

Abstract
Natural product extracts are a rich source of bioactive compounds. As a result, the screening of natural products for the identification of novel biologically active metabolites has been an essential part of several drug discovery programs. It is estimated that more than 70% of all drugs approved from 1981 and 2006, were either derived from or structurally similar to nature based compounds indicating the necessity for the development of a rapid method for the identification of novel compounds from plant extracts. The screening of biological matrices for the identification of novel modulators is nevertheless still challenging. In this review we discuss current techniques in phytochemical analysis and the identification of biologically active components.
1 Introduction
Natural product extracts can be considered an invaluable source of biologically active secondary metabolites. It has been clearly demonstrated that the screening of natural products, for bioactive compounds, can be a successful approach for the discovery of new drug leads. Unfortunately screening natural products for biologically active compounds, is laborious, time consuming and costly. Further, the concentration differences between different secondary metabolites poses additional problems. This has resulted in the development of high-throughput and efficient analytical approaches enabling identification of compounds with the desired biological effect. The predominant approach used is dereplication with hyphenated techniques for the chemical profiling of crude plant extracts, which has been extensively reviewed and therefore is not discussed in great detail.1 In this review, particular attention has been paid to recent developments in the use of analytical techniques for the identification of novel active components from plant extracts including advanced hyphenated techniques in phytochemical analysis, ultrafiltration HPLC-MS, on-line HPLC-biochemical detection, cellular membrane affinity chromatography and ligand fishing using protein coated magnetic beads. The limitations and advantages of each of these methods will also be discussed.
2 Phytochemical analysis
Phytochemical characterization of secondary metabolites is the first step in the majority of screening techniques for the identification of biologically active metabolites. It is an initial chemical screening, followed by biological/pharmacological tests to determine which components have the desired activity. The chromatographic fingerprint obtained prior to the biological screening is used for subsequent comparisons with fingerprints of individual fractions that have the desired biological activity, thereby reducing possible false-positive results. It also enables structural identification of many secondary metabolites present in the complex matrix, in a single run (dereplication of complex matrices).2–7
HPLC is the most extensively used separation technique for phytochemical characterization of botanical extracts and has been coupled with numerous detectors, giving the opportunity to analyze different classes of plant metabolites. For example, HPLC has been hyphenated with ultraviolet-photodiode array detection (HPLC-DAD) and mass spectrometry (HPLC-MS). Online hyphenation of mass spectrometry to HPLC has been a milestone in the analysis of complex plant extracts, as well as, in drug discovery programs. The greatest advantage has arisen from the development of high resolution mass spectrometers (HRMS) using the Orbitrap or hybrid quadrupole-time of flight (Q-TOF) mass spectrometers, allowing for the direct identification of the molecular formula of the secondary metabolites. More recently, the advent of UHPLC has gained increasing use in phytochemical characterization of plant extracts, as it results in shorter analysis time, lower use of samples and solvents and increased peak capacity.8 Full phytochemical characterization of complex extracts, as well as structure determination of secondary metabolites, can be further enhanced by the use of relatively novel and advanced hyphenated techniques, like: LC-NMR, LC-NMR-MS, LC-SPE-NMR, LC-DAD/MS-SPE-NMR and finally LC-HRMS-SPE-NMR. Hyphenated techniques have been shown to be extremely useful in the dereplication step, aimed at detection and identification of new biologically active metabolites.8
HPLC-NMR hyphenation can be regarded as an ideal combination of a versatile separation technique with a universal detector, providing structural information on separated compounds. Initial attempts to hyphenate HPLC with NMR, were made in the 1970s, however, due to analytical limitations, it was not commonly used until the late 1990’s.9 NMR spectroscopy while a universal detection method, requires sufficient concentration to be detectable, which is a limitation considering the concentration of secondary plant metabolites present in eluates. In addition, the acquisition time of NMR spectrum is another limitation.9 To circumvent these limitations in HPLC-NMR analysis, the sample injection volume is increased and columns with increased lengths and larger inner diameters are used. An alternative, is the use of on-flow HPLC-NMR, with direct coupling of HPLC and NMR. In this case, the use of deuterated solvents is necessary and typically D2O is used, while non-deuterated organic modifiers are used to reduce costs. On-flow HPLC-NMR is suitable for detection and identification of highly abundant secondary plant metabolites, however due to low S/N ratios the analyses results in poor NMR spectra.9 The real breakthrough came with the advent of on-line solid-phase extraction (SPE) add-on to the HPLC-NMR systems. The use of this new hyphenation eliminated the problem connected to the use of only those HPLC solvents, which are suitable for NMR analysis.9 In this approach, following the chromatographic separation and detection by DAD/MS, the compounds of interest are retained on SPE cartridges, dried and subsequently eluted with a deuterated solvent into an NMR probe (Fig. 1). LC-NMR hyphenation, as well as “hypernations” with other detection techniques, have been thoroughly reviewed.6,9,10
Fig. 1.

Schematic of an integrated UHPLC-UV-MS-SPE-NMR system. Reproduced with permission from Fig. 2, L. W. Sumner et al., Nat. Prod. Rep., 2015, 32, 212–229.
Most of the recently reported HPLC-HRMS-SPE-NMR hyphenation has been proven to constitute a powerful platform for direct structural and biological activity characterization of metabolites, from a crude extract. The usefulness of this approach has been confirmed by several applications and some of the most recent examples are described. In one study, 88 plant extracts were screened for identification of inhibitors of necrosis enzymes (hyaluronidase, phospholipase A and protease enzymes) in four snake venoms, using HPLC-HRMS-SPE-NMR.11 The plant extracts that were traditionally used to treat snake bites were chosen and tested for biological activity using microplate-based bioassays. Of the extracts tested, twenty-two exhibited greater than 90% inhibition in the hyaluronidase assay against at least one venom at 0.1 mg ml−1. All twenty-two extracts underwent high-resolution hyaluronidase inhibition profiling, which indicated that, the tannin metabolites were responsible for the observed activity. The results were consistent with previous reports describing tannins as antidotes against snake venoms. High-resolution hyaluronidase inhibition profiling showed four extracts, out of the tested twenty-two samples, contained peaks of hyaluronidase inhibitors that were not tannins, namely: Clausena excavata Burm.f., Androsace umbellata (Lour.) Merr., Oxalis corniculata L. and Trachelospermum jasminoides (Lindl.) Lem. These four extracts were subsequently analyzed using HPLC-HRMS-SPE-NMR approach, in order to identify the novel inhibitors. After chromatographic separation, 1% of the flow was directed to Bruker microTOF-Q II mass spectrometer equipped with an electrospray ionization (ESI) interface and the remaining eluate was diluted with water and trapped onto previously preconditioned SPE cartridges using a Prospect 2 SPE-unit.11 Following the trapping step, the cartridges were dried with a stream of nitrogen and eluted into a NMR probe, using methanol-d4. This approach identified four non-tannin inhibitors, namely: ansiumamide B from Clausena excavata Burm.f., myricetin 3-O-β-D-glucopyranoside from Androsace umbellata (Lour.) Merr., and vitexin and 4′,7-dihydroxy-5-methoxyflavone-8-C-β-D-glucopyranoside from Oxalis corniculata L., all of which were previously unknown to be effective in preventing tissue necrosis.
In a similar approach the HPLC-HRMS-SPE-NMR was used to identify natural fungicides from Uvaria chamae P. Beauv., targeting the fungal plasma membrane (PM) H+-ATPase.12 U. chamae was chosen to screen for potential fungicides, as it is traditionally used for its antibiotic effects. U. chamae ethyl acetate stem bark extract was first analyzed using high-resolution H+-ATPase assay. In this study, 300 μg defatted extract was chromatographed on C18 column using a gradient elution and 160 fractions of 88 μL aliquots were collected between 5 and 35 min. All the fractions were tested for inhibitory effect against the fungal plasma membrane (PM) H+-ATPase and for growth inhibition of Saccharomyces cerevisiae and Candida albicans. The activity profiles were constructed by plotting the results of the bioassay, for each well, against respective retention times. Subsequently, in order to identify compounds exerting anti-fungal activity, HPLC-HRMS-SPE-NMR assay was performed, using the same settings, as in the previous example. This approach led to identification of a series of uncommon o-hydroxybenzylated flavanones and chalcones, i.e., chamanetin, isochamanetin, isouvaretin, uvaretin, dichamanetin, and diu-varetin. The identified compounds were isolated and their IC50 values, for inhibition of the PM H+ATPase, and growth inhibition of Saccharomyces cerevisiae Meyen ex E.C. Hansen and Candida albicans (C.P.Robin) Berkhout, were determined.
In another approach the HPLC-HRMS-SPE-NMR was coupled with a microplate-based high-resolution profiling assay, to screen Radix Scutellariae for antidiabetic constituents exerting three different biological activities (aldose reductase/α-glucosidase/radical scavenging high-resolution inhibition profile).13 Scutellaria baicalensis Georgi was previously reported to be effective in the treatment of diabetes-related microvascular complications. In the first step of the analysis, high-resolution aldose-reductase and α-glucosidase inhibition profiles and radical scavenging profile biochromatograms were obtained and correlated with Scutellaria chromatograms (at 280 nm), in order to pinpoint peaks of biologically active metabolites. Active compounds were then identified using HPLC-HRMS-SPE-NMR hyphenation. In this study 480 μg of crude extract was chromatographed on the C-18 column using a gradient elution. 1% of the eluate was directed to a microTOF-Q II mass spectrometer, while the rest was directed to a diode-array detector and finally to a SPE unit. Five consecutive injections were performed before the SPE cartridges were dried with nitrogen gas and adsorbed compounds eluted into NMR probes, with acetonitrile-d4. Using this assay, baicalein was identified as an α-glucosidase inhibitor and baicaelein and skullcapflavone II were identified as aldose reductase inhibitors. Ganhuangemin, viscidulin III, baicalin, oroxylin A 7-O-glucuronide, wogonoside, baicalein, wogonin, and skullcapflavone II were identified as most potent radical scavengers.
While these studies demonstrate the versatility and success of phytochemical analysis in the identification of novel ligands for therapeutic targets, this approach is labor-intensive and time consuming. However, the greatest limitations of this approach is that it is heavily dependent on the concentration of the secondary metabolites. As a result, most compounds identified using these approaches tend to be the more abundant secondary metabolites and not necessarily the most active.
3 Identification of bioactive components
In this section recent developments in the use of analytical techniques as an alternative for bioassay-guided fractionation will be discussed. These bioactive detection approaches will be divided into four general categories: (1) ultrafiltration (2) bio-affinity chromatography (3) cellular membrane affinity chromatography and (4) ligand fishing.
3.1 Ultrafiltration
Ultrafiltration HPLC-MS approach was originally developed by van Breemen et al., in 1997, in response to a growing need, to screen combinatorial libraries for new possible drug leads.14 Initially, newly synthetized compounds were tested individually using classical bioassays, which led to the extensive effort to increase throughput. van Breeman et al. adjusted the concept of membrane based separations which was commonly used for processing biological macromolecules, to screen complex mixtures for biologically active compounds. This technique was predominantly used for the isolation of active compounds for synthetic or combinatorial libraries,14 in addition to studying thermodynamic and kinetic ligand–protein binding parameters. In recent years, the technique has been adapted for the detection and identification of active compounds from botanical extracts. The use of ultrafiltration HPLC-MS applied to screening plant extracts for active compounds has been extensive. For example, cyclooxygenase-2 (COX-2) inhibitors (10-gingerol, 8-shogaol and 10-shogaol) were identified from Zingiber officinale Roscoe,15 estrogen receptor-β ligands (genistein, daidzein, biochanin A) from Trifolium pratense L.,16 tyrosinase inhibitors: quercetin-3-O-(6-O-malonyl)-β-D-glucopyranoside and kaempferol-3-O-(6-O-malonyl)-β-D-glucopyranoside from Morus alba L.,17 α-glucosidase inhibitors (quercetin-3-O-rha-(1-4)-glc-rha and C-glycosylflavones (vitexin-2′′-O-glucoside, vitexin-2′′-O-rhamnoside and vitexin)) were identified from Crataegus oxyacantha L. extract (Fisch.) Bge.18 are just a few examples. Selected studies of the aforementioned applications will be discussed in more detail.
The ultrafiltration-HPLC approach consists of three steps: incubation, ultrafiltration and HPLC analysis.14,17 A rate limiting factor for this approach is the high concentration of the protein needed, as it should be similar to the binding affinity (Kd) of the weakest ligand to be tested.19 In the first step, the sample (botanical extract) is incubated with the target protein for a period of time (15 min to 2 h) at 25 °C, however, some reports have also used 37 °C.17,20,21 Multiple buffers can be used including phosphate buffer, Tris–HCl and ammonium acetate and the addition of organic solvents (10% isopropanol) is necessary if large amounts of non-specific interactions present.22 Non-specific binding to the protein or system are addressed by incubating the sample with either the denatured protein or no protein. After the incubation, ultrafiltration is carried out typically using a 10 000 molecular weight cutoff (MWCO) regenerated cellulose ultrafiltration membrane, allowing for the separation of unbound compounds from ligands that are bound to the protein. The ultrafiltration cell, which contains the ligand–protein complex, is subsequently washed with ammonium acetate buffer to remove any unbound compounds14 and until the levels are below background.19 Subsequently, the aqueous-organic buffer is run through the chamber to dissociate the ligand–protein interactions. The bound compounds are either analyzed directly by the mass spectrometer or trapped on an HPLC column and analyzed by HPLC-MS (Fig. 2).
Fig. 2.

(a) Scheme of ultrafiltration HPLC-DAD-MS assay for screening for tyrosinase inhibitors. Reproduced with permission from Fig. 1, Z. Yang et al., Anal. Chim. Acta, 2012, 719, 87–95. (b) Scheme of pulsed ultrafiltration-mass spectrometry (PUFMS) to screen chemical mixtures for compounds that bind to a macromolecular receptor. The ultrafiltration membrane traps a receptor in solution, but allows low molecular weight compounds to pass through. Bound ligands are eluted from the chamber by destabilizing the ligand–receptor complex with an organic solvent or pH change. The ligands are characterized with MS. Reproduced with permission from Fig. 1, B. M. Johnson et al., Mass Spec. Rev., 2002, 21, 76–86.
There are two operational modes for ultrafiltration: an online approach, also termed pulsed ultrafiltration mass spectrometry (PUF-MS) and an off-line mode. In the off-line mode the ultra-filtrates from the ultrafiltration chamber are manually injected into HPLC-MS system (Fig. 2a),17 while for PUF-MS, the chamber is part of the analytical system, with the ultra-filtrate injection onto the MS being fully automated (Fig. 2b).19 The ultrafiltration cell is a flow-through chamber composed of polysulfone or polyetheretherketone (PEEK).19 The chamber used in the van Breemen et al. report, contained an in-line solvent filtration unit (Upchurch Scientific), in which a filtration disk was replaced with an ultrafiltration membrane with a 10 000 MWCO (Amicon).14
Ultrafiltration studies may result in false-positives resulting from non-specific enzyme–ligand interactions, as a result a novel LC-MS ultrafiltration approach was developed to address this problem with an enzyme channel blocking assay (ECB).23 In this approach, a channel blocking ligand (high affinity competing ligand) is added during the incubation step, preventing new potential ligands from entering the orthosteric binding site. Chromatographic fingerprints obtained for samples, run with and without competitive ligands are compared to distinguish between specific binding ligands and non-specific binders. For example, febuxostat, a selective xanthine oxidase inhibitor, was used as the channel blocking ligand, to screen Flos Chrysanthemi for possible inhibitors.23 The addition of febuxostat allowed for the identification of seven selective inhibitors for xanthine oxidase using a UF-LC-MS approach. Another approach to prevent excessive binding of compounds to the ultrafiltration membrane was carried out by using a blocking protein.24 For example, glutathione S-transferase (GST) was demonstrated to reduce non-specific binding and also effectively decreased background signals.
One of the initial groups to demonstrate the usefulness of the ultrafiltration approach was Bolton and co-workers.16 In this study, human estrogen receptors ERβ was targeted as it may be a suitable target for the treatment of menopausal syndromes or estrogen induced cancer. Of the eight botanical extracts tested against ERβ, Trifolium pretense L. (red clover) had the highest affinity. As a result, it was further screened against ERβ estrogen receptor using an ultrafiltration approach. Recombinant ERβ was incubated with Trifolium pretense L. for 2 hours and filtered through a cellulose ultrafiltration membrane (30 000 MWCO).16 The filter was washed with ammonium acetate buffer to remove unbound compounds, and the ligand–protein complex was disrupted with a methanol : water (90 : 10) solution. After removal of the solvent, under reduced pressure, the retained compounds were analyzed by means of HPLC-MS. The results of the study, identified daidzein, genistein and biochanin A as estrogen receptor β ligands. This study was the first to confirm these isoflavonoids as responsible for the estrogenic activity of red clover extracts.
Another example was the development of an α-glucosidase inhibition assay coupled with ultrafiltration LC-DAD-ESI-MSn approach.18 α-Glucosidase, an enzyme present in the small intestine, is a popular target as α-glucosidase inhibitors can reduce postprandial hyperglycemia by delaying digestion and absorption of carbohydrates making it a therapeutic target in type 2 diabetes. In the first step, 20 μg Crataegus oxyacantha L. (hawthorn) flavonoid leaf extract was incubated for 30 minutes at 37 °C with α-glucosidase in ammonium acetate buffer [10 mM, pH 6.8]. Then the incubation mixture was filtered through the regenerated cellulose ultrafiltration membrane (10 000 MWCO), by centrifugation for 10 minutes at room temperature. After washing the filter with ammonium acetate, the protein-bound ligands were disrupted by the addition of methanol : water (50 : 50, v/v; pH 3.3). The solution was then centrifuged for 15 minutes, and the filtrates were dried under vacuum. After reconstitution in methanol : water (50 : 50, v/v) the α-glucosidase ligands were analyzed by LC-MS. Using this approach four new inhibitors were identified in Crataegus oxyacantha L. leaf extract: quercetin-3-O-rha-(1-4)-glc-rha and the following C-glycosylflavones: vitexin-2′′-O-glucoside, vitexin-2′′-O-rhamnoside and vitexin. Based on the obtained results, other C-glycosylflavones were also tested for possible inhibition of α-glucosidase. It was concluded that the 5,7,4-trihydroxyflavone structure was crucial for the inhibitory activity and that the additional C-3-OH substitute on the B-ring enhanced the activity. The inhibition was weakened in case of C-glycosylations at C-6 or C-8.
Another recent example of the success of ultrafiltration LC-MS was demonstrated with quinone reductase-2 (QR-2). This protein has been targeted as a new antimalarial and antitumor target. Extracts of Humulus lupulus L. and of marine sediment bacteria were screened against QR-2 for the identification of novel inhibitors.21 In this study, 2 μg of Humulus lupulus L. extract was incubated with 12 μg of human recombinant QR-2 in Tris buffer [100 mM, pH 7.5] containing 10% glycerol, 50 mM KCl, and 1 mM EDTA at room temperature for 2 h. Following incubation, the solution was filtered through the regenerated cellulose membrane (10 000 MWCO), by centrifugation at 13 000 × g for 7 minutes at 4 °C. The membrane was washed three times with ammonium acetate buffer and centrifuged at 13 000 × g for 7 minutes and the QR-2-ligand complexes were then disrupted with methanol and centrifuged. The QR-2 ligands were dried under the stream of nitrogen gas, followed by reconstitution in 50% aqueous methanol and analyzed by LC-MS. This approach led to the identification of tetrangulol methyl ester from an extract of Actinomyces sp. from marine sediment and xanthohumol and xanthohumol D from Humulus lupulus L. as QR-2 ligands.
In an ECB approach, activated and denatured tyrosinase, an enzyme responsible for synthesis of melanin and a therapeutic target for Parkinson’s disease, was incubated with Morus alba L. leaves extract,17,25 with and without resveratrol (a known ligand). The mixtures were filtered through cellulose membrane (10 000 MWCO) and the ultrafiltrates were collected and injected for analysis by HPLC-DAD-MS. Initially, the fingerprint of Morus alba L. leaf extract was compared to the fingerprint obtained from the eluate after ultrafiltration with the active tyrosinase. To confirm that the retained compounds were not due to nonspecific binding, the fingerprint was compared with the fingerprint of the eluate obtained with the denatured tyrosinase. To confirm that the identified compounds bound to the active site of tyrosinase, the study was repeated in the presence of resveratrol. A decrease in the peak areas on the fingerprint indicated that the identified compounds in the ultrafiltration study were binding to the active site of tyrosinase. In this study, quercetin-3-O-(6-O-malonyl)-β-D-glucopyranoside and kaempferol-3-O-(6-O-malonyl)-β-D-glucopyranoside, were identified as new tyrosinase inhibitors.
More recently, Yang et al. developed an interesting approach aimed at the identification of mitochondria-targeted natural ligands,26 as mitochondrial dysfunction is known to lead to a number of diseases, including cancer, neurodegenerations, diabetes, ischemia-reperfusion injury and many others.26 Polygoni Cuspidati Rhizoma et Radix and Scutellariae Radix have been previously reported to modify mitochondrial functions and to exert antitumor, cardio-, hepatic- and neuro-protection effects.27,28 As a result, extracts of Polygonum cuspidatum (Houtt.) Ronse Decr and Scutellaria baicalensis Georgi were incubated with isolated rat cardiac muscle mitochondria suspension at 37 °C for 60 minutes.29 The incubation step was followed by ultrafiltration through regenerated cellulose membrane (10 000 MWCO), and multiple washes with ammonium acetate buffer [50 mM, pH 7.5] and centrifuged at 14 000 × g. The bound mitochondrial ligands were eluted by disrupting the mitochondria–ligand complex via ultrasonication in aqueous methanol.26 The ultrafiltrate was dried and reconstituted for analysis by LC-MS. This study identified piceid, polygonimitin B, epigallocatechin gallate, 3,5,4′-trihydroxystilben-3-O-(6′′-galloyl)-glucoside, emodin-1-O-glucoside, resveratrol, torachryson-8-O-glucoside, emodin-8-O-glucoside, emodin-8-O-(6′-malonyl)-glucoside and emodin as active compounds from Polygonum cuspidatum (Houtt.) Ronse Decr and baicalin, 5,7,8-trihydroxy-flavanone-(7 or 8)-O-glucuronide, 2′,5,7-trihydroxy-6-methoxyflavanone-(7 or 8)-O-glucuronide, oroxylin A-7-O-glucuronide, wogonoside, baicalein, 5,7-dihydroxy-2′,3′,6,8-tetramethoxyflavone, wogonin and oroxylin A from Scutellaria baicalensis Georgi.
While ultrafiltration has been successfully used in phytochemical analysis as evidenced above, one of its most significant limitations is that it can only be used with purified proteins and is not suitable for transmembrane proteins, as a large amount of non-specific interactions, would result in false positives. Further, for the cytosolic protein, ultrafiltration assays require the use of relatively high amount of proteins, which practically limits the studies to commercially available enzymes/proteins. While, this approach has been successfully used to fish out active compounds, similar dereplication, this approach typically identifies the compounds that are more abundant in the complex matrices, and overlooks low-affinity ligands. Further, in most cases, the proteins targets cannot be reused, however, this is not always the case. van Breemen et al. reported that human serum albumin could have been used two more times after the initial run,14 thereby allowing the recovery of receptor molecules, allowing the reuse of protein targets, in further runs.
3.2 Bioaffinity chromatography
Apart from being a powerful tool for detection and identification of secondary plant metabolites, HPLC hyphenated techniques, have been proved to be useful in the screening of complex matrices for biologically active secondary metabolites. Generally, all the HPLC-based bioaffinity techniques can be ascribed to one of the following categories: post-chromatographic biochemical detection assays (on-line mode, at line approach and off-line mode)30 or enzyme-immobilized HPLC reactors (silica and monolith-based supports).
In on-line HPLC-biochemical detection techniques (Fig. 3), the eluate from the analytical column is split and part of it is directed to the mass spectrometer (fingerprint), while the rest of the eluate is mixed with the target protein (continuous flow) and allowed to interact in the first reaction coil. Subsequently, the eluate is mixed with a substrate specific for the targeted enzyme in the second coil. The product of the enzymatic reaction is measured using UV, MS or fluorescence detectors. Enzyme inhibitors, present in the analyzed sample will prevent the formation of the reaction product and therefore a reduced peak is observed in the biological profile. In case of non-enzymatic proteins, active components compete with a fluorescently-labelled ligand in the second coil. Successful binding of a plant metabolite is observed by a decrease in fluorescence signal. The simultaneous analysis of the fingerprint and the biological activity allows the identification of the peaks with the desired activity, allowing for a more rapid isolation than dereplication. A few limitations with this approach include the fact that solvent and buffer choice must be compatible with the protein to be tested, it has complex instrumentation, limited reaction time between compounds and protein, and that the activity is strongly influenced by gradient elution and finally that it requires the use of large amounts of protein. The limited reaction time between potential ligands and target protein has been addressed with at-line HPLC-bioassays. In this approach, all the biochemicals that are necessary to complete the assay are added on-line post-column and the eluate is collected in microplates instead of being directed to the reaction coil, and subsequently handled off-line to complete the assay.30 In the off-line assay, the fractions of eluate are collected, and UV or MS data are recorded simultaneously. The obtained fractions are usually dried and re-dissolved and biological tests are performed. This approach does not pose as many limitations as the on-line assays, as there is no need for sophisticated equipment, no restrictions regarding post-column reaction time or in the solvents used in the chromatographic step. Structural elucidation of active metabolites is usually completed using NMR spectra, recorded off-line. The off-line approaches have been used more often, when compared to on-line assays. All these approaches have been successfully used for many proteins including acetylcholine esterase,31 estrogen receptor,32 glutathione-S-transferase, serine protease and free radical scavengers30 and has been extensively reviewed by Poteratt and Hamburger and will be briefly discussed.
Fig. 3.
Typical on-line setup for HPLC post-column receptor assays. Reproduced with permission from Fig. 2, O. Potterat and M. Hamburger, Nat. Prod. Rep., 2013, 30, 546–564.
An on-flow post-column bioassay was developed to screen natural products for the identification of novel inhibitors of the angiotensin-converting enzyme (ACE),33 as ACE inhibitors are currently used in the treatment of hypertension. The authors developed an HPLC-biochemical detection-MS assay, to fish for ACE inhibitors in hydrolyzed milk samples. The analyzed samples were first chromatographed on a C18 (5μ, 100 Å particle) analytical column at 1 ml min−1. The eluate was split post-column, and 50 μl min−1 was directed for biochemical screening, while 200 μl min−1 was introduced to QTOF micro mass spectrometer for fingerprinting with the remaining 750 μl min−1 directed to waste. For the biochemical screening the eluate was mixed with ACE (0.0375 U ml−1 dissolved in Tris buffer [200 mM, pH 7.5] containing 300mMNaCl, 0.5% Tween). The mixture was mixed for 1 min in the first reaction coil, after which it was mixed with ortho-aminobenzoic acid–phenylalanine–arginine–lysine–dinitrophenol–proline (a fluorescence substrate) and incubated for 2 min in the second coil. The fluorescence enhancement was monitored continuously at an excitation and emission wavelength of 320 and 420 nm, respectively. Any compound that reduced the rate of enzymatic substrate conversion, demonstrated by negative peaks in the bioactivity chromatogram, was identified as an ACE inhibitor. Using this approach, twenty ACE inhibitory compounds were detected and identified in hydrolyzed milk protein samples.
An on-flow post-column HPLC technique was developed to screen a chemical library for inhibitors of rat cytosolic glutathione-S-transferases (cGSTs) and purified human GST P1,34 as the inhibitors of glutathione-S-transferases were found to sensitize cancer cells to anticancer drugs and may be used as adjuvants in chemotherapy. However, this approach has not been demonstrated on plant extracts to date. In order to identify specific inhibitors of glutathione-S-transferase, a mixture of potential inhibitors was first separated on C18 column, using gradient elution. After which, the eluate was split 1 : 2 : 7, to the GST P1 enzyme affinity detection (EAD) system, the cGSTs EAD system, and the UV detector, respectively. The enzyme and the substrate (mono-chlorobimane) were delivered post column and allowed to interact with the eluate in the reaction coils. The substrate is converted to glutathione-bimane, which fluoresces, as a result, the presence of inhibitors would result in negative peaks in the resulting bioactivity fingerprint.
Another approach was carried out by Zhao and co-workers. In this study, they developed a capillary microreactor where they co-immobilized adenosine deaminase (ADA) and xanthine oxidase (XOD).35 Both enzymes were immobilized onto the surface of gold nanoparticle (AuNPs). Gold nanoparticles have the advantage of having large surface area, compatibility with proteins and can be covalently attached to free surface amines due to the strong affinity of gold for amines. A capillary was then primed with sodium hydroxide, washed with deionized water and then a 5.6% w/v solution of polyethyleneimine (PEI, MW 70 000, 30% w/v aqueous solution) was filled up to 0.5 cm to produce a positively charged coating on the surface of the capillary. The unreacted PEI was washed and the protein coated AuNPs were injected onto the capillary and kept at 4 °C for 2 h. The non-immobilized protein coated Au-NPs were then washed and the capillary microreactor was ready for analysis. In this study, they screened both enzymes simultaneously with a series of known inhibitors and demonstrated that this method can be used for high throughput screening, using capillary electrophoresis. Initially, the capillary was rinsed with borate running buffer [25 mM, pH 8.0]. Subsequently, the substrate solution (0.375 mM of adenosine (ADA) and xanthine (XOD)) was injected and the products were separated from the un-reacted substrates with the application of 22 kV. After separation the products were quantified by UV. For the inhibition studies, the natural product extract was run in conjunction with the substrates and incubated for 2.5 min in the capillary. The peak area of the product with inhibitor was compared to the peak area of the product without inhibitor to determine the level of inhibition. While this method did not determine which compound was the active component from the natural product, it was able to identify which plant extract had activity in a ‘yes’ ‘no’ manner. Of the twenty extracts screened against ADA and XOD for inhibitory activity, Rhizoma Chuanxiong, Cortex Phellodendri, Rhizoma Alpiniae officinarum, and Ramulus Cinnamomi inhibited XOD activity and Rhizoma Chuanxiong and Indigo naturalis inhibited ADA activity.
Several other examples exist where they coupled immobilized enzymes with CE to screen natural products.36 Due to the versatility of monolithic supports, they have become the support of choice for enzyme immobilization. In this study, α-glucosidase was immobilized onto the surface of the poly(-glycidyl methacrylate-co-ethylene dimethacrylate) poly(GMA-co-EDMA) monolith modified by AuNPs.36 The resulting α-glucosidase microreactor was preconditioned with phosphate buffer [10 mM, pH 7.0], then a substrate solution p-nitrophenyl-α-D-glucopyranoside (pNPG) with or without an inhibitor was injected onto the microreactor and the enzymatic activity was determined by measuring the peak area of the product by capillary electrophoresis. The addition of acarbose, an α-glucosidase inhibitor, resulted in an expected decrease in the product formation. As a result, a series of eleven natural products were tested for inhibitory activity against α-glucosidase. Of the eleven extract tested, natural indigo had no effect and Radix Paeoniae rubrae and Puerariae lobatae inhibited α-glucosidase activity by the greatest extent.36 Similar to the previous studies, this method only identified which extracts had the desired activity and did not identify the active components.
While there are several advantages of bio-affinity chromatography, including the direct identification of a pharmacologically active component, the process involved for screening plant extracts is nevertheless still challenging. If active compounds are not retained for a significant amount of time by the immobilized protein, the risk of co-eluting active and inactive compounds is high. In addition, running crude plant extracts through the protein-based stationary phase column may prove detrimental to the column. In addition, an additional limitation is that the mobile phase has to be predominantly aqueous (typically >95%) to ensure proper ligand–protein interactions, which may result in increased non-specific binding.
3.3 Cellular membrane affinity chromatography
In the previous sections, enzymes or cytosolic proteins have been predominantly discussed. However, about half of all drug discovery programs are targeting transmembrane proteins for the identification of novel drugs illustrating their importance.37
The first immobilization of a transmembrane protein was carried out by Lundahl’s group in 1995. In these studies, the online study of glucose transport was carried out by the immobilization of GLUT1 transporter,38,39 demonstrating that transmembrane protein-based stationary phases could be used as a tool to study ligand–protein interactions. Following Lundahl’s initial work, two major approaches were developed for the immobilization of transmembrane proteins from cells from cultured cells or tissues: cellular membrane affinity chromatography (CMAC) and Cellular Membrane Chromatography (CMC). In both methods, the immobilization of the transmembrane protein is carried out through adsorption. While covalent immobilization is preferred, in the case of transmembrane proteins, the integrity of the boundary lipids is of paramount importance, as a result adsorption is desirable.
The preparation of the CMAC has been previously discussed in great detail.37 Briefly, a cell line or tissue, containing the targeted protein is homogenized, centrifuged at low speed to remove nuclei followed by high speed centrifugation to yield the cellular membranes. The pellet is then solubilized using a mild detergent. A detergent with a high critical micellar concentration (~mM) and low aggregate number per micelle (c.f. CHAPs, cholate, n-octyl β-glucopyranoside) is preferred as strong detergents strip all the boundary lipids from the transmembrane protein, which may result in the immobilization of a non-functional protein. The solubilized solution is then mixed with Immobilized Artificial Monolayer stationary phase (Regis Technologies). The immobilized artificial membrane (IAM) liquid chromatographic stationary phase was developed by Pidgeon40 and is a silica particle (12 μ with 300 Å pores) to which a monolayer of phospholipid analogues with functional head groups (choline), have been covalently coupled, mimicking the environment of the cell membrane. The detergent is removed through the process of dialysis over 24–48 hours, resulting in the adsorption of the transmembrane proteins onto the surface of the IAM stationary phase. A similar protocol is followed for the immobilization of cellular membranes from tissues.41 The advantage of this approach is that solubilized tissues from healthy and diseased state can be compared, potentially identifying novel active compounds specific for the diseased state. The immobilization of tissues results in the co-immobilization of several receptor types and may be used to identify active components for multiple targets. Similarly, the immobilization of homogenized cell line and tissue has also been carried out onto the surface of a 100 μm id open tubular capillary. This approach is necessary when the ligands are lipophilic, for example substrates of the Pgp transporter42 or cannabinoids for the CB1 and CB2 receptors,43 because in these cases, the nonspecific interactions between the ligand and the IAM stationary phase are greater than the specific interactions. For example, for the Pgp transporter, the experimental run time was reduced greater than 90% when immobilized on the open tubular format versus the IAM format.42 A large number of transmembrane proteins have been immobilized using this method including GPCRs: cannabinoid receptors,43 P2Y1 purinergic receptor,44 LGICs: α3β4, α3β4α5, α4β2, α4β4, α7 nicotinic receptors,45–47 g-amino-butyric acid receptors and N-methyl-D-aspartate receptors;41 ATP binding cassette (ABC) transporters: Pgp, BCRP, MRP1;48,49 and Solute Carrier transmembrane proteins including OAT, OCT1, OCT1R488M and OCT1G465R.50–52
In cellular membrane chromatography (CMC), cell membranes are suspended in buffer and ruptured by ultrasonic procedure.53 The resulting homogenate is centrifuged at low speed to remove nuclei followed by high speed centrifugation to yield cell membranes.54,55 The cell membrane is resuspended in buffer and slowly added to the activated silica over 30 min,53,56 resulting in a CMC column. Similar to the CMAC method, this has been carried out with cell lines,53 muscarinic receptors55 and fibroblast growth factor receptor 4;56 tissue: cyno-blood vessel57 and rabbit arteriae aorta.58 These are desirable approaches for the identification of the active components from plant extracts.
The protein-based stationary phases were initially used to characterize the binding characteristics of the immobilized protein. A comparison was done between the binding affinities determined by frontal affinity chromatography and standard binding techniques including binding assays, calcium fluorescent assays, monolayer efflux assays, rubidium efflux assays and surface plasmon resonance.45,50,53,55,56,59–61 Frontal affinity chromatography is a chromatographic method, where a constant concentration of a marker ligand is placed in the mobile phase with varying concentrations of a displacer under dynamic equilibrium conditions.62 In this method, the initial flat portion is followed by a vertical breakthrough, which reflects the specific interactions with the immobilized protein, ending in a plateau, which represents saturation of the binding sites on the immobilized protein.37 The inflection point of the breakthrough curves is directly related to the concentration of the applied ligand and the association equilibrium constants between the ligand and immobilized protein can be calculated.50 It was also demonstrated that the retention of the tested ligands correlated with the binding affinity of the compound,55,60 and that a chromatographic displacement assay, where the log k′ values of the ligand determined in the presence of varying concentrations of a marker ligand for the targeted protein, can be used to calculate the binding affinity.53
A recent example of this application was the study for the identification of a novel dual acting CB1 antagonist/CB2 agonist for the treatment of Type 1 diabetes.63 CB1 antagonists have been shown to be effective in preventing β-cell apoptosis, while CB2 agonist could reduce T-cell activation which would prevent the autoimmune response.63 As phytocannabinoids are one of the major groups of CB receptor-directed drugs currently in clinical use, botanical extracts are therefore a logical source for the identification of novel ligands with cannabinoid activity. As a result, membrane fragments of the KU-812 cell line were immobilized onto the surface of an open tubular capillary, resulting in the CB1/CB2-OT column.43 After the characterization of the agonist binding site, the column was used to screen a series of plant extracts to identify one, which may have the desired activity. Zanthoxylum clava-herculis L. (Rutaceae), an extract rich in alkylamides, was tested against the CB1/CB2 receptors for activity. Alkylamides are a logical choice as they were previously shown to have high affinity for CB2 receptor.64 A simple yes/no experiment was conducted to determine whether the extract had the desired activity. A single frontal chromatographic experiment was carried out with 0.25 nM [3H]-Win-551212 as the marker ligand in the absence or presence of 1% Zanthoxylum extract (Fig. 4). A significant displacement was observed for Zanthoxylum extract indicating the presence of phytocannabinoids. This demonstrates that a single chromatographic run can identify whether a botanical extract has any active components for the targeted immobilized protein. The study was subsequently expanded to a classic dereplication approach and identified a novel alkylamide with the desired dual activity.63
Fig. 4.
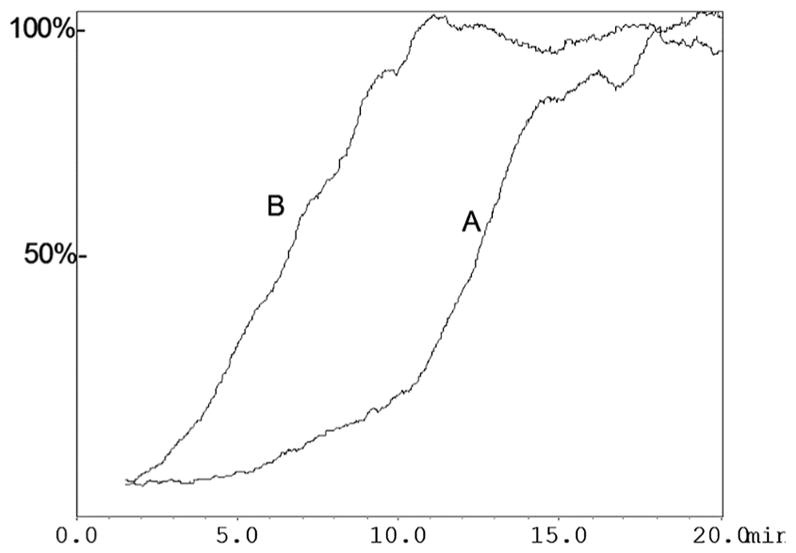
Frontal Affinity Chromatogram showing the displacement of 0.25 nM [3H]-Win 551212 (A) in the presence of 1% Zanthoxylum clavaherculis (B) at a flow rate of 50 ml min−1 and the mobile phase composed of ammonium acetate [10 mM, pH 7.4] and methanol (90 : 10, v/v). Reproduced with permission from Fig. 3, Moaddel et al., Anal. Biochem., 2011, 412, 85–91.
In another example, He and colleagues immobilized membranes from human epidermal squamous cells (A431 cell line) using CMC, in order to identify antagonists for EGFR from complex matrices.53 They screened against Radix Sophorae flavescentis against the EGFR column and identified oxymatrine and matrine as novel antagonists of EGFR.
Another approach that was adapted with CMAC columns called missing peak chromatography. In this study tobacco smoke condensates were screened against the α3β4 nicotinic receptor.65,66 This was carried out by collecting similar fractions from both the α3β4 (+) nAChR-IAM (CMAC(+)) column and the α3β4 (−) nAChR-IAM CMAC(−) column, which is the column resulting from the non-transfected cell line. As a result, a peak that is missing in the fingerprint of a CMAC(+) fraction, but present in the corresponding CMAC(−) (control) fraction is indicative of a specific binder. The “missing peaks” can be isolated from the initial mixture using an analytical or semi-preparative C18 column and the structure determined and tested for activity. The use of a control column is a very important aspect when using CMAC or CMC, as the retention time of a compound on a trans-membrane column is the summation of all the specific and non-specific interactions and not necessarily a reflection of affinity. Using this approach reduces the amount of false-positive results. The results of the study identified nicotine, anatabine, N′-nitrosonornicotine, nornicotine, anabasine, n-methyl-g-oxo-3-pyridinebutanamide and (1′s,2′s)-nicotine 1′-oxide as α3β4 nAChR ligands,65 demonstrating the applicability of missing peak chromatography to study complex matrices.
In a continuation from the previous study, the α3β4α5 nAChR has been studied due to its role in smoking cessation and addiction.66 As very few ligands have been identified as selective α3β4α5 ligands, and several alkaloids including nicotine, anatabine, anabasine are nAChR ligands, a screening study against plant extracts was carried out. An aqueous-alcoholic solution of Lycopodium clavatum L., which contains a large amount of alkaloids and Trigonella foenum graecum L., whose only alkaloid is trigonelline were tested. Both extracts were screened against the α3β4 and α3β4α5 nAChRs using 60 pM [3H]-epibatidine as the marker ligand for the agonist binding site and 1 μM MCM as the marker ligand for the non-competitive inhibitor binding site. 0.5% of the Lycopodium clavatum L. extract caused a significant displacement of 60 pM [3H]-epibatidine for both subtypes, while a similar concentration of Trigonella foenum graecum L. had no effect. While studying the non-competitive inhibitor binding site, the concentration of the plant extracts was increased to 5%. In this case, Lycopodium clavatum L. extract caused a significant displacement for mecamylamine on the α3β4 nAChR, with no displacement for the α3β4α5 nAChR. This demonstrates an added advantage of this method of screening, in that it is capable of identifying subtype selective ligands, which is important in any drug discovery program.
CMAC offers significant advantages relative to other screening methods for transmembrane proteins. These advantages include the direct identification of active components, the immobilization of the target transmembrane protein in its natural lipid environment (allowing for proper functioning of the immobilized protein) and the identifications of subtype selective ligands. However, the method has limitations including the mobile phase being predominantly aqueous and the requirement to run the corresponding control column (CMAC(−)), to confirm the absence of non-specific interaction. Also, when immobilizing cell membranes, the co-immobilization of several receptor types will be carried out. While this could be an advantage, allowing for the simultaneous screening of multiple proteins, it could also result in specific retention of components for a yet unidentified immobilized protein, further necessitating a CMAC(−) column that is only lacking the targeted protein.
3.4 Ligand fishing
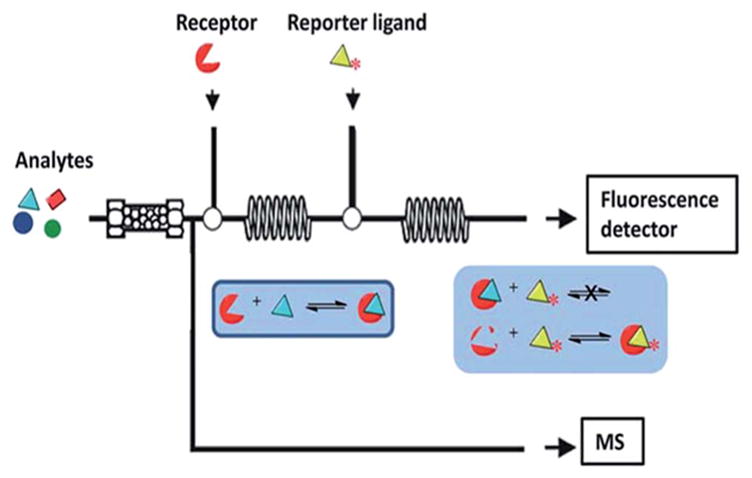
The identification of active components through bioaffinity chromatography, while having several advantages has several limitations as described above. In order to circumvent these problems, another plausible approach is the use of protein-coated magnetic beads. In this technique, the protein-coated beads are immersed directly into the extract and any compounds with an affinity for the immobilized protein are retained while non-binders remain in the supernatant. The combination of this technique with chromatographic and spectroscopic techniques allows for the detection and identification of new potential drugs. This screening technique is known as ligand fishing.67–71 The success of a ligand fishing experiment is dependent on the ability to detect the bound ligand.72 In this approach, the targeted protein is immobilized onto the surface of magnetic beads and the active ligands are subsequently fished out by suspending the protein-coated beads directly in a crude plant extract (Fig. 5).
Fig. 5.

Ligand fishing approach. Protein coated magnetic beads are incubated with a plant extract, washed and then eluted in an aqueous organic buffer. The elution buffer is then analyzed by HPLC-MS.
One of the greatest advantages of using magnetic particles is the possibility of identification of low-affinity ligands, which have been usually overlooked, when using other screening techniques, as well as low abundant secondary metabolites which have high affinity for the targeted protein.70,73,74 Several proteins have been used for ligand fishing including: the enzymes protein tyrosine phosphatase 1B and α-glucosidase extracted several compounds from Pericarpium Granati (pomegranate peel) with activity towards both enzymes;75 pancreatic lipase coated magnetic nanoparticles successfully extracted 3 flavan-3-ol from oolong tea extract;76 AChE coated magnetic beads identified active components from Melodinus fusiformis Champ. ex Benth;70 active components from Trigonella foenum-graecum L. against SIRT6 were identified using SIRT6 coated magnetic beads;69,71 the use of magnetic nanoparticles as a high throughput screening method to determine whether a compound has an affinity for the immobilized estrogen receptor in a ‘yes’ or ‘no’ method73 and ligand fishing with LC-HRMS-SPE-NMR was also carried out with α-glucosidase coated magnetic beads for the identification of active components from Eugenia catharinae O.Berg.74
The initial report on the use of protein coated magnetic beads for ligand fishing was carried out for human serum albumin (HSA).67 In this study, Moaddel et al., demonstrated that a protein coated magnetic bead could be used to fish out known binders from a mixture of binders and non-binders. In order to consider non-specific binding, the study was repeated with control beads that did not contain the immobilized HSA. A rate limiting step of ligand fishing experiments is the amount of protein that is required for the successful fishing experiments, typically 50 μg of protein is required. In this experiment, the HSA coated MBs were incubated with a sample mixture of six compounds: warfarin, AZT and naproxen (HSA ligands) and nicotine, fenoterol and labetolol (non-binders). The beads were incubated with the sample mixture for 15 min and then separated. The beads were then subsequently washed twice with ammonium acetate buffer [10 mM, pH 7.4] and eluted with ammonium acetate buffer [10 mM, pH 7.4] containing 10% ACN. The supernatant was removed and analyzed. It was clearly demonstrated that the majority of the known ligands for HSA were retained and eluted with the elution buffer (warfarin, AZT and naproxen; 51%, 60% and 74% respectively), while the non-binders were predominantly present in the initial supernatant (nicotine, fenoterol, labetolol; 47%, 71%, 52%, respectively).
A few examples of ligand fishing studies in complex matrices are discussed. As AChE inhibitors are currently one of the few therapies approved for the treatment of Alzheimer’s disease, the identification of novel ligands that could modulate AChE activity is of great therapeutic importance. Further, a number of known inhibitors of AChE have been derived from plant extracts, including galanthamine, currently in clinical use was extracted from Galantus nivalis L. species;77 Huperzine A (HupA), an alkaloid from the extract of Lycopodium genus;78 and indole alkaloids from Ervatamia hainanensis Tsiang,79 making AChE an ideal target for fishing experiments in plant extracts. In a recent study, plant extracts from Melodinus genus were investigated for AChE activity, as they are known to be a good source of alkaloids.70–78 As with any ligand fishing experiment, the validation of the immobilized AChE was carried out by determining the effect of known inhibitors to the ratio of product to substrate. Similarly, for plant extracts, an initial screen was carried out to determine which extract had the desired activity on AChE. Of the five extracts prepared from the leaves of Melodinus fusiformis Champ. ex Benth. species the total alkali JSC-E (TA-JSC-E) extracts was the most effective at inhibiting AChE activity. As a result, only the TA-JSC-E was used for the subsequent ligand fishing studies. Prior to carrying out the ligand fishing experiment, a set of known binders (galanthamine, tacrine and the coumarin derivative) and non-binders (ketamine and labetalol) were incubated with the AChE-MB to optimize the parameters including incubation time, temperature, washes and elution buffers. It was clearly demonstrated that only the binders were retained by the AChE-MB, while the non-binders were present in the loading buffer. A parallel experiment was carried out with control magnetic beads without AChE and it was shown that 90% of all compounds tested (binders and non-binders) remained in the loading buffer. The ligand fishing experiment was then carried out with the TA-JSC-E extract. 2.0 μg mL−1 of TA-JSC-E extract was incubated with the AChE-MB with vigorous shaking for 30 s and set on the magnetic separator for 2 min. The beads were retained and then washed twice with ammonium acetate [15 mM; pH 8.0] with vigorous shaking for 10 s. The bound material was then eluted with ammonium acetate [15 mM, pH 8.0] containing 20% of methanol and shaken at 300 rpm for 10 min. The elution buffer was subsequently injected and monitored by HPLC-MS and compared to the fingerprint of the T. A.-JSC-E, to identify which peaks were retained. As over 30 m/z ions were detected, multiple fractions were collected to determine whether any of the retained compounds could inhibit AChE activity. Of the compounds isolated, a compound with the m/z of 156 was isolated and demonstrated to inhibit AChE activity by greater than 40% and had a Ki of 200 nM.
Another example of the advantages of ligand fishing experiments was demonstrated with SIRT6 protein. The SIRT6 protein, a master regulator of glucose homeostasis and a target for the treatment of obesity and insulin resistant diabetes,80–82 had limited known binders. Therefore, the identification of new compounds that could modulate SIRT6 activity could be of great therapeutic importance. Using protein-coated magnetic beads and a candidate approach several novel inhibitors for the SIRT6 protein were identified.69 A key point that was demonstrated in this study, is the immobilization procedure is of paramount importance. The SIRT6 protein was immobilized by both the C-terminus and N-terminus, and it was demonstrated that the immobilization through the N-terminus resulted in an inactive protein, which was consistent with previous studies reporting that the catalytic site was near the N-terminus.83 As a result, only the C-terminus immobilized SIRT6 protein was used for the ligand fishing studies. As previous studies had demonstrated that T. foenum-graecum L. seed extracts could decrease blood glucose levels84–86 and had favorable effects on serum lipids86,87 and improved glycemic control in Type 2 diabetes, this extract was chosen to screen against the SIRT6 protein. Initial studies demonstrated that 1% fenugreek seed extract (Trigonella foenum-graecum L.) inhibited the deacetylation activity of SIRT6 (CT)-MB by >50%, and through a candidate approach identified quercetin and vitexin as novel SIRT6 inhibitors,69 with quercetin subsequently being demonstrated to being a weak activator as well.88 As a result, a ligand fishing experiment was prepared for the identification of novel inhibitors for the SIRT6 protein. Initially, the conditions for the ligand fishing experiments were optimized using known inhibitors, including quercetin, naringenin and vitexin (strong, moderate and weak binders).61,69,71 A 1% solution of T. foenum-graecum L. was incubated with SIRT6-MB and control MBs for 15 min. The MBs were washed twice with ammonium acetate buffer [10 mM, pH 7.4] and eluted with ammonium acetate buffer [10 mM, pH 7.4] containing 20% methanol for 15 min. From the extract of T. foenum-graecum L., only 18 compounds were retained by the SIRT6-MBs. Of these 18 compounds, several were known inhibitors including: vitexin, naringenin, quercetin and isovitexin. None of these compounds were retained in the control MB. Of the remaining 14 compounds, fractions were isolated and tested by frontal chromatography to determine whether they had an affinity for the SIRT6 protein and if so, did they compete with quercetin for the quercetin binding site. Of the remaining 14 compounds two compounds were identified as high affinity SIRT6 ligands. One was identified as orientin and displaced quercetin, while the other compound (m/z 556), bound to an allosteric site. This study demonstrates a limitation of ligand fishing, similar to other methods, the possibility of retaining false positives exists, however, in light of the considerable reduction in the number of identified hits from a complex matrix, makes this approach worthwhile.
Another prominent target from botanical matrices is the membrane-bound α-glucosidase enzyme.74,75 As discussed above, this enzyme has been targeted through multiple approaches as it controls post-prandial hyperglycemia,13,18,74,75 and its inhibition is a current therapeutic strategy for managing blood glucose level in diabetic patients.89 α-Glucosidase was immobilized through the N-terminus onto the magnetic beads (AG-MB) and the activity of the immobilized enzyme was tested to confirm that it was immobilized in an active conformation. This was carried out by incubating the AG-MBs with a standard substrate p-NPG and determining the ratio of the catalytic product para-nitrophenol (p-NP) and substrate p-NPG. The results demonstrated that α-glucosidase was immobilized in its active conformation and that its activity was inhibited by acarbose (α-glucosidase inhibitor). As a result, the optimization of the ligand fishing experiments was carried out using an equimolar concentration of luteolin (binder) and caffeine, ferulic acid (non binders), and it was clearly demonstrated that only luteolin was retained from the AG-MB.74 The optimized ligand fishing assay was then carried out with Eugenia catharinae O.Berg, a native Brazilian plant belonging to the Myrtaceae Family, as this family is used in traditional folk medicine for treatment of diabetes.90–92 In this study, the ligand fishing experiment was combined with HPLC-HRMS-SPE-NMR platform for direct structural identification of constituents from crude extracts at analytical-scale HPLC conditions.93,94 Crude ethyl acetate extract of E. catharinae O.Berg were incubated with ammonium acetate buffer [10 mM, pH 7.4] containing 5% methanol for 10 min and then washed three times with ammonium acetate buffer [10 mM, pH 7.4] and three sequential elutions with the elution buffer [90% methanol]. The retained compounds (elution buffer) were targeted in the HPLC-HRMS-SPE-NMR analysis using the retention time and m/z information. By comparing the LC-HRMS chromatograms for the loading buffer (S0) and first elution fraction (S5), 4 compounds were identified (5, 10, 12 and 14) as α-glucosidase inhibitors (Fig. 6). These peaks were subsequently trapped on SPE cartridges and eluted into NMR tubes for NMR analysis. The peaks were directly identified with the HPLC-HRMS-SPE-NMR platform to be myricitrin (peak 5), myricetin (peak 10), quercetin (peak 12) and kaempferol (peak 14).74 The retention of these peaks into the elution fraction, clearly demonstrate the power of this method as all these metabolites were previously known α-glucosidase inhibitors. In addition, compared to a dereplication technique, a further advantage of the method is clearly demonstrated with the isolation of myricetin (peak 10). This was a minor peak in the plant extract and would have most likely been missed using classic dereplication techniques. A more careful examination of quercetin (peak 12) also offers a good insight into the process of ligand fishing. The crude extract (Fig. 7) HRMS showed the co-elution of quercetin and another metabolite at the retention time of quercetin (peak 12). However, after examination of the elution fraction (S5), which represents only the retained compounds, only quercetin (peak 12) was observed by HRMS, while the co-eluting metabolite was absent. The HPLC-HRMS-SPE-NMR analysis revealed that the minor co-eluent metabolite in peak 12 was 4-hydroxy-3-methoxy-5-pentylphenyl-O-α-D-glucopyranoside. This study identified four α-glucosidase inhibitory ligands and demonstrated the selectivity of the method.
Fig. 6.

Overlaid base peak chromatograms acquired of different solutions (S0–S7) obtained from AGN-TCMB-based ligand fishing from an ethyl acetate extract of E. catharinae. Reproduced with permission from Fig. 5, S. G. Wubshet et al., J. Nat. Prod., 2015, 78, 2657–2665.
Fig. 7.
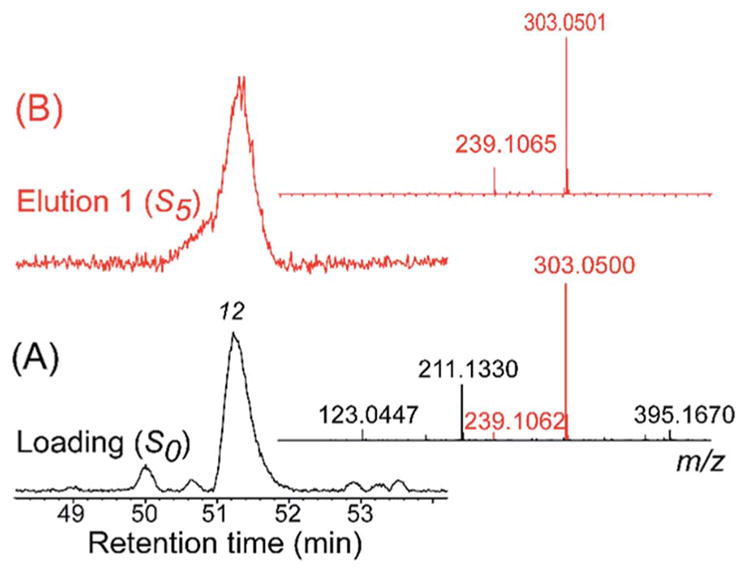
Expanded (50–54 min) base peak chromatogram of crude ethyl acetate extract of E. catharinae loaded on the magnetic beads for ligand fishing (A, black) and the eluents from the fishing experiment (B, red). Inserted to the right are the corresponding MS spectra of peak 12 from the two samples. Reproduced with permission from Fig. 6, S. G. Wubshet et al., J. Nat. Prod., 2015, 78, 2657–2665.
As typical ligand fishing experiments are followed by preparative-scale isolation of the ligands or analytical-scale LC-MS based identification, the combination of ligand fishing with the HPLC-HRMS-SPE-NMR platform offers a significant advantage for the direct identification of the retained metabolite. These studies clearly demonstrate the advantage and versatility of protein coated magnetic beads for the identification of active components from plant extracts. The results of this study indicate that the protein coated magnetic beads are a viable option for ‘fishing’ out and identifying an active compound from a complex matrix. A limitation of this method, is that to date, it has been limited to cytosolic proteins. The transmembrane protein coated beads, result in retention of a large amount of non-specific ligands, due to the presence of the boundary lipids and make this a non-viable option.
4 Conclusions
While all these methods have been shown to be very effective, each one comes with its own advantages/disadvantages, in terms of complexity, time, solvent compatibility, equipment, labor-intensive, etc. Complex mixtures contain large numbers of structurally diverse compounds and can therefore result in false positives, false negatives, missed binders, in addition to the presence of allosteric modifiers or non-competitive inhibitors that may elicit a conformational change in the targeted protein and result in a change in the binding characteristics of the protein. While each method has its limitations, certain methods have been more successful at the isolation and identification of active components from complex matrices. Ligand fishing, for example, is a relatively new methodology and has already shown significant promise in the identification of active compounds from the plant matrices.68–74
Acknowledgments
This work was supported by funds from the NIA Intramural Research Program.
Biographies

Lukasz Ciesla obtained his PhD at the Medical University of Lublin in 2011. After defending his thesis he worked 18 months at the Department of Plant Biochemistry and Crop Quality, Institute of Soil Science and Plant Cultivation, Poland under the supervision of prof. W. Oleszek. From 2012–2014 he worked in Foundation for Polish Science project “Multidisciplinary development of drugs acting on selected neuronal receptors” in prof. K. Jozwiak team. He is a laureate of the program SKILLS-Mentoring, mentor: Prof. Christian Zidorn, University of Innsbruck, Austria. Currently he works as a visiting fellow at the National Institute on Aging, Baltimore, USA.

Ruin Moaddel is currently a Staff Scientist in the Laboratory of Clinical Investigation at the National Institute on Aging, Baltimore, MD. He obtained his PhD at Northeastern University in 1999 followed by postdoctoral studies at Georgetown University. His focus for the past few years has been on the use of cellular membrane affinity chromatography columns for the identification of active components in complex matrices, including tobacco smoke condensates and on the development and application of protein-coated magnetic beads for ligand fishing. Specifically, he has focused on the isolation and identification of biologically active secondary metabolites for SIRT6 protein.
References
- 1.Newman DJ, Cragg GM. J Nat Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clarkson C, Stærk D, Honoré Hansen S, Jaroszewski JW. Anal Chem. 2005;77:3547–3553. doi: 10.1021/ac050212k. [DOI] [PubMed] [Google Scholar]
- 3.Lambert M, Stærk D, Hansen SH, Sairafianpour M, Jaroszewski JW. J Nat Prod. 2005;68:1500–1509. doi: 10.1021/np0502037. [DOI] [PubMed] [Google Scholar]
- 4.Clarkson C, Stærk D, Hansen SH, Smith PJ, Jaroszewski JW. J Nat Prod. 2006;69:527–530. doi: 10.1021/np050504g. [DOI] [PubMed] [Google Scholar]
- 5.Wolfender JL, Queiroz EF, Hostettmann K. Expert Opin Drug Discovery. 2006;1:237–260. doi: 10.1517/17460441.1.3.237. [DOI] [PubMed] [Google Scholar]
- 6.Wolfender JL, Marti G, Thomas A, Bertrand S. J Chromatogr A. 2015;1382:136–164. doi: 10.1016/j.chroma.2014.10.091. [DOI] [PubMed] [Google Scholar]
- 7.Cakova V, Urbain A, Antheaume C, Rimlinger N, Wehrung P, Bonté F, Lobstein A. Phytochem Anal. 2015;26:34–39. doi: 10.1002/pca.2533. [DOI] [PubMed] [Google Scholar]
- 8.Gaudencio SP, Pereira F. Nat Prod Rep. 2015;32:779–810. doi: 10.1039/c4np00134f. [DOI] [PubMed] [Google Scholar]
- 9.Wolfender JC. In: High Performance Liquid Chromatography in Phytochemical Analysis. Hajnos MW, Sherma J, editors. CRC Press; 2010. pp. 287–330. [Google Scholar]
- 10.Sumner LW, Lei Z, Nikolau BJ, Saito K. Nat Prod Rep. 2015;32:212–229. doi: 10.1039/c4np00072b. [DOI] [PubMed] [Google Scholar]
- 11.Liu Y, Staerk D, Nielsen MN, Nyberg N, Jäger AK. Phytochemistry. 2015;119:62–69. doi: 10.1016/j.phytochem.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 12.Kongstad KT, Wubshet SG, Kjellerup L, Winther AM, Staerk D. Fitoterapia. 2015;105:102–106. doi: 10.1016/j.fitote.2015.06.013. [DOI] [PubMed] [Google Scholar]
- 13.Tahtah Y, Kongstad KT, Wubshet SG, Nyberg NT, Jønsson LH, Jäger AK, Qinglei S, Staerk D. J Chromatogr A. 2015;1408:125–132. doi: 10.1016/j.chroma.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 14.van Breemen RB, Huang CR, Nikolic D, Woodbury CP, Zhao YZ, Venton DL. Anal Chem. 1997;69:2159–2164. doi: 10.1021/ac970132j. [DOI] [PubMed] [Google Scholar]
- 15.van Breemen RB, Tao Y, Li W. Fitoterapia. 2011;82:38–43. doi: 10.1016/j.fitote.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu J, Burdette JE, Xu H, Gu C, van Breemen RB, Bhat KPL, Booth N, Constantinou AI, Pezzuto JM, Fong HHS, Farnsworth NR, Bolton JL, Agric J. Food Chem. 2001;49:2472–2479. doi: 10.1021/jf0014157. [DOI] [PubMed] [Google Scholar]
- 17.Yang Z, Zhang Y, Sun L, Wang Y, Gao X, Cheng Y. Anal Chim Acta. 2012;719:87–95. doi: 10.1016/j.aca.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 18.Li H, Song F, Xing J, Tsao R, Liu Z, Liu S. J Am Soc Mass Spectrom. 2009;20:1496–1503. doi: 10.1016/j.jasms.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 19.Johnson BM, Nikolic D, van Breemen RB. Mass Spectrom Rev. 2002;21:76–86. doi: 10.1002/mas.10020. [DOI] [PubMed] [Google Scholar]
- 20.Mulabagal V, Calderon AI. Anal Chem. 2010;82:3616–3621. doi: 10.1021/ac902849g. [DOI] [PubMed] [Google Scholar]
- 21.Choi Y, Jermihov K, Nam SJ, Sturdy M, Maloney K, Qiu X, Chadwick LR, Main M, Chen SN, Mesecar AD, Farnsworth NR, Pauli GF, Fenical W, Pezzuto JM, van Breemen RR. Anal Chem. 2011;83:1048–1052. doi: 10.1021/ac1028424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao H, Zhou S, Zhang M, Feng J, Wang S, Wang D, Geng Y, Wang X. J Pharm Biomed Anal. 2016;120:235–240. doi: 10.1016/j.jpba.2015.12.025. [DOI] [PubMed] [Google Scholar]
- 23.Song HP, Zhang H, Fu Y, Mo HY, Zhang M, Chen J, Li P. J Chromatogr B: Anal Technol Biomed Life Sci. 2014;961:56–61. doi: 10.1016/j.jchromb.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 24.Chen X, Li LX, Chen S, Xu YT, Xia Q, Guo Y, Liu X, Tang YT, Zhang TJ, Chen Y, Yang C, Shui WQ. Anal Chem. 2013;85:7957–7965. doi: 10.1021/ac401732d. [DOI] [PubMed] [Google Scholar]
- 25.Wang Z, Hwang SH, Huang B, Lim SS. J Chromatogr B: Anal Technol Biomed Life Sci. 2015;1002:319–328. doi: 10.1016/j.jchromb.2015.08.030. [DOI] [PubMed] [Google Scholar]
- 26.Yang XX, Xu F, Wang D, Yang ZW, Tan HR, Shang MY, Wang X, Cai SQ. J Chromatogr A. 2015;1413:33–46. doi: 10.1016/j.chroma.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 27.Li J, Khan I. Acta Hortic. 2006;720:157–169. [Google Scholar]
- 28.Wang JX, Xiao XH, Li GK. J Chromatogr A. 2008;1198–1199:45–53. doi: 10.1016/j.chroma.2008.05.045. [DOI] [PubMed] [Google Scholar]
- 29.Jiang XS, Dai J, Sheng QH, Zhang L, Xia QC, Wu JR, Zeng R. Mol Cell Proteomics. 2005;4:12–34. doi: 10.1074/mcp.M400079-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.Potterat O, Hamburger M. Nat Prod Rep. 2013;30:546–564. doi: 10.1039/c3np20094a. [DOI] [PubMed] [Google Scholar]
- 31.Ingkaninan K, de Best CM, van der Heijden R, Hofte AJP, Karabatak B, Irth H, Tjaden UR, van der Greef J, Verpoorte R. J Chromatogr A. 2000;872:61–73. doi: 10.1016/s0021-9673(99)01292-3. [DOI] [PubMed] [Google Scholar]
- 32.Schobel U, Frenay M, Van Elswijk DA, McAndrews JM, Long KR, Olson LM, Bobzin SC, Irth H, Biomol J. Screening. 2001;6:291–303. doi: 10.1177/108705710100600503. [DOI] [PubMed] [Google Scholar]
- 33.van Elswijk DA, Diefenbach O, van der Berg S, Irth H, Tjaden UR, van der Greef J. J Chromatogr A. 2003;1020:45–58. doi: 10.1016/j.chroma.2003.08.055. [DOI] [PubMed] [Google Scholar]
- 34.Kool J, Eggink M, van Rossum H, van Liempd SM, van Elswijk DA, Irth H, Commandeur JNM, Meerman JHN, Vermeulen NPE. J Biomol Screening. 2007;12:396–405. doi: 10.1177/1087057107299527. [DOI] [PubMed] [Google Scholar]
- 35.Lin P, Zhao S, Lu X, Ye F, Wang H. J Sep Sci. 2013;36:2538–2543. doi: 10.1002/jssc.201300315. [DOI] [PubMed] [Google Scholar]
- 36.Zhang A, Ye F, Lu J, Zhao S. Food Chem. 2013;141:1854–1859. doi: 10.1016/j.foodchem.2013.04.100. [DOI] [PubMed] [Google Scholar]
- 37.Moaddel R, Wainer IW. Nat Protoc. 2009;4:197–205. doi: 10.1038/nprot.2008.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Q, Lundahl P. Biochemistry. 1995;34:7289–7294. doi: 10.1021/bi00022a001. [DOI] [PubMed] [Google Scholar]
- 39.Brekkan E, Lundqvist A, Lundahl P. Biochemistry. 1996;35:12141–12145. doi: 10.1021/bi9603231. [DOI] [PubMed] [Google Scholar]
- 40.Pidgeon C, Venkataram UV. Anal Biochem. 1989;176:36–47. doi: 10.1016/0003-2697(89)90269-8. [DOI] [PubMed] [Google Scholar]
- 41.Moaddel R, Cloix JF, Ertem G, Wainer IW. Pharm Res. 2002;19:104–107. doi: 10.1023/a:1013619802766. [DOI] [PubMed] [Google Scholar]
- 42.Moaddel R, Bullock P, Wainer IW. J Chromatogr B Anal Technol Biomed Life Sci. 2004;799:255–263. doi: 10.1016/j.jchromb.2003.10.054. [DOI] [PubMed] [Google Scholar]
- 43.Moaddel R, Rosenberg A, Spelman K, Frazier J, Frazier C, Nocerino S, Brizzi A, Mugnaini C, Wainer IW. Anal Biochem. 2011;412:85–91. doi: 10.1016/j.ab.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moaddel R, Calleri E, Massolini G, Frazier C, Wainer IW. Anal Biochem. 2007;364:216–218. doi: 10.1016/j.ab.2007.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moaddel R, Jozwiak K, Whittington KC, Wainer IW. Anal Chem. 2005;77:895–901. doi: 10.1021/ac048826x. [DOI] [PubMed] [Google Scholar]
- 46.Moaddel R, Jozwiak K, Wainer IW. Med Res Rev. 2007;27:723–753. doi: 10.1002/med.20091. [DOI] [PubMed] [Google Scholar]
- 47.Moaddel R, Oliveira RV, Kimura T, Hyppolite P, Juhaszova M, Bernier M, Wainer IW. Anal Chem. 2008;80:48–54. doi: 10.1021/ac701943b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moaddel R, Hamid R, Patel S, Wainer IW, Bullock P. Anal Chim Acta. 2006;578:25–30. doi: 10.1016/j.aca.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 49.Habicht KL, Singh N, Khadeer MA, Shimmo R, Wainer IW, Moaddel R. J Chromatogr A. 2014;1339:80–85. doi: 10.1016/j.chroma.2014.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kimura T, Perry J, Anzai N, Pritchard J, Moaddel R. J Chromatogr B: Anal Technol Biomed Life Sci. 2007;859:267–271. doi: 10.1016/j.jchromb.2007.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moaddel R, Yamaguchi R, Ho P, Patel S, Wainer IW. J Chromatogr B: Anal Technol Biomed Life Sci. 2005;818:263–268. doi: 10.1016/j.jchromb.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 52.Moaddel R, Bighi F, Yamaguchi R, Patel S, Ravichandran S, Wainer IW. Anal Biochem. 2010;401:148–153. doi: 10.1016/j.ab.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang S, Sun M, Zhang Y, Du H, He L. J Chromatogr A. 2010;1217:5246–5252. doi: 10.1016/j.chroma.2010.06.037. [DOI] [PubMed] [Google Scholar]
- 54.He L, Wang S, Yang G, Zhang Y, Wang C, Yuan B, Hou X. Drug Discoveries Ther. 2007;1:104–107. [PubMed] [Google Scholar]
- 55.Hou J, Yuan BX, Lang-Chong HE, Yang GD, Mi M. Chin J Pharmacol Toxicol. 2003;17:70–73. [Google Scholar]
- 56.Zhang T, Han S, Huang J, Wang SJ. J Chromatogr B Anal Technol Biomed Life Sci. 2013;912:85–92. doi: 10.1016/j.jchromb.2012.10.029. [DOI] [PubMed] [Google Scholar]
- 57.Zhao HR, Yang GD, He LC, Yang YJ. Chin Pharm J. 2000;35:13–15. (in Chinese with English abstract) [Google Scholar]
- 58.Liang MJ, He LC. Chin J Anal Chem. 2004;32:83–86. [Google Scholar]
- 59.Cooper MA. J Mol Recognit. 2004;17:286–315. doi: 10.1002/jmr.675. [DOI] [PubMed] [Google Scholar]
- 60.Singh N, Sarangan R, Norton DD, Fugmann SD, Moaddel R. Anal Biochem. 2013;436:78–83. doi: 10.1016/j.ab.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ravichandran S, Singh N, Donnelly D, Migliore M, Johnson P, Fishwick C, Luke BT, Martin B, Maudsley S, Fugmann SD, Moaddel R. J Mol Graphics Modell. 2014;49:38–46. doi: 10.1016/j.jmgm.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ng ESM, Chan NWC, Lewis DF, Hindsgaul O, Schriemer DC. Nat Protoc. 2007;2:1907–1917. doi: 10.1038/nprot.2007.262. [DOI] [PubMed] [Google Scholar]
- 63.Dossou KSS, Devkota KP, Morton C, Egan JM, Lu G, Beutler JA, Moaddel R. J Nat Prod. 2013;76:2060–2064. doi: 10.1021/np400478c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spelman K, Wetschler MH, Cech NB. J Pharm Biomed Anal. 2009;49:1141–1149. doi: 10.1016/j.jpba.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maciuk A, Moaddel R, Haginaka J, Wainer IW. J Pharm Biomed Anal. 2008;48:238–246. doi: 10.1016/j.jpba.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ciesla L, Okine M, Rosenberg A, Dossou KSS, Toll L, Wainer IW, Moaddel R. J Chromatogr A. 2016;1431:138–144. doi: 10.1016/j.chroma.2015.12.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moaddel R, Marszall M, Bigi F, Yang Q, Duan X, Wainer IW. Anal Chem. 2007;79:5414–5417. doi: 10.1021/ac070268+. [DOI] [PubMed] [Google Scholar]
- 68.Marszall MP, Moaddel R, Kole S, Gandhari M, Bernier M, Wainer IW. Anal Chem. 2008;80:7571–7575. doi: 10.1021/ac801153h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yasudo M, Wilson DR, Fugman S, Moaddel R. Anal Chem. 2011;83:7400–7407. doi: 10.1021/ac201403y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vanzolini KL, Jiang Z, Zhang X, Vieira LCC, Gonçalvez Corrêa A, Cardoso CL, Cass QB, Moaddel R. Talanta. 2013;116:647–652. doi: 10.1016/j.talanta.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Singh N, Spelman K, Ravichandran S, Fugmann S, Moaddel R. J Chromatogr B Anal Technol Biomed Life Sci. 2014;968:105–111. doi: 10.1016/j.jchromb.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lackmann M, Bucci T, Mann RJ, Kravets LA, Viney E, Smith F, Moritz RL, Carter W, Simpson RJ, Nicola NA, Mackwell K, Nice EC, Wilks AF, Boyd AW. Proc Natl Acad Sci U S A. 1996;93:2523–2527. doi: 10.1073/pnas.93.6.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jonker N, Kretschmer A, Kool J, Fernandez A, Kloos D, Krabbe JG, Lingeman H, Irth H. Anal Chem. 2009;81:4263–4270. doi: 10.1021/ac9000755. [DOI] [PubMed] [Google Scholar]
- 74.Wubshet SG, Brighente IM, Moaddel R, Staerk D. J Nat Prod. 2015;78:2657–2665. doi: 10.1021/acs.jnatprod.5b00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qing LS, Tang N, Xue Y, Liang J, Liu YM, Liao X. Anal Methods. 2012;4:1612–1615. doi: 10.1039/C2AY25320H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhu YT, Ren XY, Yuan L, Liu YM, Liang J, Liao X. Food Chem. 2015;173:521–526. doi: 10.1016/j.foodchem.2014.10.087. [DOI] [PubMed] [Google Scholar]
- 77.Junior CV, Bolzani VDS, Furlan M, Fraga CAM, Barreiro EJ. Quim Nova. 2004;27:655–660. [Google Scholar]
- 78.Yang Y, Xi-Qiang L, Chun-Ping T, Sheng Y. Chin J Nat Med. 2012;10:1–12. [Google Scholar]
- 79.Zhan ZJ, Yu Q, Wang ZL, Shan WG. Bioorg Med Chem Lett. 2010;20:6185–6187. doi: 10.1016/j.bmcl.2010.08.123. [DOI] [PubMed] [Google Scholar]
- 80.Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack D, Guimaraes A, Marinelli B, Widstrom JD, Nir T, Clish CB, Vaitheesvaran B, Illiopoulos O, Kurland L, Dor Y, Weissleder R, Shirihai OS, Ellisen LW, Espinosa JM, Mostoslavsky R. Cell. 2010;40:280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dominy JE, Jr, Lee Y, Jedrychowski MP, Chim H, Jurczak MJ, Camporez JP, Ruan HB, Feldman J, Pierce K, Mostoslavsky R, Denu JM, Clish CB, Yang X, Shulman GI, Gygi SP, Puigserver P. Mol Cell. 2012;48:900–913. doi: 10.1016/j.molcel.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mullaicharam AR, Deori G, Uma Maheswari R. Res J Pharm, Biol Chem Sci. 2013;4:1304–1313. [Google Scholar]
- 83.Lee WJ, Chen YR, Tseng TH. Oncol Rep. 2011;25:583–591. doi: 10.3892/or.2010.1097. [DOI] [PubMed] [Google Scholar]
- 84.Yadav M, Lavania A, Tomar R, Prasad GB, Jain S, Yadav H. Appl Biochem Biotechnol. 2010;160:2388–2400. doi: 10.1007/s12010-009-8799-1. [DOI] [PubMed] [Google Scholar]
- 85.Abdel-Barry JA, Abdel-Hassan IA, Jawad AM, Al-Hakiem MH. East Mediterr Health J. 2000;6:83–88. [PubMed] [Google Scholar]
- 86.Kassaian N, Azadbakht L, Forghani B, Amini M. Int J Vitam Nutr Res. 2009;79:34–39. doi: 10.1024/0300-9831.79.1.34. [DOI] [PubMed] [Google Scholar]
- 87.Bordia A, Verma SK, Srivastava KC. Prostaglandins, Leukotrienes Essent Fatty Acids. 1997;56:379–384. doi: 10.1016/s0952-3278(97)90587-1. [DOI] [PubMed] [Google Scholar]
- 88.Rahnasto-Rilla M, Kokkola T, Jarho E, Lahtela-Kakkonen M, Moaddel R. ChemBiochem. 2016;17:77–78. doi: 10.1002/cbic.201500482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mooradian A, Thurman J. Drugs. 1999;57:19–29. doi: 10.2165/00003495-199957010-00003. [DOI] [PubMed] [Google Scholar]
- 90.Bokesch HR, Wamiru A, Le Grice SFJ, Beutler JA, McKee TC, McMahon JB. J Nat Prod. 2008;71:1634–1636. doi: 10.1021/np8002518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Grangeiro MS, Calheiros-Lima AP, Martins MF, Arruda LF, Garcez-do-Carmo L, Santos WC. J Ethnopharmacol. 2006;108:26–30. doi: 10.1016/j.jep.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 92.Omar R, Li L, Yuan T, Seeram NP. J Nat Prod. 2012;75:1505–1509. doi: 10.1021/np300417q. [DOI] [PubMed] [Google Scholar]
- 93.Staerk D, Kesting JR, Sairafianpour M, Witt M, Asili J, Emami SA, Jaroszewski JW. Phytochemicals. 2009;70:1055–1061. doi: 10.1016/j.phytochem.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 94.Schmidt B, Jaroszewski JW, Bro R, Witt M, Stærk D. Anal Chem. 2008;80:1978–1987. doi: 10.1021/ac702064p. [DOI] [PubMed] [Google Scholar]