

Abstract
Background & Aims
Microbial dysbiosis and aberrant host–microbe interactions in the gut are believed to contribute to the development and progression of Crohn’s disease (CD). Microbiome studies in CD typically have focused on microbiota in feces or superficial mucosal layers of the colon because accessing DNA from deeper layers of the bowel is challenging. In this study, we analyzed the deep tissue microbiome in patients who underwent surgical resection of the small intestine.
Methods
Paraffin blocks were obtained from 12 CD patients undergoing ileocecal resection, and healthy ileum samples (inflammatory bowel disease–free controls) were obtained from 12 patients undergoing surgery for right-sided colon cancer. Diseased and healthy-appearing ileum was identified using microscopy, and paraffin blocks were macrodissected using a core needle to specifically isolate DNA. Illumina Whole Genome Sequencing was used for microbial sequence identification and subsequent taxonomic classification using the PathSeq tool.
Results
We observed significant differences between the microbiome of CD samples vs inflammatory bowel disease–free controls, including depletion of Bacteroidetes and Clostridia. Notably, microbial composition at the phyla level did not differ markedly between healthy and diseased areas of CD patients. However, we observed enrichment of potentially pathogenic organisms at the species level.
Conclusions
Our study showed dysbiosis within deeper layers of the ileum of CD patients, specifically enrichment of enterotoxigenic Staphylococcus aureus and an environmental Mycobacterium species not described previously. Future studies with larger cohort sizes are warranted to confirm these findings. Studies would benefit from effective microbial DNA extraction methods from paraffin sections and host nucleic acid depletion approaches to increase microbial read coverage.
Keywords: Crohn's Disease, Deeper Mucosal Layers, Microbial Dysbiosis
Abbreviations used in this paper: CD, Crohn's disease; FDR, false-discovery rate; IBD, inflammatory bowel disease; LDA, linear discriminant analysis
Summary.
Our study shows that the in situ microbial community of the diseased bowel in patients with Crohn's disease is distinct from inflammatory bowel disease–free individuals. In Crohn's disease patients, the microbial composition at the phyla level did not differ markedly between healthy and diseased areas, but at the species level an enrichment of potentially pathogenic organisms was observed in the diseased ileum.
Microbial communities in the gut lumen of patients with Crohn’s disease (CD) are distinct from those observed in healthy controls and include depletion of commensal phyla such as Firmicutes and Bacteroidetes.1, 2 Although the etiology of this dysbiosis is unclear, it is thought to trigger intestinal inflammation.3 Microbiome studies in CD typically have focused on the microbiota in feces or in the superficial mucosal layers of the colon because accessing tissue from the deeper layers of the bowel is challenging.
To identify potential pathogens we took an in situ approach to determine invasive tissue microbiota. We analyzed the deep tissue microbiome in patients who underwent surgical resection of the small intestine. Inflamed (diseased or involved) and uninflamed (healthy or uninvolved) areas of the resection specimens were selected from the same patient for analysis (Supplementary Table 1). Specifically, samples were obtained from 12 patients who underwent ileocecal resections at Massachusetts General Hospital (Boston, MA) for treatment of Crohn’s disease and who were part of the Prospective Registry in Inflammatory Bowel Disease (IBD) study at Massachusetts General Hospital. We selected patients who had surgery early in the course of the disease (<5 years after diagnosis). Healthy ileum samples were obtained from patients undergoing a similar surgery for right-sided colon cancer and were designated as IBD-free controls. Deeper sections of the small bowel were macrodissected from these surgical specimens. The submucosal lymphoid areas, granulomas, and lymphoid reactions around fissures were identified by histologic examination of the tissue by a pathologist and co-investigator (A.K.B.). The diseased (involved) area was identified and cored on paraffin blocks (Supplementary Figure 1B and C) using a 1.5-mm dermal punch as described previously.4 Punch biopsies were performed using sterilized, disposable needles and samples were placed directly into sterile Eppendorf tubes for further processing. Healthy-appearing (uninvolved) ileum from these CD patients and IBD-free ileum from patients undergoing surgery for right-sided colon cancer were cored similarly. DNA extraction and preparation of bar-coded DNA sequencing libraries were performed as previously described.4 Microbial sequence identification and subsequent taxonomic classification was performed using the PathSeq tool.5
Supplementary Figure 1.

H&E sections of small intestine showing (A) relative abundance levels of various Helicobacter species in pilot set. Results show accurate identification of H pylori in a H pylori–positive gastric submucosal sample. (B and C) Representative H&E section before and after macrodissection of diseased area of the small bowel is shown. (B) The sequenced portion of the bowel wall is shown as a circle and was removed from the paraffin section by the biopsy needle. (C) The rest of the bowel wall otherwise is intact.
Microbial DNA was identified in the deeper bowel layers in all 3 experimental groups: involved (n = 12 samples) and uninvolved (n = 11 samples; 1 surgical resection did not contain an uninvolved segment) regions from ileal segments of 12 CD patients, and IBD-free control (n = 12 samples) ileal segments from 12 right-sided colon cancer patients. The microbiomes of all 3 groups were dominated by 4 phyla (Actinobacteria, Firmicutes, Bacteroidetes, and Proteobacteria). The relative abundance of these phyla, however, varied between the groups (Figure 1A). Reads corresponding to viral, archaeal and fungal sequences were virtually absent (Supplementary Tables 2 and 3). At the phylum level, the microbial community was similar in involved and uninvolved regions but differed markedly from the microbiomes of IBD-free patients (Figure 1A). In particular, we observed depletion of commensal bacterial phyla such as Bacteroidetes in the involved and uninvolved regions compared with IBD-free ileum. The depletion in Bacteroidetes was especially significant (P = .0002; involved vs IBD-free ileum), with nearly 10-fold greater relative abundance in IBD-free ileum (10.22% mean relative abundance, compared with 1.15% and 0.82% mean relative abundance in involved and uninvolved regions, respectively). The relative abundance of Firmicutes conversely was higher in the involved regions vs IBD-free ileum (P = .0204), whereas abundance of the 2 other dominant phyla, Actinobacteria (P = .2189) and Proteobacteria (P = .1005), was not significantly different in involved vs IBD-free ileal specimens (Figure 1A). Here, it is important to note that the CD patients in this study were prescribed metronidazole and levofloxacin during the course of their treatment. Metronidazole specifically targets anaerobic and microaerophilic bacteria, including Bacteroidetes and members of the Firmicutes phylum containing the class Clostridia. We therefore cannot rule out the possibility that antibiotic treatment may have contributed to the depletion of members of these phyla in patients with Crohn’s disease. Although all patients were treated with antibiotics, the duration of treatment varied. We decided to compare longer-term (>2 wk) to short-term (≤2 wk) treatment in the involved (antibiotic treatment > 2 wk, n = 7 samples; antibiotic treatment ≤ 2 wk, n = 5 samples) and uninvolved (antibiotic treatment >2 wk, n = 7 samples; antibiotic treatment ≤ 2 wk, n = 4 samples) regions. We observed neither a significant difference in the presence of Bacteroidetes or Clostridiales nor a clear enrichment of aerobic organisms and depletion of anaerobic or microaerophilic organisms in the longer-term treated group compared with short-term treatment (data not shown). Because we did not have an untreated cohort we could not determine reliably the consequences of antibiotic treatment on the microbiome in this study. However, Gevers et al6 previously investigated the influence of antibiotic treatment for mucosal samples from CD patients and noted a similar pattern of microbial dysbiosis between treated and untreated patients although the dysbiosis was more pronounced in the treated group.
Figure 1.

(A) Relative abundance of bacterial phyla in involved and uninvolved Crohn’s and IBD-free ileum segments. (B) Box plots showing relative abundance of Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria phyla. (C) Unsupervised hierarchical clustering of bacterial genus level relative abundance from 3 sample groups (red, involved; pink, uninvolved; green, IBD-free). AU, approximately unbiased; BP, bootstrap probability.
We next performed unsupervised hierarchical clustering analysis based on genus-level bacterial abundance. Our results support the hypothesis that IBD-free ileum is significantly different than CD ileum, from both involved and uninvolved regions (Figure 1B). There are few reports on the microbiome of involved and uninvolved gut tissue in CD patients, although a study of colonic specimens has reported differences in the CD-associated microbiota between these regions.7
A metagenomic analysis of the 3 groups was performed using the linear discriminant analysis effect size (LEfSe) tool.8 Analysis of the microbial community diversity showed that no phyla were enriched significantly in involved vs uninvolved regions, although the genera Staphylococcus and Delftia were increased modestly in inflamed regions using a linear discriminant analysis (LDA) cut-off score of 3.0 or greater (Figure 2A). At the species level resolution, we noted a significant enrichment (LDA score, ≥3.0) of Staphylococcus aureus in involved (average relative abundance, 0.45%) vs uninvolved tissue (average relative abundance, 0.05%). Because LEfSe analysis does not perform multiple hypotheses correction in identifying features significantly enriched in a group, we performed comparative marker selection using GENE-E (available: http://www.broadinstitute.org/cancer/software/GENE-E), to identify significantly enriched features in each group. Comparing involved (n = 12) vs uninvolved (n = 11) regions confirmed a marked but statistically nonsignificant enrichment of S aureus in the involved samples with a false-discovery rate (FDR) of 0.19 (P = .06) and a fold increase of 7.76. Analysis of the S aureus sequencing reads showed the presence of an S aureus pathogenicity island in the involved tissue from 2 patients (CD-I-82 and CD-I-56). The S aureus pathogenicity islands are a large family of mobile, phage-related, genetic elements, most of which carry genes for 1 or more superantigens/enterotoxins.9 A recent study reported infection with enterotoxigenic methicillin-resistant S aureus in a Crohn’s patient with active colitis, which was resolved after targeted antibiotic treatment.10 Superantigens secreted by these strains are potent T-cell mitogens that lead to a massive release of proinflammatory cytokines,11 and several reports have highlighted the ability of enterotoxigenic strains to trigger intestinal inflammation.10, 12, 13, 14 These observations suggest that enterotoxigenic strains, coupled with compromised barrier function in intestinal epithelia, may contribute to localized lesions in CD.
Figure 2.

Bacterial taxa in involved vs uninvolved regions of Crohn’s patients, and involved regions of Crohn’s patients vs IBD-free patients. (A) LDA and effect size measurements identify a modest enrichment of Staphylococcus and Delftia genera in involved vs uninvolved ileal regions in Crohn’s patients. Red, taxa enriched in involved regions; pink, taxa enriched in uninvolved tissues. Only taxa exceeding an LDA threshold of 3.0 (red dotted line) were considered for analysis. (B) A cladogram of the most enriched taxa in involved ileum of Crohn’s patients vs IBD-free individuals. Red, taxa enriched in involved regions; green, taxa enriched in IBD-free tissues. LDA and effect size measurements for involved segments vs IBD-free are shown in Supplementary Figure 2.
We observed that the microbial community in the involved ileum was markedly different from the IBD-free ileum (Figure 2B and Supplementary Figure 2), including enrichment of some Mycobacterium species in the involved tissue. Mycobacterium species have been implicated previously in CD, specifically Mycobacterium avium.15 Here, we observed an increase in Mycobacterium abscessus (average relative abundance, 0.09% and 0.002% in involved and normal tissue, respectively) (Supplementary Figures 2 and 3).16 Comparative marker selection with GENE-E confirmed the enrichment of M abscessus in involved samples compared with IBD-free samples, with a FDR of 0.044 (P = .02) and a fold increase of 53. Our results show a bacterial imbalance in CD that is not restricted to mucosal surfaces, but also occurs in the deeper intestinal layers. We showed a marked depletion of Bacteroidetes in Crohn’s patients, regardless of the presence of inflammation, which is consistent with studies suggesting that depletion of commensals such as Bacteroidetes (Supplementary Figure 4) can induce an aberrant immune response.2 The Firmicutes phylum, including the Bacilli class, is enriched with a LDA score of 3.0 or greater in involved regions compared with IBD-free ileum, with enrichment of Streptococcus and Bacillus genera. We observed depletion of the Clostridia class, specifically the Clostridiales order, in involved ileum, which also is consistent with the literature.6 An enrichment of potential bacterial pathogens emerged at the species level, including S aureus and M abscessus. Although this finding is intriguing and refuels the debate on the role of Mycobacteria in CD, further studies are required to confirm or negate their role in CD. Given that Mycobacterium species and S aureus are notoriously difficult-to-lyse organisms,17, 18, 19 it is possible that we may in fact be underestimating their abundance in these samples. Moving forward, we intend to apply more effective lysis methods to investigate their prevalence in CD more rigorously. This approach may help reduce the intra-individual variation in isolation of bacterial nucleic acids from paraffin sections. Notably, our analysis of the deeper diseased ileum did not show an enrichment of the bacterial families Enterobacteriaceae, Pasteurellaceae, Veillonellaceae, and Fusobacteriaceae previously observed in the upper mucosal layers.6 Although members of these families, such as Fusobacteria, have the potential to be invasive organisms, it is possible that they are not invasive in CD and remain at the mucosal interface. Interestingly, organisms belonging to the bacterial families that Gevers et al6 noted as being enriched in the mucosal layers, including obligate anaerobes (all Veillonellaceae members and specific members of Fusobacteriaceae), were absent in our analysis of the deeper ileal layers. Rather, we observed aerobic (M abscessus) or aerotolerant (S aureus) species. A potential explanation for this is that inflammation within the deeper layers can result in increased oxygenated blood supply to these sites, promoting the growth of these species. It is possible that oxygen tension may determine which microbial species can colonize, and ultimately contribute to, pathology.
Supplementary Figure 2.
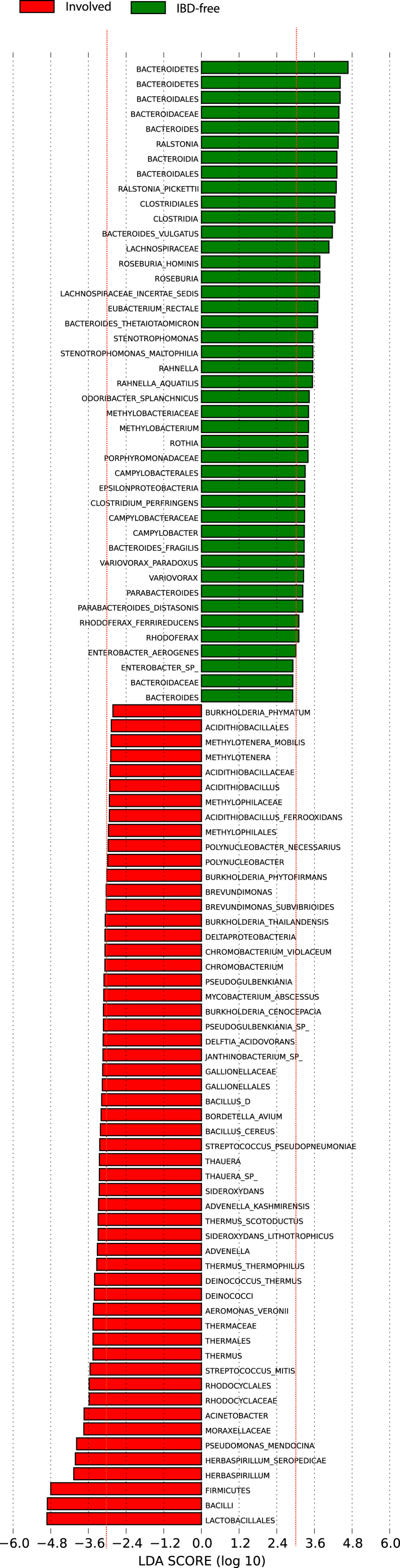
LDA coupled with effect size measurements identifies significant depletion of Bacteroidetes phyla and Clostridia class in Firmicutes phyla when compared with involved ileum segments with IBD-free ileum. In addition, there was significant enrichment of Bacilli class in Firmicutes phyla when comparing involved ileum segments with IBD-free. Involved enriched taxon have a negative score (red), and taxa enriched in IBD-free tissue have a positive score (green). Only taxa meeting an LDA threshold of 3.0 (red dotted line) were considered for analysis.
Supplementary Figure 3.

Relative abundance of various Mycobacterium species in the cohort.
Supplementary Figure 4.

Box plots show comparison of Bacteroidetes, Firmicutes, and Proteobacteria phyla between our study and the Gevers et al7study.
In summary, the in situ microbial community of patients with CD is distinct from IBD-free individuals. We show the enrichment of an enterotoxigenic strain of S aureus in diseased areas of the bowel as well as low levels of an environmental mycobacterial species in CD. These microbial communities may perpetuate stimulation of the host immune response, contributing to the ileal Crohn’s phenotype and necessitating surgical intervention. Our study had limitations, including a smaller cohort size and the use of DNA extraction methods from paraffin sections that are designed for host, rather than microbial, cell lysis and nucleic acid extraction. To define the contribution of the pathogens identified in this study on the initiation and progression of Crohn’s disease, further investigation is warranted with a larger cohort size and optimized DNA extraction methods for intracellular pathogens in conjunction with sophisticated host nucleic acid–depletion approaches.
Acknowledgments
The authors thank the Center for the Study of Inflammatory Bowel Disease, Massachusetts General Hospital (Boston, MA).
Footnotes
Current address of A.S.B. and V.M.: Department of Medicine, Department of Genetics, Stanford University, Stanford, California
Current address of A.I.O.: Department of Epidemiology, University of Alabama at Birmingham, Birmingham, Alabama
Conflicts of interest The authors disclose no conflicts.
Funding This work was funded by a Harvard Institute of Translational Immunology (HITI) pilot grant from the Helmsley Charitable Trust, a computational grant from Amazon Web Services and a grant from Center for the Studies on Inflammatory Bowel Disease (DK4331).
Contributor Information
Matthew Meyerson, Email: Matthew_Meyerson@dfci.harvard.edu.
Vijay Yajnik, Email: vyajnik@mgh.harvard.edu.
Supplementary Materials and Methods
Sample Selection and Pathologic Review
All patient samples were selected using a clinically annotated patient registry, (Prospective Registry in IBD Study at Massachusetts General Hospital), in which more than 1000 patients are enrolled with detailed treatment history, IBD genetics, and have consented to banking DNA, blood, and tissue. Twenty-seven formalin-fixed, paraffin-embedded ileal resection samples were identified for further investigation. The inclusion/exclusion criteria place an emphasis on selecting patients with preferably less than 6 months of exposure to antibiotics and immunosuppressive agents, which could alter the invasive gut microbiome. IBD-free ileum from right hemicolectomy specimens (obtained from colorectal carcinoma surgeries) were selected as negative controls. Institutional review board approval was obtained through the Massachusetts General Hospital and the Broad Institute. All patient samples were de-identified. All samples were reviewed histologically by a co-investigator who is a gastrointestinal pathologist with a specialization in IBD (A.K.B.).
In pilot experiments, we selected 2 submucosal CD samples, 2 deep fissure macrodissected CD samples, 2 submucosal biopsy specimens from patients with appendicitis, and 1 Helicobacter pylori–positive gastric submucosal sample (positive control sample) to validate our approach.
DNA Extraction, Library Construction, and Sequencing
DNA was extracted from all formalin-fixed, paraffin-embedded samples using the Ambion Recoverall kit (ThermoFisher Scientific, Waltham, MA), as previously described.1 DNA was quantified using both a Nanodrop apparatus and the Qubit quantification system (ThermoFisher Scientific). DNA was sheared using a Covaris apparatus (Covaris, Woburn, MA) to an average fragment length of 350–500 bp. Sequencing libraries were prepared using the Illumina TruSeq kit per the manufacturer’s instructions (Illumina, San Diego, CA). Samples were pooled and multiplexed before sequencing on the Illumina V3 HiSeq platform (6 samples per lane, for an average depth of 3× human whole genome sequencing coverage).
PathSeq and Statistical Analysis
The PathSeq2 algorithm was used to perform computational subtraction of human reads, followed by alignments of residual reads to human reference genomes and microbial reference genomes (which included bacterial, viral, archaeal, and fungal sequences; downloaded from National Center for Biotechnology Information in June, 2012). These alignments resulted in taxonomic classification of reads into bacterial, viral, archaeal, and fungal sequences in whole genome sequencing data.
Briefly, PathSeq is used to remove low-quality reads followed by subtraction of human reads by mapping reads to a database of human genomes (downloaded from NCBI in November 2011) using BWA3 (release 0.6.1, default settings), MegaBLAST (release 2.2.25; cut-off E-value, 10-7; word size, 16), and BLASTN4 (release 2.2.25; cut-off E-value, 10-7; word size, 7; nucleotide match reward, 1; nucleotide mismatch score, -3; gap open cost, 5; and gap extension cost, 2). Only sequences with perfect or near-perfect matches to the human genome were removed in the subtraction process. In addition, low-complexity and highly repetitive reads were removed using Repeat Masker5 (version open-3.3.0; libraries dated 2011-04-19).
Taxonomic classification is performed by residual read alignment using MegaBLAST (release 2.2.25; cut-off E-value, 10-7; word size, 16) to a database of bacterial sequences and followed by BLASTN (release 2.2.25; cut-off E-value, 10-7; word size, 7) to human reference gnome and microbial reference genomes.
After the taxonomic classification of nonhuman DNA sequencing reads the relative abundance value for each bacterial organism was calculated as follows by using reads that maps with 90% or greater sequence identity and 90% or greater query coverage. Classifications were performed at the domain, then phylum, then genus, and then species level, requiring unique alignments (ie, reads with equivalent E-values to multiple taxa were removed from analysis).
At the species level, relative abundance for each organism was calculated as follows: relative abundance = (number of unique alignment positions in genome × 1,000,000)/(number of total aligned bacterial reads × genome size). The relative abundance values then were per-sample normalized such that the total relative abundance for each sample summed to 1. The resulting normalized relative abundance matrix was analyzed on LEfSe (online version available on August 2015)6 between different sample group labels (uninvolved, involved, and IBD-free samples) with default parameters. However, we considered the bacteria that had a LDA score of 3.0 or greater for further analysis. In addition, the Wilcoxon rank-sum test was used to calculate the P value for phyla level relative abundance between groups. Unsupervised hierarchical clustering was performed using the pvclust R package (available: https://cran.r-project.org/web/packages/pvclust/index.html) with 1000 bootstrap iterations, Ward’s clustering method, and otherwise default parameters on genus level bacterial relative abundance values.
Comparative marker selection was used to complement LEfSe analysis via GENE-E (http://www.broadinstitute.org/cancer/software/GENE-E/) with default settings and 10,000 permutations. This analysis was performed between involved (n = 12) and uninvolved (n = 11) samples, and between involved (n = 12) and IBD-free (n = 12) samples. This analysis returned a FDR, a fold change, and a P value based on a permutation test.
Results of Sequencing and PathSeq Analysis
PathSeq was used to assign a taxonomic classification of each sequence based on the organism of origin. Results from pilot experiments (complete data not shown) used to validate our approach shows accurate identification of H pylori in H pylori–positive gastric submucosal samples (Supplementary Figure 1A). We obtained a median of 39.3 million human DNA sequencing reads (range, 14.5–188.8 million), 2330 bacterial sequencing reads (range, 244–19445 reads), and 8660 unmapped sequencing reads (range, 2110–78466 reads) per Crohn’s disease or IBD-free sample. Microbial reads included those corresponding to known bacteria, archaea, fungi, and viruses. The number of human reads, microbial reads, and unmapped reads are presented in Supplementary Table 2. The relative abundance for each bacterium at the species level is presented in Supplementary Table 4.
Supplementary Material
References
- 1.Frank D.N. Proc Natl Acad Sci U S A. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Round J.L. Nat Rev Immunol. 2009;9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chassaing B. Gastroenterology. 2011;140:1720–1728. doi: 10.1053/j.gastro.2011.01.054. [DOI] [PubMed] [Google Scholar]
- 4.Bhatt A.S. N Engl J Med. 2013;369:517–528. doi: 10.1056/NEJMoa1211115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kostic A.D. Nat Biotechnol. 2011;29:393–396. doi: 10.1038/nbt.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gevers D. Cell Host Microbe. 2014;15:382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sepehri S. Inflamm Bowel. 2007;13:675–683. doi: 10.1002/ibd.20101. [DOI] [PubMed] [Google Scholar]
- 8.Segata N. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Novick R.P. Plasmid. 2003;49:93–105. doi: 10.1016/s0147-619x(02)00157-9. [DOI] [PubMed] [Google Scholar]
- 10.Bettenworth D. World J Gastroenterol. 2013;19:4418–4421. doi: 10.3748/wjg.v19.i27.4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Proft T. Clin Exp Immunol. 2003;133:299–306. doi: 10.1046/j.1365-2249.2003.02203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu J. Gastroenterology. 2003;125:1785–1795. doi: 10.1053/j.gastro.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 13.Benjamin M.A. Infect Immun. 1998;66:2193–2199. doi: 10.1128/iai.66.5.2193-2199.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baca-Estrada M.E. Clin Exp Immunol. 1995;99:398–403. doi: 10.1111/j.1365-2249.1995.tb05564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Behr M.A. Inflamm Bowel Dis. 2006;12:1000–1004. doi: 10.1097/01.mib.0000228183.70197.dd. [DOI] [PubMed] [Google Scholar]
- 16.Byrd T.F. Infect Immun. 1999;67:4700–4707. doi: 10.1128/iai.67.9.4700-4707.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willner D. PLoS One. 2012;7:e34605. doi: 10.1371/journal.pone.0034605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vandeventer P.E. J Clin Microbiol. 2011;49:2533–2539. doi: 10.1128/JCM.02171-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stuhlmeier R. J Clin Pathol. 2003;56:782–785. doi: 10.1136/jcp.56.10.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 1.Bhatt A.S. N Engl J Med. 2013;369:517–528. doi: 10.1056/NEJMoa1211115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kostic A.D. Nat Biotechnol. 2011;29:393–396. doi: 10.1038/nbt.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li H. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Altschul S.F. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smit AFA, et al. Available from: http://www.repeatmasker.org. Accessed 2011.
- 6.Segata N. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gevers D. Cell Host Microbe. 2014;15:382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.