127:8–10. [DOI] [PubMed] [Google Scholar]
- 48. Assassi S, Mayes MD, Pedroza C, Chang JT, Furst DE, Crofford LJ, et al. Immunoablation followed by autologous stem cell transplantation in systemic sclerosis patients decreases significantly the interferon signature [abstract]. Arthritis Rheumatol 2015;67 Suppl 10 URL: http://acrabstracts.org/abstract/immunoablation-followed-by-autologous-stem-cell-transplantation-in-systemic-sclerosis-patients-decreases-significantly-the-interferon-signatrue/. [Google Scholar]
- 49. Brkic Z, van Bon L, Cossu M, van Helden‐Meeuwsen CG, Vonk MC, Knaapen H, et al. The interferon type I signature is present in systemic sclerosis before overt fibrosis and might contribute to its pathogenesis through high BAFF gene expression and high collagen synthesis. Ann Rheum Dis 2015;75:1567–73. [DOI] [PubMed] [Google Scholar]
- 50. Pincus T, Sokka T, Wolfe F. Premature mortality in patients with rheumatoid arthritis: evolving concepts [editorial]. Arthritis Rheum 2001;44:1234–6. [DOI] [PubMed] [Google Scholar]
- 51. Fried TR. Shared decision making—finding the sweet spot. N Engl J Med 2016;374:104–6. [DOI] [PubMed] [Google Scholar]