

Abstract
The final step in the biosynthesis of the sesquiterpenoid antibiotic pentalenolactone (1) is the highly unusual cytochrome P450-catalyzed, oxidative rearrangement of pentalenolactone F (2), involving the transient generation and rearrangement of a neopentyl cation. In Streptomyces arenae this reaction is catalyzed by CYP161C2 (PntM), with highly conserved orthologues being present in at least 10 other Actinomycetes. Crystal structures of substrate-free PntM, as well as PntM with bound substrate 2, product 1, and substrate analogue 6,7-dihydropentalenolactone F (7) revealed interactions of bound ligand with three residues, F232, M77, and M81 that are unique to PntM and its orthologues and absent from essentially all other P450s. Site-directed mutagenesis, ligand-binding measurements, steady-state kinetics, and reaction product profiles established there is no special stabilization of reactive cationic intermediates by these side chains. Reduced substrate analogue 7 did not undergo either oxidative rearrangement or simple hydroxylation, suggesting that the C1 carbocation is not anchimerically stabilized by the 6,7-double bond of 2. The crystal structures also revealed plausible proton relay networks likely involved in the generation of the key characteristic P450 oxidizing species, Compound I, and in mediating stereospecific deprotonation of H-3re of the substrate. We conclude that the unusual carbocation intermediate results from outer shell electron transfer from the transiently generated C1 radical to the tightly paired heme-•Fe3+-OH radical species. The oxidative electron transfer is kinetically dominant as a result of the unusually strong steric barrier to oxygen rebound to the neopentyl center C-1si which is flanked on each neighboring carbon by syn-axial substituents.
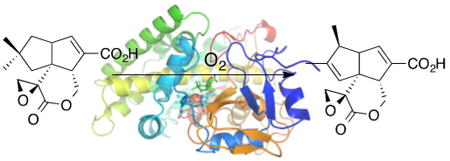
Graphical abstract
Introduction
Cytochrome P450 enzymes are responsible for a remarkable range of controlled oxidations at sp3 carbon centers that have little or no parallel in laboratory organic chemistry, including regiospecific and stereospecific insertion of oxygen into unactivated C–H bonds and oxidative cleavage of C–C bonds.1 Such reactions are catalyzed by both catabolic P450s, whose primary biological function is degradation of a broad range of foreign organic compounds2a or mobilization of environmental organic chemicals as sources of energy and carbon,2b as well as by metabolic P450s that catalyze substrate-specific reactions as part of multistep biosynthetic pathways.3
We recently reported the cloning and biochemical characterization of a pair of orthologous cytochrome P450s, designated PntM (CYP161C2) and PenM (CYP161C3), that catalyze the final step in the biosynthesis of the sesquiterpenoid antibiotic pentalenolactone (1) in Streptomyces arenae and S. exfoliatus UC5319, respectively (Scheme 1).4 The conversion of pentalenolactone F (2) to pentalenolactone (1) involves an oxidative rearrangement that is completely unprecedented in the already vast repertoire of P450-catalyzed transformations. Isotopic labeling studies have established that the net 2-electron oxidation involves the stereospecific removal of H-1si of 2, syn-1,2-migration of the 2si methyl group (C-12), and antarafacial loss of H-3re to generate the characteristic (1S)-1,2-dimethyl-cyclopent-2-ene moiety of pentalenolactone (1).5
Scheme 1.

Oxidative rearrangement of pentalenolactone F (2) to pentalenolactone (1)
Cytochrome P450-catalyzed reactions normally involve the generation of caged substrate radicals, resulting from abstraction of a substrate hydrogen atom by the highly reactive ferryl oxygen of porphyrin+•∼Fe4+=O (Compound I), followed by rapid oxygen rebound that transfers the hydroxyl group to the original reacting carbon atom (Scheme 2).6 The transition state for the abstraction of the hydrogen atom of the substrate by the reactive oxygen atom of Compound I involves a linear geometry of the reacting O--H–C array, while oxygen rebound requires a rapid rotation of the initially-formed, Fe-bound OH group so as to allow approach and recombination with the paired carbon radical.7 P450-catalyzed rearrangements are rare, with prior examples limited to the formation of isomeric hydroxylation products derived from either 1) an allylic radical8 or 2) a cyclopropylcarbinyl radical whose rapid, thermodynamically-driven ring-opening generates the corresponding 3-butenyl radical.9 Such ring opening reactions have in fact been exploited as radical clocks that have established the rate constants for oxygen rebound within the initially-generated [Fe3+-OH]/carbon radical pairs to be ∼1010–1011 s-1.9 Even in such artificially engineered systems, however, the proportion of ring-opened rearrangement products is typically 1-15% or less. Functionalized cyclopropane derivatives bearing substituents that favor, and which are therefore diagnostic of, carbocation-based rearrangements have also indicated that less than 2% of the overall oxidation product mixture results from an alternative ring opening of a cyclopropylcarbinyl cation.10-12 Such carbocations are most likely formed by a stepwise process involving oxidation of the initially formed carbon radical by rapid electron transfer to the strongly oxidizing [Fe3+-OH] species, in competition with the usually favored oxygen rebound reaction.
Scheme 2.

P450-catalyzed hydrogen abstraction, oxygen rebound, and competing electron transfer.
Neopentyl radicals, such as that which would be formed by conventional P450-catalyzed oxidation at C-1 of 2, do not undergo skeletal rearrangement.13 By contrast, neopentyl cations, typically generated by SN1 solvolysis reactions, undergo extremely rapid, coupled Wagner-Meerwein rearrangement such that they can neither be trapped nor directly observed.14 To explain the PntM-catalyzed oxidative rearrangement of 2, we therefore have proposed a mechanism involving P450-mediated generation of the C-1 neopentyl cation intermediate 6 (Scheme 1).4 Circumstantial support for such a carbocationic mechanism comes from the isolation from large-scale fermentations of isomeric rearrangement products, pentalenolactones A (3), B (4), and P (5), that presumably are formed by alternative deprotonation of the intermediates generated by the rearrangement of the initially formed neopentyl cation 6.15
The apparent ability of PntM to catalyze an exclusive carbocation-based oxidative rearrangement clearly presents an intriguing mechanistic puzzle. To unravel this conundrum, we have now determined the 2.00 – 2.28 Å resolution crystal structures of wild-type PntM in both substrate-free form and individually complexed with substrate 2, product 1, and the reduced substrate analogue 6,7-dihydropentalenolactone F (7). We have also determined the 2.05 Å structures of four active site PntM mutants with bound substrate 2. We have also determined the steady-state kinetic parameters, substrate binding constants, and reaction product profiles for the wild-type and mutant proteins with native substrate 2, as well as for wild-type PntM incubated with reduced substrate analogue 7. The combined results exclude any special electronic features in either the substrate pentalenolactone F (2) or the PntM protein itself that might favor a carbocation over a radical mechanism, and also rule out any unusual mode of substrate–protein binding. We conclude instead that the conversion of 2 to the neopentyl carbocation intermediate 6 is a consequence of tightly constrained, conventional binding of pentalenolactone F, combined with the exceptional degree of steric hindrance to attack at C-1 of 2 that prevents access of the transiently-generated heme-bound hydroxyl group to the si face of the C-1 radical, thus favoring competing electron transfer to the paired, strongly oxidizing [Fe3+OH] radical.
Results
Incubation of PntM with 6,7-dihydropentalenolactone F (7)
We initially speculated that the carboxylate-conjugated 6,7-double bond of the natural substrate 2 might provide anchimeric stabilization of a C-1 carbocation in the derived intermediate 6 (Scheme 3a). To probe this idea, we tested whether the reduced analogue 6,7-dihydropentalenolactone F (7) would undergo simple hydroxylation at C1 when incubated with PntM, in competition with the usual oxidative rearrangement. The requisite sample of 7 was readily prepared by hydrogenation of pentalenolactone F methyl ester (2-Me) followed by mild basic hydrolysis (Scheme 3b). GC-MS analysis established that the reduction was stereospecific, generating a 12:1 mixture of diastereomers. The major diastereomer was subsequently assigned as (6R)-7, based on the best fit to the electron density in the 2.03 Å crystal structure of PntM complexed with 7, discussed below (Figure 4). Binding of 7 to PntM resulted in a normal Type I P450 binding spectrum,1,4 with the characteristic hypsochromic UV shift from the low-spin, ligand-free 420 nm absorption to the high-spin 390 nm substrate-bound state (Figure S4). Analysis of the concentration dependence of the UV difference spectra gave a Kd of 170±17 μM for 7, nearly 25 times weaker than binding of the natural substrate 2 (Kd 7.2±0.8 μM) (Table 1). The analogue 7 was recovered completely unchanged upon extended aerobic incubation with PntM, NADPH, and the ferredoxin/NADPH:ferredoxin oxidoreductase, as established by capillary GC-MS analysis of the organic extract, with no trace of the analogous products of either oxidative rearrangement, 6,7-dihydropentalenolactone (8), or direct hydroxylation, (9) (Scheme 3b).
Scheme 3.

6,7-Dihydropentalenolactone F (7)
Figure 4.
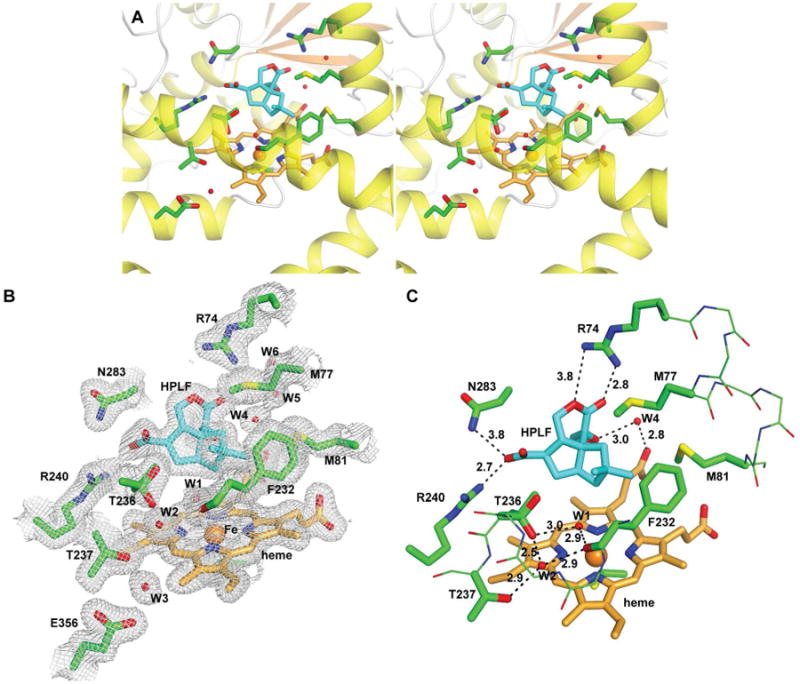
Structure of PntM with bound substrate analogue 7. A, stereo diagram showing heme/analogue 7-binding site. 7 is shown as sticks with carbon atoms in cyan. B, final 2mFo – DFc electron density map of the active site region (contoured at 1 σ). C, close-up view showing relevant water molecules and H-bonds positioning 7 in the active site.
Table 1.
Binding of pentalenolactone F (2) and 6,7-dihydropentalenolactone F (7) to wild-type PntM and mutants.
| Protein | KD (μM) | Protein | KD (μM) |
|---|---|---|---|
|
| |||
| PntM - wt | 7.2±0.8 (2) | PntM - wt | 170±17 (7) |
| PntM-F232A | 270±85 (2) | PntM-M81A | 340±90 (2) |
| PntM-F232G | 225±45 (2) | PntM-M81C | 43±3 (2) |
| PntM-F232L | 160±12 (2) | PntM-M77S | 115±8 (2) |
| PntM-M81C-BME | 31±1 (2) | ||
PntM Structures
The structure of substrate-free PntM was solved to a resolution of 2.00 Å by molecular replacement, using as a template the polyene macrolide epoxidase PimD (PDB ID 2X9P). By soaking individual PntM crystals in crystallization buffer containing pentalenolactone F (2), pentalenolactone (1), or 6,7-dihydropentalenolactone F (7), we were also able to obtain structures of PntM complexed with substrate (2) (2.03 Å), product (1) (2.28 Å), and the reduced substrate analogue (7) (2.03 Å) (Table S1). The topology of PntM corresponds to the classic P450 fold, characterized by the long I-Helix from D219 to R250 that spans the distal face of the heme cofactor. (Figure 1) The bent equatorial D-Helix encompassing residues T88 – A129 wraps around nearly half the circumference of the protein just below the latitude of the heme plane, while the long B-C loop (D55-S71) and C-Helix (A72-M81) as well as the conserved F-Helix, (L157 – F171, G-Helix (N176 – V199), and short F-G loop (D172 – D175) control access by substrate to the distal face of the heme cofactor. The hexacoordinate Fe3+ of the heme cofactor is liganded on the proximal face by the thiolate of the conserved C347, while the axial water ligand (W1) on the distal face is slightly displaced by ∼0.9 Å from the vertical axis by the hydroxyl group of a bound bicine molecule, which originates from the crystallization buffer (Figure 1C).16 Water W1 is held in place by a network of hydrogen bonds involving the side chain hydroxyl of T236, the backbone carbonyl oxygen of F232 and a second molecule of water, W2, that is wedged into the expanded kink of the I-helix.17 The carboxyl group of the bicine is anchored by a pair of H-bonds to NH2 of R240 (2.8 Å) and to water molecule W25 (2.8 Å) which is also H-bonded to NH1 of R240 (2.8 Å), one helical turn of the I-Helix downstream of T236. The bicine is itself buttressed by van der Waals interactions with the side chains of F232, M77 and M81, the latter two residues being located at the C-terminus of the C-Helix.
Figure 1.
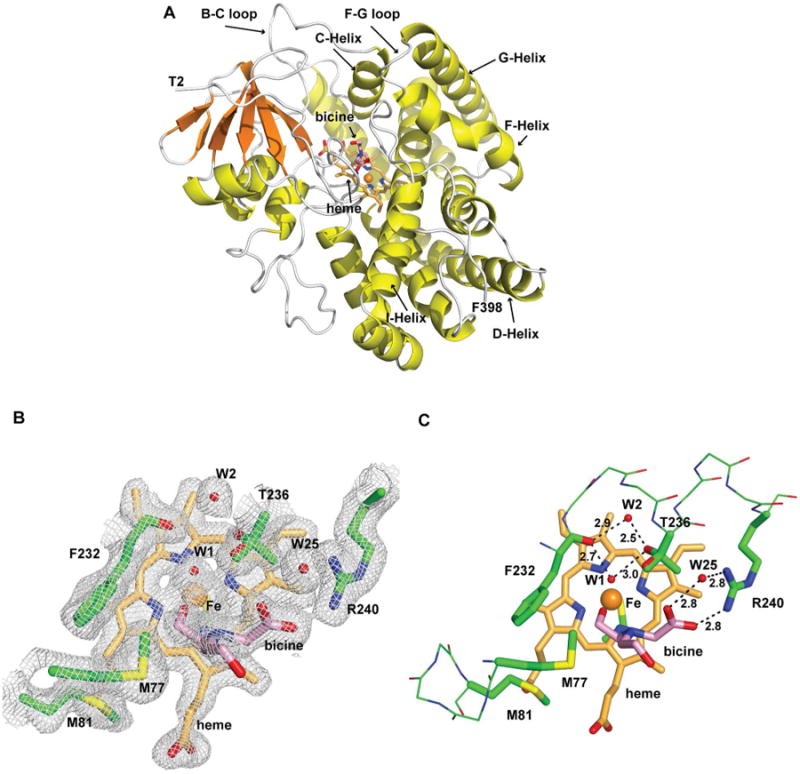
Structure of substrate-free PntM. A, Schematic representation of the structure of PntM with bound bicine. Bicine is shown as sticks with carbon atoms in pink, the heme cofactor is shown as sticks with carbon atoms in bright orange and the heme-bound Fe3+ is shown as an orange sphere. T2 is the N-terminal amino acid and F398 is the C-terminal amino. B, Final 2mFo – DFc electron density map of the active site region (contoured at 1 σ). C, close-up view, including relevant water molecules (small red spheres) and H-bonds (distances in Å) that position bicine in the active site.
The topology of PntM undergoes minimal changes upon binding of either substrate 2, product 1, or substrate analogue 7 (Figures 2, 3, 4, and S10), with the calculated rmsd of the Cα carbons ranging from 0.13 – 0.22 Å compared to PntM with bound bicine. Significantly, the binding of the substrate, pentalenolactone F (2), precisely orients the scissile C1–H-1si bond so that it is pointed directly towards the site that would be occupied by the strongly oxidizing ferryl oxygen of porphyrin+•/Fe4+=O (Compound I) (Figure 2). The bound substrate 2 is held tightly in place by an interlocking network of ionic, hydrogen bond, and van der Waals interactions. The C13 carboxylate of 2 is positioned by a pair of bifurcating H-bonds to the NH2 of R240 (2.7 Å) and to the side chain amide NH2 of N283 (3.3 Å), while the lactonic carbonyl and ester oxygen atoms of 2 are hydrogen-bonded to both NH1 and NH2 of R74 at distances of 2.9 Å and 4.0 Å, respectively. The C3 methylene of substrate 2 is sandwiched between the sidechains of M77 and M81, while the C14 and C15 geminal methyl groups of 2 abut the aromatic face of F232, which itself contacts the M81 side chain.
Figure 2.
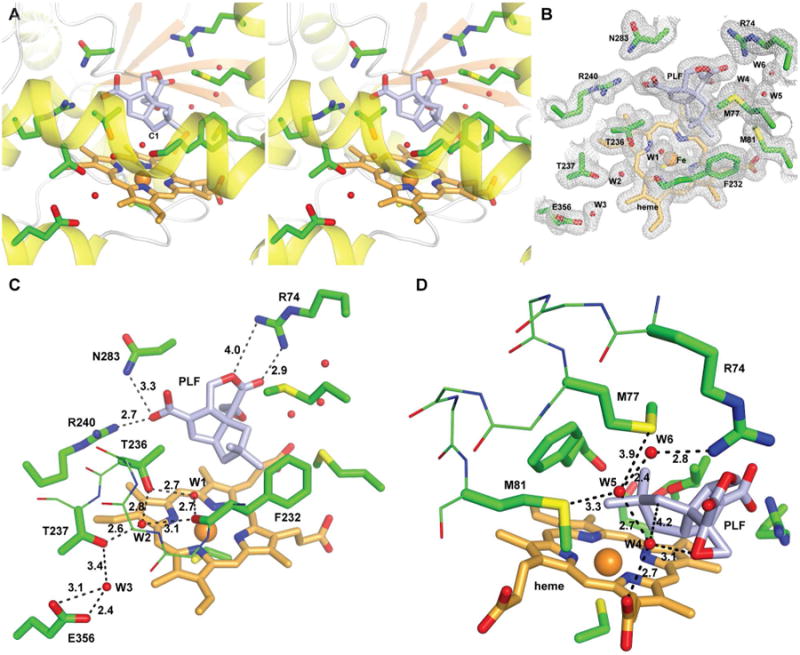
Structure of PntM with bound substrate 2. A, stereo diagram showing heme/substrate 2-binding site. 2 is shown as sticks with carbon atoms in light blue. B, final 2mFo – DFc electron density map of the active site region (contoured at 1 σ). C, close-up view showing water relay 1 (W1, W2, and W3) and H-bonds positioning substrate 2. D, close-up view showing water relay 2 (W4, W5, W6).
Figure 3.
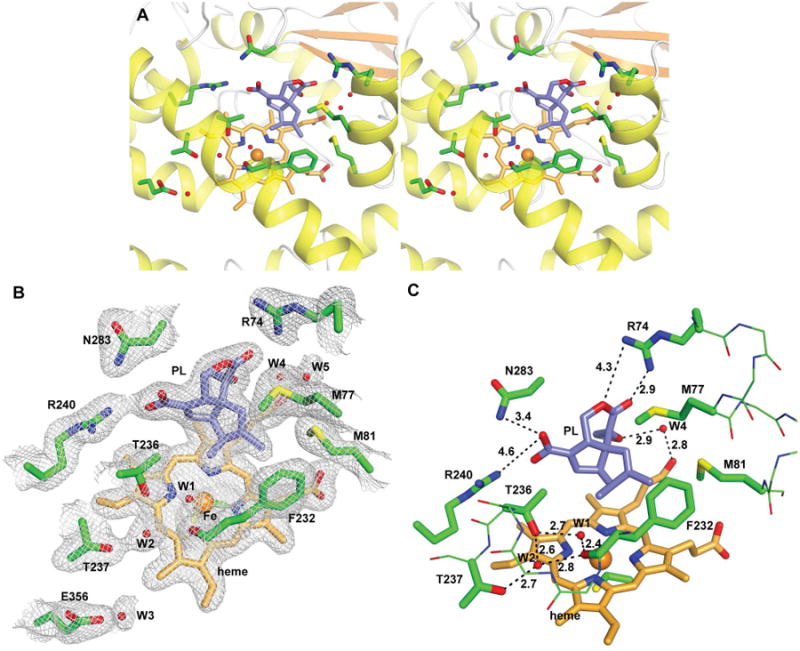
Structure of PntM with bound product 1. A, stereo diagram showing heme/product 1-binding site. 1 is shown as sticks with carbon atoms in white/blue. B, final 2mFo – DFc electron density map of the active site region (contoured at 1 σ). C, close-up view showing relevant water molecules and H-bonds positioning product 1 in the active site.
Amino acid F232 forms part of a highly unusual F232GYET236 sequence at the I-Helix kink that replaces the nearly universally conserved A/Gn-1GnXXTn+3 motif characteristic of the vast majority of P450s. A BLAST search of the non-redundant protein databases using PntM as the query reveals that the 10 closest matches (sequence identity 74-89% over 398 aa; E value 0.0), each of which harbor the FGYET motif, are all likely PntM orthologs found at identical loci within known or probable pentalenolactone biosynthetic gene clusters (Figure S9). Each of these 10 orthologs also displays the same R74XXM77XALM81 substrate-binding motif.
By contrast, the next 88 best protein matches (sequence identity 39-49%) all contain the canonical A/Gn-1GnXXTn+3 I-Helix motif, while none retain the R74XXM77XALM81 sequence character istic of the PntM orthologs.
During the catalytic cycle of a prototypical P450, generation of the key oxidant requires delivery of two protons to the distal oxygen atom of the intermediate reduced ferrous-dioxygen (peroxyferric) complex, followed by release of a molecule of water and formation of Compound I. Since this oxygen atom is buried 12-15 Å from the surface of the P450 protein, it is not directly accessible to the external medium. Delivery of the requisite protons therefore requires a proton relay chain, with the hydroxyl group of the conserved I-Helix threonine frequently acting as the penultimate proton donor/acceptor, as in the prototypical monoxygenase P450cam.3,18 A presumptive proton relay network (Water relay 1) is readily apparent in the structure of substrate-bound PntM (Figure 2C). Thus water W1, laterally displaced from the axial sixth ligand position of the heme iron by the binding of pentalenolactone F (2), is H-bonded to both the hydroxyl group of T236 (2.7 Å) and the carbonyl oxygen of F232 (2.7 Å), while these two oxygen atoms are each in turn H-bonded (2.8 and 3.1 Å) to water W2, which resides in the expanded groove of the I-helix. W2 is further H-bonded to the side chain hydroxyl of T237 (2.6 Å), which is in turn H-bonded to water W3 (3.4 Å), held in place by H-bonds (2.4 and 3.1 Å) to the carboxylate oxygen atoms of E356. In the actual intermediate Fe2+-O2-pentalenolactone F complex, it is of course likely that water W1 is either further shifted by the oxygen atom of the bound O2, or displaced altogether, with the remainder of the proton relay network remaining essentially intact.19 Notably, the well-characterized proton relay networks in two other well-studied P450s, Pseudomonas putida cytochrome P450cam, which catalyzes the C5 exo-hydroxylation of camphor,18,20 and Saccharopolyspora erythraea P450eryF,3b,21 which catalyzes the C6-hydroxlation of 6-deoxyerythronolide B, the parent macrolide aglycone of erythromycin A, are each anchored by homologous glutamate residues, P450cam-E366 and P450eryF-E360, respectively, which occupy identical sites in the corresponding L-Helix of each protein.20,21 Indeed, this glutamate is nearly universally conserved in all P450s, being found in >90% of the top 100 BLAST matches to the PntM sequence. Although the conserved glutamates do not appear to have a continuous H-bonded link with bulk water at the protein surface, it has been proposed that the requisite proton delivery may involve concerted dynamic fluctuations of amino acid chains and bound waters.3b,22
The PntM-catalyzed oxidative rearrangement of pentalenolactone F (2) to pentalenolactone (1) is completed by deprotonation of the labile H-3re proton from the rearranged neopentyl cation (Scheme 1). From the crystal structure of PntM with bound substrate 2, the only suitable Brønsted base is the previously cited water W4 which is positioned 4.2 Å from C3 of 2, just off the in-line axis of the C3- H-3re bond (Figure 2D). Water W4, which is H-bonded to the epoxide oxygen of 2 (3.1 Å) is connected to the external medium by another well-organized proton relay system (water relay 2) also involving waters W5 and W6. The latter water, which is exposed to the external medium, is anchored by an H-bond (2.8 Å) to NH1 of the previously mentioned R74, while W5 may also be held in place by H-bonds to the sidechain sulfides of M77 and/or M81. Notably, R74, M77, and M81 are themselves located at successive turns of the C-Helix, and together play a major role in the binding and orientation of the substrate, as noted above. Indeed, it is conceivable that protonation of water W6 causes repulsion of the positively charged guanidino side chain of R74, thus displacing Helix C and facilitating release of the product pentalenolactone (1).
In the structure of PntM with bound product pentalenolactone (1) (Figure 3), the rmsd of the Cα carbons compared to substrate-bound PntM is 0.25 Å, while there is insignificant change in the positions of either M77, M81, or F232 (Figure S10D). The binding of 1 is unchanged from that of the substrate 2, other than the necessary alteration in the conformation and substitution pattern of C1-C3 due to the migration of the β-methyl group to C1 and the introduction of the 2,3-double bond. Water W1 remains H-bonded to both the side chain hydroxyl group of T236 (2.7 Å) and the backbone carbonyl oxygen of F232 (2.4 Å), while the latter two oxygen atoms are H-bonded (2.6 and 2.8 Å, respectively) to W2 in the I-Helical groove (Figure 3C). On the other face of bound 1, water W4 remains H-bonded to both the epoxide oxygen (3.0 Å) and the propionate carboxyl oxygen (2.8 Å).
When PntM is bound to 6,7-dihydropentalenolactone F (7), the relative positions of the side chains of F232, M77, M81 are unperturbed compared to PntM with the bound natural substrate 2, as are the positions of the three bound water molecules, W1, W2, and W4 (Figures 4 and S10F). As a consequence of the reduction of the 6,7-double bond, the C13-carboxylate of 7 is slightly twisted with respect to the carboxylate of 2, while still held in place by H-bonds to R240 (2.7 Å) and N283 (3.8 Å). The altered conformation of 7 also results in a ca. 0.3 Å clockwise rotation of C1 away from the iron of the heme cofactor, providing a possible rationale for the absence of any detectable oxidative rearrangement or hydroxylation products.
Site-directed mutagenesis of active site PntM residues
The protein structural studies and sequence comparison described above established that PntM harbors a constellation of active site residues, F232, M77, and M81, unique among all known P450s, that snugly cradle the bound substrate, pentalenolactone F (2) (Figure 2). Given the proximity of all three residues to C1, C2, and C3 of the substrate, the loci for the generation, rearrangement, and deprotonation of the proposed neopentyl cation intermediate, we examined the possibility that the side chains of these three amino acid residues might electronically stabilize the various carbocation intermediates through quadrupole-cation or dipole-cation interactions, analogous to well-established role of aromatic residues in the active sites of terpene synthases that stabilize and chaperone the reactive carbocation intermediates genrerated by the ionization and cyclization of allylic diphosphates to complex terpenes.23
To probe the catalytic roles of F232, M81, and M77, the F232A, F232G, F232L, F232Y, and F232H mutants of PntM, as well as the M81A, M81C, and M77S variants, were prepared by site-directed mutagenesis. Each mutant was purified to homogeneity, as monitored by SDS-PAGE (Figure S1), and the MD of each protein was directly verified by LC-ESI-MS (Table S4). The LC-MS analysis revealed that the original M81C protein preparation had actually been obtained as the disulfide conjugate with β-mercaptoethanol (M81C-BME). The native M81C mutant was then readily prepared by carrying out the purification of recombinant protein using buffers that did not contain β-mercaptoethanol. In substrate binding assays, the M81C and M81C-BME mutants had KD values for substrate 2 that were only 4-6-fold greater than that of wild-type PntM, while the majority of the remaining mutants bound 2 with 15-50-fold lower affinity than did PntM, except for F232Y and F232H, which did not bind 2 at all (Table 1, Figures S5 and S6).
The steady-state kinetic parameters and product profiles for the six active PntM mutants were then determined, using a calibrated GC-SIM-MS assay to quantitate the time-dependent formation of 2 (Table 2, Figures S7 and S8). Significantly, the F232L mutant exhibited only a 7-fold reduction in kcat/Km compared to wild type Pnt M, reflecting a 20-fold decrease in kcat that was partially offset by a modest 3-fold reduction in Km. Importantly, GC-MS analysis of the total product mixture indicated formation of only the natural oxidative rearrangement product, pentalenolactone (1), with no detectable products corresponding to simple hydroxylation at C1 or any other site. This result conclusively rules out any special orbital stabilization of the carbocation intermediates by the aromatic ring F232 and indicates that the role of the aromatic side chain is to precisely position the substrate 2 and derived intermediates. Notably, the F232A mutant, which carries the canonical I-helix Ala residue, still generates exclusively pentalenolactone, albeit with a >60-fold lower kcat/Km than wild type PntM. The M81C mutant showed a modest 10-fold reduction in kcat/Km compared to wild-type, with exclusive formation of the oxidative rearrangement product 1, while the M81C-BME, M81A, and M77S mutants were all 5-15-fold less active but with no change in product profile.
Table 2.
Steady-state kinetic parameters for PntM and mutants.
| PntM/mutant | kcat (min-1) | kcat/Km (min-1μM-1) | Km (μM) | R2 |
|---|---|---|---|---|
| Wild-type | 1.8±0.3 | 5.0×10-2 | 36±14 | 0.95 |
| F232A | 0.035±0.002 | 8.2×10-4 | 43±5 | 0.99 |
| F232L | 0.090±0.006 | 7.5×10-3 | 12.0±3 | 0.96 |
| M81A | 0.004±0.001 | 3.7×10-4 | 37±6 | 0.98 |
| M81C | 0.092±0.003 | 4.9×10-3 | 19±3 | 0.99 |
| M77S | 0.019±0.001 | 1×10-3 | 19±4 | 0.96 |
| M81C-BME | 0.085±0.25 | 3.0×10-4 | 280±110 | 0.99 |
The structures of the PntM F232L, M77S, M81A, M81C, and M81C-BME mutants with bound pentalenolactone F (2) were solved to resolutions of 2.06-2.12 Å (Table S2, Figure S11). None of these five mutants exhibited notable changes in the either the overall protein topology or the positions of the amino acid side chains, the heme cofactor, bound 2, or the key bound water molecules W1–W4, except for the mutated residues and a few minor changes, some of which are noted below. In PntM_F232L, the M77 side chain is now observed as a mixture two conformers, one (55%) unchanged from the wild-type and the other (45%) in which the sulfur atom has been rotated 2.2 Å towards the space vacated by the replacement of the phenyl ring of F232 by the isopropyl group of L232, which itself is closely superimposed on the corresponding atoms of the F232 side chain (Figure S11B). In both conformers, the position of the M77 methyl remains unchanged. In the M81C mutant, the only significant change is that a new water molecule is found in the site previously occupied by the terminal methyl of M81 (Figure S11D). Finally, in the M81C-BME mutant, the only significant change is that the terminal hydroxyl group of the covalently linked β-mercaptoethanol has replaced water W5 and is hydrogen-bonded (2.5 Å) to water W4, which has been very slightly displaced from its original position in the wild type protein (Figure S11J).
Discussion
The combined protein structure and site-directed mutagenesis studies conclusively rule out any special role of the F232, M77, or M81 residues at the active site of PntM in promoting or favoring the formation of cationic intermediates instead of, or in addition to, radical intermediates in the oxidative rearrangement of pentalenolactone F (2) to pentalenolactone (1), while the reduced substrate analogue studies do not support anchimeric assistance by the 6,7-double bond of substrate 2. These crystallographic studies do establish that the enzyme tightly controls the geometry of binding of both the substrate and product through a complex network of reinforcing H-bond and van der Waals interactions, so as to precisely position the target C1-Hsi bond of the substrate in line with the reactive iron-oxo intermediate, Compound I, while providing a strategically placed water molecule (W4) as part of a dedicated proton relay system that can accept the labile proton from C3-Hre. The key issue then becomes: What controls the exclusive formation of the key carbocation intermediate from the initially generated carbon radical?
Ortiz de Montellano and Groves have provided a critical analysis of the use of radical clock substrates and the apparent competition between P450-catalyzed formation of radical rebound products and electron-transfer processes that result in the formation of cation-derived products.24 Drawing on the extensive prior studies by Kochi25 of the kinetics, mechanism, and product profiles for the oxidation of a wide range of organic radicals by a series of organometallic complexes of increasing reduction potential, they have proposed that in P450-catalyzed reactions there are two competing processes that follow initial formation of the caged [Fe3+-OH]/carbon radical pair. The first, which is characteristic of the vast majority of P450s, involves a rapid oxygen rebound reaction, which normally occurs at a rate of ∼1010-1011 s-1, in which the iron-bound hydroxyl group is transferred within the caged radical pair to the substrate carbon atom, resulting in generation of the typical hydroxylated product from the nominally unreactive C-H bond (Scheme 2). In competition with this oxygen rebound process, there is also the possibility of a thermodynamically favorable electron transfer from the carbon radical to the strongly oxidizing heme •Fe3+-OH species. This latter process is normally too slow to compete with hydroxylation, or at best represents no more than 1-2% of the overall reaction flux in the P450-catalyzed oxidation of specifically engineered radical clock substrates. As pointed out by Kochi, such an electron transfer, which has been assigned a second order rate constant of ∼108 M-1s-1, is an outer sphere process that depends only on the difference between the reduction potential of the oxidizing •Fe3+-OH species and the ionization potential of the carbon radical.25 The radical ionization potential is itself insensitive to the degree of β-branching. Indeed, the rate of electron transfer has been shown not to be affected by substitution adjacent to the radical center.25 By contrast, Kochi has clearly demonstrated that the inner sphere ligand transfer process, which corresponds to oxygen rebound, is extremely sensitive to steric effects, including those associated with β-branching, with neopentyl radicals undergoing ligand transfer substantially more slowly than isobutyl radicals.25 Thus steric hindrance retards oxygen rebound while having no effect on the (normally kinetically silent) rate of electron transfer associated with oxidative formation of the carbocation. While none of the ingenious radical clock probes investigated by Ortiz de Montellano9 or by Newcomb10 suppress oxygen rebound by more than 2% relative to cation formation, the C1si face of pentalenolactone F represents an extreme, naturally occurring case in which the initially generated C1 radical is buttressed on both sides, by the axial C2si-methyl (C-12) and the C7-vinylidene substituents. (Figure 5). Oxygen rebound within the Heme-•Fe3+-OH/pentalenolactone F-C1 radical pair is therefore likely to be strongly retarded (probably by a factor >104) such that the otherwise kinetically insignificant electron transfer to generate the C1 carbocation would now become the dominant, indeed essentially exclusive, kinetic process (Scheme 2).26 Rapid rearrangement of the resulting C1 neopentyl cation followed by deprotonation of H-3re gives pentalenolactone (1), with competitive formation of the minor rearrangement products, pentalenolactones A (3), B (4), and P (5) (Scheme 1).
Figure 5.
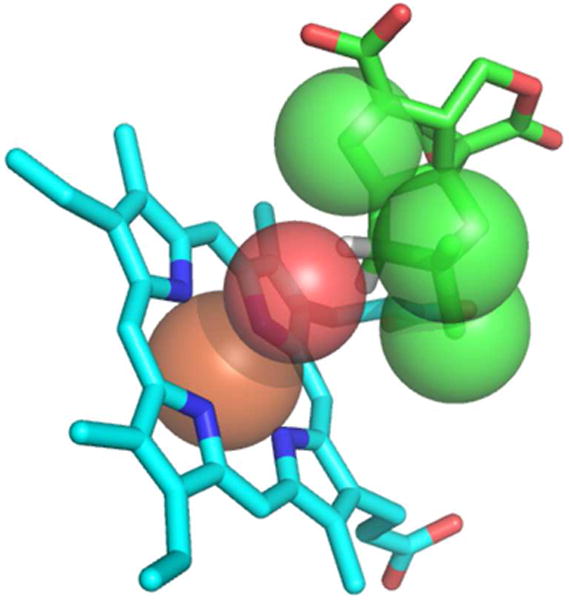
Model showing steric hindrance to oxygen rebound to the C1si face of pentalenolactone F.
Pentalenic acid (10) is a commonly occurring shunt metabolite of pentalenolactone biosynthesis that results from hydroxylation of the early pathway intermediate, 1-deoxypentalenic acid (11). The formation of 10 involves stereospecific removal of H-1re with net retention of configuration, in contrast to the abstraction of H-1si of 2 in the oxidative rearrangement catalyzed by PntM.5 The responsible P450, CYP105D7 (SAV_7469) has been cloned and characterized from S. avermitilis.27 Although the recently determined structure of CYP105D7 (PDB ID 4UBS) has two molecules of diclofenac, rather than the natural substrate 11, bound in the active site,28 alignment with the structure of PntM carrying bound pentalenolactone F (2) reveals that the binding region for the carboxylate of 11 lies on the wall of the active site cavity of CYP105D7 that is opposite to that established for carboxylate binding by PntM. Binding of 1-deoxypentalenic acid (11) to CYP105D7 could therefore readily be accommodated by a ca. 180° rotation with respect to the heme compared to the binding of 2 to PntM, resulting in orientation of the target C1-Hre bound directly toward the predicted position of the reactive oxo atom of Compound I in CYP105D7. Importantly, al-though C1 of 1-deoxypentalenic acid is also formally a neopentyl center, it is flanked on the targeted re face by only one CH3 substituent, attached to C2, while the other neighboring β-carbon, C8, carries a proton on the re face. This reduced degree of steric hindrance at C1 of 11 compared to that on the si face of 2 is apparently sufficient to permit normal oxygen rebound to occur during CYP105D7-catalyzed oxidation, allowing formation of the canonical hydroxylation product, pentalenic acid (10), in direct contrast to the exclusive electron transfer and oxidative rearrangement catalyzed by PntM.
Conclusions
The PntM-catalyzed conversion of pentalenolactone F (2) to pentalenolactone (1) involves the rearrangement of an oxidatively-generated neopentyl cation that is unique among all known P450-catalyzed reactions. The combined protein structural, site-directed mutagenesis, ligand-binding, steady-state kinetic, and product profile data rule out any unusual features of the protein active site, the heme cofactor, or the mode of substrate binding, while also excluding any special electronic interactions between substrate and active site residues or within the substrate itself that might stabilize carbocation intermediates. Severe steric inhibition of normal oxygen rebound favors competing internal electron transfer, leading to generation of the highly unusual neopentyl carbocation intermediate that undergoes rapid rearrangement and deprotonation.
Experimental Methods
Materials
Isopropylthio-β-galactopyranoside (IPTG), ampicillin, and kanamycin were purchased from Thermo Scientific. Chloroform-D (D, 99.8%) was purchased from Cambridge Isotope Laboratories, Inc. All other chemical reagents were purchased from Sigma-Aldrich and utilized without further purification. DNA primers were synthesized by Integrated DNA Technologies. Restriction enzymes, Q5® high-fidelity DNA polymerase, and T4 DNA ligase were purchased from New England Biolabs Inc (NEB) and used according to the manufacturer's specifications. Competent Escherichia coli 10-beta and BL21(DE3) cloning and expression strains were purchased from NEB. The pre-charged 5-mL HisTrap™ FF column, Hiload™ 16/600 Superdex™ 200 column, and Hiprep™ Q FF 16/10 column were purchased from GE Healthcare Life Sciences. Amicon Ultra Centrifugal Filter Units (Amicon Ultra-15 and Amicon Ultra-4 (30,000 MWCO) and Steriflip® Vacuum-driven Filtration System (0.22 μm, 50mL) were purchased from Millipore. Costar® 96-Well Microplates were purchased from Thermo Scientific. The Thrombin CleanCleave™ Kit was purchased from Sigma-Aldrich. CrystalEX second generation Corning® 96-well plates and Cryschem 24-well sitting drop plates were purchased from Hampton Research. Plasmids pET28a-penM and pET28a-pntM used for protein expression and the strain S. exfoliatus ZD22 used for isolation of pentalenolactone F were prepared by Dr. Dongqing Zhu, as previously described.4
Methods
General methods were as previously described.29 Growth media and conditions used for E. coli and Streptomyces strains and standard methods for handling strains were those described previously, unless otherwise noted.30 All DNA manipulations performed following standard procedures.30b DNA sequencing was carried out at the U. C. Davis Sequencing Facility, Davis, CA or by Genewiz. All proteins were handled at 4 °C, unless otherwise stated. Protein concentrations were determined according to the method of Bradford,31 using a Tecan Infinite M200 Microplate Reader with bovine serum albumin as the standard. Protein purity and size was estimated using both SDS-PAGE gel electrophoresis and an ATKA FPLC System. Accurate protein molecular weight was determined by ESI-MS on an Agilent 6530 Accurate-Mass Q-TOF LC/MS equipped with a Phenomenex Jupiter C4 column (50 mm × 2.00 mm, 5 μm). Substrate binding assays were carried out on the Tecan Microplate Reader. 1H and 13C NMR spectra were obtained on a Bruker Avance III HD Ascend 600 MHz spectrometer. GC-MS analyses were carried out using an Agilent Technologies 5977A MSD with an Agilent Technologies 7890B GC system.
Isolation of pentalenolactone F (2)
Streptomyces exfoliatus ZD22, from which the penM gene had been deleted, was grown on Mannitol soya flour (MS) agar for 6 days, as previously described.4 The spores were harvested and used to inoculate a seed culture in a liquid medium containing 2.5% Pharmamedia and 2.5% bactodextrose. After incubation at 28 °C for 3 days, 10 mL of seed culture was added to 1 L fermentation medium containing 0.2% NaCl, 0.5% CaCO3, 1% corn gluten meal, 0.1125% bactodextrose, 0.2% blackstrap molasses, and 2% corn starch, pH 7.2. After another 3 days shaking at 28 °C, the culture was centrifuged and the cells were washed with water and then discarded. The combined aqueous layers were acidified to pH 2.6 with HCl and extracted with chloroform. The organic extract was dried over Na2SO4, concentrated in vacuo, and the crude product was dissolved in 3 mL of methanol and 7 mL of benzene. For methylation, 0.5 mL trimethylsilyldiazomethane (TMS-CHN2) was added and the solution was stirred at room temperature for 30 min. A mixture of pentalenolactone F methyl ester (2-Me) and 9-epi- pentalenolactone F methyl ester (∼2:1) was obtained after flash silica gel column chromatography of the crude methyl esters (600 mL benzene:ethyl acetate/10:1). The concentrated mixture of isomeric methyl esters (260 mg) was applied to a 20 cm × 20 cm Silica gel 60 F254 TLC plate which was developed with benzene:ethyl:acetate (5:1). The band containing 2-Me (Rf 0.55-0.65) was scraped off and eluted with benzene to recover purified 2-Me (170 mg) which was identical to authentic 2-Me by 1H and 13C NMR and GC-MS.
6,7-Dihydropentalenolactone F methyl ester (7-Me)
Purified 2-Me (100 mg) was dissolved in anhydrous methanol (50 mL) in a 250-mL high-pressure hydrogenation bottle. After adding 20 mg of 5% Pd/C powder, the bottle was filled with hydrogen gas (22 psi) and shaken for 24 h. The mixture was filtered through Celite and then concentrated under reduced pressure. The hydrogenated products were separated on a silica gel column eluted with ∼900 mL benzene:ethyl acetate (15:1). 7-Me 1H NMR (600 MHz, CDCl3) δ 1.03 (s, 3H), 1.15 (s, 3H), 1.41 (dd, J = 13.0, 7.5 Hz, 1H), 1.65 (dd, J = 13.0, 8.2 Hz, 1H), 1.74 (d, J = 13.9 Hz, 1H), 1.85 (d, J = 13.9 Hz, 1H), 2.04 (m, 1H), 2.24 (m, 1H), 2.32 (m, 1H), 2.81 (q, J = 8.7 Hz, 1H), 3.00 (m, 1H), 3.01 (d, J = 5.1 Hz, 1H), 3.11 (d, J = 5.1 Hz, 1H), 3.75 (s, 3H), 4.18 (dd, J = 12.0, 7.4 Hz, 1H), 4.64 (dd, J = 12.0, 6.2 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 29.56, 30.56, 35.34, 41.26, 45.13, 47.50, 48.69, 50.32, 51.05, 51.13, 52.27, 56.57, 58.25, 68.43, 170.39, 174.22. HR-ESI-MS [M+H]+ 7-Me m/z 295.1548 (calculated for C16H23O5 : 295.1545). (Figures S2 and S3).
Hydrolysis of methyl esters to pentalenolactone (1), pentalenolatone F (2), and 6,7-dihydropentalenolactone F (7)
The methyl esters 1-Me, 2-Me, and 7-Me were each dissolved in 4:1 THF:H2O and 2 eq. LiOH was added to the solution. After 30 min at 0 °C, the reaction mixture was warmed to room temperature and incubated an additional 2 h. After acidification with 2 N HCl to pH 2, the quenched reaction mixture was extracted 3× with CHCl3 and the combined organic extracts were concentrated to dryness in vacuo. The typical yields were quantitative and the resulting carboxylic acids 1, 2, and 7 were dissolved in buffer and directly used without further purification.
Substrate binding assays with wild-type PntM and mutants
Wild-type or mutant PntM (17 mg/mL) was diluted 20× in 50 mM Tris-HCl (pH 8.0) to a final concentration of 19 μM. For binding assays with wild-type PntM, 2 (72 mM in THF) or 7 (86 mM in THF) were each diluted into the same buffer to final concentrations of 50 μM, 25 μM, 10 μM, 2.5 μM, and 1 μM for 2 and 200 μM, 100 μM, 50 μM, 25 μM, 12.5 μM, and 3.13 μM for 7. For binding assays with mutants of PntM, the dilution series for 2 encompassed 2 mM, 1 mM, 500 μM, 250 μM and 200 μM. Diluted protein (135 μL) and diluted 2 or 7 (15 μL) were mixed in 96-well plates. At least 3 sets of UV-Vis absorption spectra (350-500 nm) were recorded for each assay concentration and UV difference spectra were obtained for ligand-bound compared to ligand-free protein (Figures S4 and S5). Saturation curves were generated from the absorption maxima at 390 nm (substrate bound) and at 420 nm (substrate free) and used to calculate KD by non-linear squares fitting to the equation ΔA = ΔAmax•([L]/([L] + KD)) (n=3) (Figure S6). The reported errors are the standard deviations calculated by fitting program.
Steady-state kinetic analysis of PntM and mutants
a) GC-MS calibration curve for 1-Me
A reference series of increasing concentrations of 1-Me (4.8, 9.7, 14.5, 19.4, and 48.4 μM) was mixed with 10 μM farnesol as internal standard and analyzed by GC-MS using an Agilent 5977A GC-MS and Agilent HP-5MS column with a temperature program from 40 °C - 240 °C with an increase of 5 °C per min. For data collection, farnesol was quantitated by Selected Ion Monitoring (SIM), up to 35 min using SIM Group A, (consisting of mass fragments m/z 41.10, 55.05, 61.00, 69.05, 81.00, 93.00, 107.00). 1-Me was quantitated after 35 min using SIM Group B (consisting of mass fragments m/z 77.00, 91.00, 105.00, 115.00, 127.00, 128.00, 129.00, 130.00, 131.00, 143.00, 145.00, 157.00, 171.00, 202.00, 290.00). The SIM peak areas for the farnesol internal standard and 1-Me were individually summed and then used to calculate the calibration curve of normalized peak area versus concentration for 1-Me (Figure S7).
b) Steady-state kinetics
Kinetic assays were carried out at 30 °C using wild-type PntM (3.8 μM), and mutants F232A (4.4 μM), F232G (15.2 μM), F232L (5.5 μM), M81A (12.4 μM), M81C (7.2 μM), M81MBE (12 μM) or M77S (10.3 μM) in 20 mM Tris-HCl buffer (pH 8.0) containing 2.5% glycerol, 20 μg/mL spinach ferredoxin, 0.05 U/mL spinach ferredoxin-NADP reductase, pentalenolactone F (2) (14.4 -144 μM) and farnesol (1 μM) as internal standard in a total volume of 1.00 mL. The reactions were initiated by addition of 1 mM NADPH, incubated for 20 min at 30 °C, then quenched with 5 μL of 2 N HCl to give a final pH ∼2. The products were extracted 2× with 1 mL dichloromethane and the combined organic extracts were dried over Na2SO4. The concentrated extract was dissolved in 100 μL methanol and treated with trimethylsilydiazomethane (1 μL, TMS-CHN2, 2 M solution in hexane) to generate the corresponding methyl ester, 1-Me. Samples were analyzed and quantitated by GC/MS using the above-described SIM method, using farnesol as internal standard and the calibration curve for 1-Me to obtain the observed initial velocities of formation of 1-Me for each incubation. The steady-state kinetic parameters were calculated by direct non-linear least squares fitting to the Michaelis-Menten equation (SigmaPlot 12.5 (Table 2, Figure S8). The recorded error limits represent the statistical deviations calculated by the data fitting program (n=4, except for F232A for which n=1).
Preparative incubation of PntM and mutants with 2 or 7 and GC-MS analysis of products
Preparative-scale incubations were carried out using wild-type PntM (3.8 μM), or with mutants F232A (4.4 μM), F232G (15.2 μM), F232L (5.5 μM), M81A (12.4 μM), M81C (7.2 μM), M81MBE (12 μM) or M77S (10.3 μM) in 20 mM Tris-HCl buffer (pH 8.0) containing 2.5% glycerol, 20 μg /mL spinach ferredoxin, 0.05 U/mL spinach ferredoxin-NADP reductase, pentalenolactone F (14.4 - 144 μM and farnesol (1 μM) as internal standard in a total volume of 4.00 mL. The reactions were initiated by adding 1 mM NADPH, then incubated for 4 h at 30 °C to maximize substrate consumption. The reaction was quenched with 16 μL of 2 N HCl to a final pH ∼2. The products were extracted 2× with 4 mL dichloromethane and the combined organic extracts were dried over Na2SO4. The concentrated extract was dissolved in 200 μL methanol and treated with trimethylsilydiazomethane (4 μL, TMS-CHN2, 2 M solution in hexane) to generate the corresponding methyl esters. GC-MS analysis using an Agilent 5977A GC-MS and Agilent HP-5MS column and temperature program from 40 - 240 °C with an increase of 5 °C per min showed the same product profile as that for oxidation of 2 by wild-type PntM, with the exclusive product being 1-Me. No additional peaks with [M+]+ m/z 308 or [M-H2O]+ m/z 290 corresponding to any hydroxylated derivative of 2-Me were detected.
Incubation of dihydopentalenolactone F (7) with PntM and GC-MS product analysis
Freshly prepared and purified PntM (10 μL, 360 μM final concentration) was added to 20 mM Tris-HCl buffer (pH 8.0, 2.5% glycerol) containing 7 (1.1 mM), 20 μL spinach ferredoxin-NADPH oxidoreductase (12.5 U/mL), 50 μL spinach ferredoxin (2.2 mg/mL) in total volume of 4 mL. The reaction was initiated by addition of 100 μL NADPH (50 mM) and incubated for 2 h at 30 °C. The reaction was quenched by addition of 20 μL 2 N HCl and the mixture was extracted (3×) with 2 mL dichloromethane. The combined organic layers were dried over anhydrous Na2SO4, concentrated and dissolved in 100 μL methanol. Trimethylsilydiazomethane (10 μL) was added to generate the methyl esters and the products were analyzed by GC-MS using the column and temperature program described above.
Preparation of PntM mutants
Site-directed mutagenesis was carried out with the Quikchange II XL site-directed mutagenesis kit (Agilent) using plasmid DNA pET28a/PntM as template and PCR primer pairs listed in Table S3. The expression and purification steps were same as those used for wild-type PntM. The mutant M81C-BME was generated during purification of PntM M81C using the standard buffer containing β-mercaptoethanol. To obtain unmodified PntM M81C, only buffers without β-mercaptoethanol used. The DNA sequences of all mutant constructs were verified directly. The purity of each mutant protein was verified by SDS-PAGE (Figure S1) and the protein MW determined by LC-ESI-HRMS (Table S4).
Purification of PntM for X-ray crystallography
Fresh clones of E. coli BL21 containing plasmid pET28a-pntM were grown on LB agar media with 50 μg/mL kanamycin. LB media (5 mL) with 50 μg/mL kanamycin was seeded with a single colony and incubated overnight. The seed culture (5 mL) was added to 500 mL Terrific Broth (TB) containing 50 μg/mL kanamycin and incubated at 37 °C until an OD >0.6, then induced with 0.4 mM IPTG. The cells were further grown at 18 °C for 24 h, harvested by centrifugation and then suspended in lysis buffer (100 mM Tris-HCl, 500 mM NaCl, 0.1 mM DTT, 2.7 mM β-mercaptoethanol, 10 mM imidazole, pH 8.0) containing 10 mg/L pepstatin, 10 mg/L PMSF, and 0.2 mg/mL benzamidine. The cells were broken using a French press at 10000 psi and separated by centrifugation at 15000g for 1 h. The supernatant was applied to a Ni-NTA column which was first washed with buffer A (100 mM Tris-HCl, 20 mM imidazole, 2.7 mM β-mercaptoethanol pH 8.0) and then eluted with buffer B (100 mM Tris-HCl, 500 mM imidazole, 2.7 mM β-mercaptoethanol pH 8.0. The collected His6-tag-PntM protein was concentrated and diluted with thrombin digestion buffer (50 mM Tris-HCl, 10 mM CaCl2, pH 8.0), then incubated with thrombin-agarose (1 mL) overnight (10 h) at 4 °C. The digested protein was reloaded onto Ni-NTA, and the flow-through protein lacking the His6-tag was then applied to a size exclusion column (Hiload 16/600, Superdex 200 pg) for further purification. The purified protein was concentrated to 16.8 mg/mL and the molecular weight and purity confirmed by LC-ESI-MS (Table S4).
Protein crystallization - wild-type PntM
Crystals of PntM were obtained at 15 °C using the sitting drop vapor diffusion method. The reservoir solution contained 1.2 M sodium citrate, 10% glycerol, 100 mM bicine, pH 9.0. Protein solution (1 μL, 16.8 mg/mL PntM, 10 mM Tris-HCl, 15 mM NaCl, and 10% glycerol) was mixed with 1 μL of reservoir solution. After growing for 7 days, crystals were flashfrozen in liquid nitrogen and stored. PntM with bound 2, 7, or 1 was obtained by soaking PntM crystals for 2 h into a 2-μL drop containing mother liquor with 2 mM 2, 7, or 1 and 5% DMSO.
Protein crystallization - PntM mutants
A cross-microseeding technique was used to get larger crystals for mutants. Small pieces of PntM crystals were crushed into tiny seeds in 500 μL mother buffer to make the seeding solution. Protein solution (1 μL, 10 mM Tris-HCl, 15 mM NaCl, and 10% glycerol) was mixed with 1 μL of seeding solution. After growing for 4 days, crystals were flashfrozen in liquid nitrogen and stored. Mutant PntM proteins complexed with 2 were obtained by soaking crystals for 2 h in a 2-μL drop containing mother liquor with 2 mM 2 and 5% DMSO.
X-ray crystallographic data collection
X-ray diffraction data for all the crystals were collected with a Rigaku Saturn 944+ CCD detector on a Rigaku FR-E Superbright rotating anode source at the Brown University Structural Biology Core Facility. All data were collected at a wavelength of 1.542 Å and 100 K. Diffraction data for PntM in space group P21212 were collected to 2.00 Å resolution with cell dimensions a = 44.46 Å, b = 164.59 Å, and c = 83.30 Å. Diffraction data to 2.03 Å for PntM with 2 were collected in space group P21212 with cell dimensions a = 44.75 Å, b = 164.48 Å, and c = 83.29 Å. Diffraction data to 2.03 Å for PntM with 7 were collected in space group P21212 with cell dimensions a = 44.53 Å, b = 164.37 Å, and c = 82.04 Å. Diffraction data to 2.28 Å for PntM with 1 were collected in space group P21212 with cell dimensions a = 44.33 Å, b = 163.76 Å, and c = 81.34 Å. Diffraction data to 2.08 Å for PntM-F232L with bound 2 were collected in space group P21212 with cell dimensions a = 44.60 Å, b = 164.15 Å, and c = 82.54 Å. Diffraction data to 2.08 Å for PntM-M77S with bound 2 were collected in space group P21212 with cell dimensions a = 44.68 Å, b = 164.60 Å, and c = 83.51 Å. Diffraction data to 2.07 Å for PntM-M81A with bound 2 were collected in space group P21212 with cell dimensions a = 44.62 Å, b = 163.86 Å, and c = 82.22 Å. Diffraction data to 2.12 Å for PntM-M81C with bound 2 were collected in space group P21212 with cell dimensions a = 44.56 Å, b = 164.23 Å, and c = 82.97 Å. Diffraction data to 2.06 Å for PntM-M81C-BME with bound 2 were collected in space group P21212 with cell dimensions a = 44.57 Å, b = 163.23 Å, and c = 83.29 Å. A single crystal was used for each data set. The diffraction images were integrated with the XDS package32 and scaled with pointless/aimless from the CCP4 suite.33 The data processing statistics are summarized in Tables S1 and S2.
Structure determination and refinement
The PntM structure was solved by molecular replacement with the program Phaser34 from the Phenix program suite35 in space group P21212 using the polyene macrolide epoxidase PimD (Pdb code 2X9P) as search model (data set PntM). The model was further rebuilt with ARP/wARP36 and manually checked and completed with Coot.37 Final crystallographic refinement was performed with the program phenix.refine.35 The other structures were subsequently solved by molecular substitution. Geometry restraints for ligands 1, 2 and 7 were obtained from PRODRG.38 The crystallographic R/Rfree factors are 0.14/0.17, 0.14/0.18, 0.16/0.19, 0.18/0.22, 0.15/0.19, 0.16/0.19, 0.15/0.18, 0.16/0.20 and 0.16/0.19 for the nine data sets: PntM, PntM with 2, PntM with 7, PntM with 1, PntM-F232L with 2, PntM-M77S with 2, PntM-M81A with 2, PntM-M81C with 2 and PntM-M81C-BME with 2, respectively. The Ramachandran statistics (most favored/outliers) are 98%/0%, 99%/0%, 98%/0%, 97%/0%, 98%/0%, 98%/0%, 98%/0%, 98%/0% and 98%/0%, for PntM, PntM with 2, PntM with 7, PntM with 1, PntM-F232L with 2, PntM-M77S with 2, PntM-M81A with 2, PntM-M81C with 2, and PntM-M81C-BME with 2, respectively. The refinement statistics are summarized in Tables S1 and S2. Protein figures were generated using Pymol.39
Supplementary Material
Scheme 4.

CYP105D7-catalyzed formation of pentalenic acid (10) by hydroxylation of 1-deoxypentalenic acid (11), with insertion of oxygen insertion of oxygen into C1-H-1re.
Acknowledgments
This work was supported by a grant from the U. S. National Institutes of Health, GM030301, to D.E.C.
Footnotes
Supporting Information. Tables of crystallographic statistics, 1H and 13C NMR spectra for 7-Me, UV difference spectra for ligand binding by wild-type PntM and mutants, ESI-MS data for PntM and mutants, steady-state kinetic plots, and GC-MS calibration plots, and additional protein structural figures. This material is available free of charge via the Internet at http://pubs.acs.org. The coordinates for substrate-free PntM (PDB ID 5L1R), PntM with bound 2 (5L1O), PntM with bound 1 (5L1P), PntM with bound 7 (5L1Q), and the PntM mutants F232L (5L1S), M77S (5L1T), M81A (5L1U), M81C (5L1V), and M81C-BME (5L1W, each with bound 2, are available from the RCSB Protein Data Bank (http://www.rcsb.org/pdb).
Author Contributions: The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.a) Ortiz de Montellano PR. Chem Rev. 2010;110:932–948. doi: 10.1021/cr9002193. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ortiz de Montellano PR, De Voss JJ. In: Cytochrome P450: Structure, Mechanism and Biochemistry. 3rd. Ortiz de Montellano PR, editor. Kluwer Elsevier; New York: 2005. pp. 183–245. [Google Scholar]
- 2.a) Denisov IG, Grinkova YV, Baylon JL, Tajkhorshid E, Sligar SG. Biochemistry. 2015;54:2227–2239. doi: 10.1021/acs.biochem.5b00079. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Poulos TL, Raag R. FASEB J. 1992;6:674–679. doi: 10.1096/fasebj.6.2.1537455. [DOI] [PubMed] [Google Scholar]
- 3.a) Cupp-Vickery JR, Poulos TL. Steroids. 1997;62:112–116. doi: 10.1016/s0039-128x(96)00168-7. [DOI] [PubMed] [Google Scholar]; b) Nagano S, Cupp-Vickery JR, Poulos TL. J Biol Chem. 2005;280:22102–22107. doi: 10.1074/jbc.M501732200. [DOI] [PubMed] [Google Scholar]; c) Sherman DH, Li S, Yermalitskaya LV, Kim Y, Smith JA, Waterman MR, Podust LM. J Biol Chem. 2006;281:26289–26297. doi: 10.1074/jbc.M605478200. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhao B, Lei L, Vassylyev DG, Lin X, Cane DE, Kelly SL, Yuan H, Lamb DC, Waterman MR. J Biol Chem. 2009;284:36711–36719. doi: 10.1074/jbc.M109.064683. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Yoshimoto FK, Guengerich FP. J Am Chem Soc. 2014;136:15016–15025. doi: 10.1021/ja508185d. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Ghosh D, Griswold J, Erman M, Pangborn W. J Steroid Biochem Mol Biol. 2010;118:197–202. doi: 10.1016/j.jsbmb.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Helliwell CA, Chandler PM, Poole A, Dennis ES, Peacock WJ. Proc Natl Acad Sci U S A. 2001;98:2065–2070. doi: 10.1073/pnas.041588998. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Tudzynski B. Appl Microbiol Biotechnol. 2005;66:597–611. doi: 10.1007/s00253-004-1805-1. [DOI] [PubMed] [Google Scholar]; i) Lin HC, Tsunematsu Y, Dhingra S, Xu W, Fukutomi M, Chooi YH, Cane DE, Calvo AM, Watanabe K, Tang Y. J Am Chem Soc. 2014;136:4426–4436. doi: 10.1021/ja500881e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu D, Seo MJ, Ikeda H, Cane DE. J Am Chem Soc. 2011;133:2128–2131. doi: 10.1021/ja111279h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cane D, Oliver J, Harrison PHM, Abell C, Hubbard B, Kane C, Lattman R. J Am Chem Soc. 1990;112:4513–4524. [Google Scholar]
- 6.Rittle J, Green MT. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 7.a) Ogliaro F, Harris N, Cohen S, Filatov M, de Visser SP, Shaik S. J Am Chem Soc. 2000;122:8977–8989. [Google Scholar]; b) Kamachi T, Yoshizawa K. J Am Chem Soc. 2003;125:4652–4661. doi: 10.1021/ja0208862. [DOI] [PubMed] [Google Scholar]
- 8.Groves JT, Adhyam DV. J Am Chem Soc. 1984;106:2177–2181. [Google Scholar]
- 9.a) Ortiz de Montellano PR, Stearns RA. J Am Chem Soc. 1987;109:3415–3420. [Google Scholar]; b) Cryle M, Ortiz de Montellano PR, De Voss JJ. J Org Chem. 2005;70:2455–2469. doi: 10.1021/jo047985d. [DOI] [PubMed] [Google Scholar]; c) Jiang Y, He X, Ortiz de Montellano PR. Biochemistry. 2006;45:533–542. doi: 10.1021/bi051840z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Newcomb M, Letadicbiadatti FH, Chestney DL, Roberts ES, Hollenberg PF. J Am Chem Soc. 1995;117:12085–12091. [Google Scholar]; b) Newcomb M, Shen R, Choi SY, Toy PH, Hollenberg PF, Vaz ADN, Coon MJ. J Am Chem Soc. 2000;122:2677–2686. [Google Scholar]
- 11.Newcomb has suggested that carbocation intermediates might not be generated by the usual Compound I intermediate but by direct insertion of HO+ into the target CH bond by the precursor hydroxyferric species (Compound 0) (Cf. ref 10). This alternative mechanism has not been generally accepted, however, nor is it supported by the results of QM/MM calculations (Cf. ref 1a, 12).
- 12.Shaik S, Cohen S, Wang Y, Chen H, Kumar D, Thiel W. Chem Rev. 2010;110:949–1017. doi: 10.1021/cr900121s. [DOI] [PubMed] [Google Scholar]
- 13.Wilt J. Free radicals. Vol. 1. Interscience; 1973. pp. 333–501. [Google Scholar]
- 14.Keating JT, Skell PS. Carbonium Ions. 1970;2:573–653. [Google Scholar]
- 15.Cane DE, Sohng JK, Williard PG. J Org Chem. 1992;57:844–852. b) Strictly speaking, the isolation of 3, 4, and 5 from cultures of S. exfoliatus is simply consistent with, but does not prove, that these minor shunt metabolites are formed by PenM. Since individually they represent less than 0.1% of the total pentalenolactones produced by S. exfoliatus, they have not been detected by GC-MS analysis of in vitro PenM- or PntM-catalyzed oxidation of 2.
- 16.Attempts to remove bound bicine by soaks in bicine-free buffers under a variety of conditions resulted in collapse of the protein crystals.
- 17.The T236 side chain is actually present as a 38:62 mixture of two conformations, in which the minor conformer has the side chain hydroxyl H-bonded to water W1, while in the major conformer the side chain hydroxyl group is hydrogen-bonded to both water W1 and the neighboring hydroxyl group of the bound bicine.
- 18.Martinis SA, Atkins WM, Stayton PS, Sligar SG. J Am Chem Soc. 1989;111:9252–9253. [Google Scholar]
- 19.The precise positions of water molecules in a typical water relay network, particularly water W1, will of course be altered by conversion of the substrate-bound complex to the O2-bound complex
- 20.a) Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]; b) Vidakovic M, Sligar SG, Li H, Poulos TL. Biochemistry. 1998;37:9211–9219. doi: 10.1021/bi980189f. [DOI] [PubMed] [Google Scholar]
- 21.Cupp-Vickery JR, Poulos TL. Struct Biol. 1995;2:144–153. doi: 10.1038/nsb0295-144. [DOI] [PubMed] [Google Scholar]
- 22.Taraphder S, Hummer G. J Am Chem Soc. 2003;125:3931–3940. doi: 10.1021/ja016860c. [DOI] [PubMed] [Google Scholar]
- 23.Christianson DW. Chem Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 24.Auclair K, Hu Z, Little DM, Ortiz De Montellano PR, Groves JT. J Am Chem Soc. 2002;124:6020–6027. doi: 10.1021/ja025608h. [DOI] [PubMed] [Google Scholar]
- 25.Rollick KL, Kochi JK. J Am Chem Soc. 1982;104:1319–1330. [Google Scholar]
- 26.In SN2 reactions, neopentyl derivatives react ∼105 more slowly than the corresponding propyl compounds. Cf Lowry T, Richardson KS. Mechanism and Theory in Organic Chemistry. Harper & Row; New York: 1976. p. 13.
- 27.Takamatsu S, Xu LH, Fushinobu S, Shoun H, Komatsu M, Cane DE, Ikeda H. J Antibiot. 2011;64:65–71. doi: 10.1038/ja.2010.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu LH, Ikeda H, Liu L, Arakawa T, Wakagi T, Shoun H, Fushinobu S. Appl Microbiol Biotechnol. 2015;99:3081–3091. doi: 10.1007/s00253-014-6148-y. [DOI] [PubMed] [Google Scholar]
- 29.Seo MJ, Zhu D, Endo S, Ikeda H, Cane DE. Biochemistry. 2011;50:1739–1754. doi: 10.1021/bi1019786. [DOI] [PMC free article] [PubMed] [Google Scholar]; Tetzlaff CN, You Z, Cane DE, Takamatsu S, Omura S, Ikeda H. Biochemistry. 2006;45:6179–6186. doi: 10.1021/bi060419n. [DOI] [PMC free article] [PubMed] [Google Scholar]; Jiang J, Tetzlaff CN, Takamatsu S, Iwatsuki M, Komatsu M, Ikeda H, Cane DE. Biochemistry. 2009;48:6431–6440. doi: 10.1021/bi900766w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.a) Kieser T, B MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. John Innes Foundation; Norwich, UK: 2000. [Google Scholar]; b) Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Third. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2001. [Google Scholar]
- 31.Bradford M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 32.Kabsch W. Acta Crystallogr, Section D, Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Collaborative Computational Project, N. Acta Crystallogr, Section D, Biol Crystallogr. 1994;50:760–763. [Google Scholar]
- 34.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. Acta Crystallogr, Section D, Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 36.Langer G, Cohen SX, Lamzin VS, Perrakis A. Nat Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. Acta Crystallogr, Section D, Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Schuttelkopf AW, van Aalten DM. Acta Crystallogr, Section D, Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 39.DeLano WL. CCP4 Newsletter on Prot Crystallography. 2002;40:82–92. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.