

Abstract
Key points
The role of the β1 strand in GABAA receptor function is unclear. It lies anti‐parallel to the β2 strand, which is known to participate in receptor activation.
Molecular dynamics simulation revealed solvent accessible residues within the β1 strand of the GABAA β3 homopentamer that might be amenable to analysis using the substituted Cys accessibility method.
Cys substitutions from Asp43 to Thr47 in the GABAA α1 subunit showed that D43C and T47C reduced the apparent potency of GABA. F45C caused a biphasic GABA concentration–response relationship and increased spontaneous gating.
Cys43 and Cys47 were accessible to 2‐aminoethyl methanethiosulphonate (MTSEA) modification, whereas Cys45 was not. Both GABA and the allosteric agonist propofol reduced MTSEA modification of Cys43 and Cys47.
By contrast, modification of Cys64 in the β2 strand loop D was impeded by GABA but unaffected by propofol.
These data reveal movement of β1 strand loop G residues during agonist activation of the GABAA receptor.
Abstract
The GABAA receptor α subunit β1 strand runs anti‐parallel to the β2 strand, which contains loop D, known to participate in receptor activation and agonist binding. However, a role for the β1 strand has yet to be established. We used molecular dynamics simulation to quantify the solvent accessible surface area (SASA) of β1 strand residues in the GABAA β3 homopentamer structure. Residues in the complementary interface equivalent to those between Asp43 and Thr47 in the α1 subunit have an alternating pattern of high and low SASA consistent with a β strand structure. We investigated the functional role of these β1 strand residues in the α1 subunit by individually replacing them with Cys residues. D43C and T47C substitutions reduced the apparent potency of GABA at α1β2γ2 receptors by 50‐fold and eight‐fold, respectively, whereas the F45C substitution caused a biphasic GABA concentration–response relationship and increased spontaneous gating. Receptors with D43C or T47C substitutions were sensitive to 2‐aminoethyl methanethiosulphonate (MTSEA) modification. However, GABA‐evoked currents mediated by α1(F45C)β2γ2 receptors were unaffected by MTSEA, suggesting that this residue is inaccessible. Both GABA and the allosteric agonist propofol reduced MTSEA modification of α1(D43C)β2γ2 and α1(T47C)β2γ2 receptors, indicating movement of the β1 strand even during allosteric activation. This is in contrast to α1(F64C)β2γ2 receptors, where only GABA, but not propofol, reduced MTSEA modification. These findings provide the first functional evidence for movement of the β1 strand during gating of the receptor and identify residues that are critical for maintaining GABAA receptor function.
Key points
The role of the β1 strand in GABAA receptor function is unclear. It lies anti‐parallel to the β2 strand, which is known to participate in receptor activation.
Molecular dynamics simulation revealed solvent accessible residues within the β1 strand of the GABAA β3 homopentamer that might be amenable to analysis using the substituted Cys accessibility method.
Cys substitutions from Asp43 to Thr47 in the GABAA α1 subunit showed that D43C and T47C reduced the apparent potency of GABA. F45C caused a biphasic GABA concentration–response relationship and increased spontaneous gating.
Cys43 and Cys47 were accessible to 2‐aminoethyl methanethiosulphonate (MTSEA) modification, whereas Cys45 was not. Both GABA and the allosteric agonist propofol reduced MTSEA modification of Cys43 and Cys47.
By contrast, modification of Cys64 in the β2 strand loop D was impeded by GABA but unaffected by propofol.
These data reveal movement of β1 strand loop G residues during agonist activation of the GABAA receptor.
Abbreviations
- DMEM
Dulbecco's modified Eagle's medium
- DTT
dithiothreitol
- HEK‐293
human embryonic kidney 293 cell
- MD
molecular dynamics
- MTS
methanethiosulphonate
- MTSEA
2‐aminoethyl methanethiosulphonate
- PhMTS
phenylmethanethiosulphonate
- pLGIC
pentameric ligand‐gated ion channel
- PTX
picrotoxin
- SASA
solvent accessible surface area
- SCAM
substituted cysteine accessibility method
- TM
transmembrane
- τw
weighted tau
- WT
wild‐type
Introduction
GABAA receptors are members of the Cys‐loop family of pentameric ligand‐gated ion channels (pLGICs). GABAA receptors are assembled from 19 different subunits. The most common synaptic GABAA receptor consists of α1, β2 and γ2 subunits (Whiting et al. 1995).
GABAA receptors have a large extracellular domain, housing the orthosteric ligand‐binding site, and four transmembrane (TM) domains (TM1–4), which contain binding sites for allosteric agonists and several non‐competitive antagonists. TM2 lines the Cl−‐selective channel pore. Similar to most pLGICs, GABAA receptors also have a large intracellular domain, mainly composed of the TM3–4 loop (Baptista‐Hon et al. 2013).
The orthosteric binding site, located at the interface between adjacent subunits (Smith & Olsen, 1995; Cromer et al. 2002), is lined by residues within six non‐continuous loops (A–F) that participate in binding (Boileau et al. 1999; Holden & Czajkowski, 2002; Wagner et al. 2004; Goldschen‐Ohm et al. 2011; Tran et al. 2011) and gating (Boileau et al. 2002; Newell & Czajkowski, 2003; Venkatachalan & Czajkowski, 2008; Szczot et al. 2014). In GABAA receptors, the primary interface contributes loops A, B and C, from the β subunit, whereas the complimentary interface contributes loops D, E and F, from the α subunit. These loops are contained within an anti‐parallel β sandwich structure, which makes up much of the N‐terminal domain.
Several pLGIC structures have recently been solved, with some in the presence of agonists providing an insight into the architecture of the orthosteric binding site. A structural model of Caenorhabditis elegans GluCl implicates β1 strand Arg37 in glutamate binding (Hibbs & Gouaux, 2011). This led to the proposal of a seventh binding loop, termed loop G. However, the equivalent β1 strand residue in Aplysia californica GluCl (Leu79) is not involved in its binding to glutamate or other amino acid agonists including GABA, suggesting that a role for the β1 strand in agonist binding may be restricted to the C. elegans GluCl (Blarre et al. 2014). Interestingly, replacement of A. californica GluCl Leu79 by Arg resulted in the abolition of glutamate evoked currents, highlighting the importance of amino acids in the β1 strand to receptor function. A comparison of GluCl structures in presumed open and closed conformations reveals that the β1 and β2 strands, as well as the β1–β2 loop, move towards the TM2–3 loop during gating (Althoff et al. 2014). This movement appears to precede the structural rearrangement of the TM domains that allow channel opening (Calimet et al. 2013). A similar conformational change has also been described in the structure of a zebrafish glycine receptor derived by cryo‐electron microscopy (Du et al. 2015).
Residues in the β1 strand of the recently solved GABAA β3 homopentamer structure are not implicated in binding its agonist, benzamidine (Miller & Aricescu, 2014). However, residues in loop D of the anti‐parallel β2 strand are involved, and this region of the α1 subunit lies within the GABA binding pocket in α1β2γ2 GABAA receptors (Boileau et al. 1999; Holden & Czajkowski, 2002). Furthermore, the loop D residue Phe64 participates in gating (Szczot et al. 2014). The use of the substituted Cys accessibility method (SCAM) revealed a series of β2 strand residues within loop D that are accessible within the binding pocket. Consistent with its role in agonist binding, residue 64 became less accessible to methanethiosulphonate (MTS) modification when receptors were activated by GABA. Residues within the α1 subunit β1 strand are probably also solvent accessible through the ligand‐binding pocket because they lie adjacent to the β2 strand. As a result, Cys‐substituted β1 strand residues may also be amenable to SCAM.
In the present study, we first examined solvent access to the GABAA β3 homopentamer using molecular dynamics (MD) simulation. We subsequently used the simulations as a guide to investigate the functional consequences of Cys substitutions at five of the equivalent β1 strand residues in the α1 subunit of α1β2γ2 GABAA receptors. The introduction of Cys at these positions enabled us to examine their accessibility in the absence and presence of receptor activation.
Methods
Cell culture and transfection
Human embryonic kidney 293 (HEK‐293) cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 100 μg ml−1 penicillin and 100 units ml−1 streptomycin at 37°C and 5 % CO2. Cells were seeded at low density in 35 mm dishes for electrophysiology. Transfections were performed by calcium phosphate precipitation, using 1 μg of total cDNA per dish, as described previously (Baptista‐Hon et al. 2013). cDNAs encoding wild‐type (WT) and mutant mouse GABAA subunits were in the pRK5 mammalian expression vector. For heteromeric expression of GABAA α1β2γ2 subunits, a 1:1:1 transfection ratio was used. cDNA encoding enhanced green fluorescent protein (in pEGFP vector, 0.1 μg) was included to identify successfully transfected cells using fluorescence microscopy. Cells were washed with media 16 h after transfection and used after 48–72 h. All tissue culture reagents were obtained from Invitrogen (Paisley, UK).
Mutagenesis of GABAA α1 subunits
Single point mutations were performed by overlap extension PCR (Heckman & Pease, 2007). PCR products were digested using SmaI restriction endonuclease and ligated into pRK5 vector. All mutagenesis reactions and ligations were verified using agarose gel electrophoresis and constructs were sequenced prior to functional characterization (Genetics Core Services, University of Dundee, Dundee, UK). All PCR and molecular cloning reagents were obtained from Fermentas (Thermo‐Fisher, Loughborough, UK).
Electrophysiology
The whole‐cell configuration of the patch clamp technique was used to record GABA‐evoked currents from HEK‐293 cells expressing WT α1β2γ2 GABAA receptors or receptors containing Cys‐substituted α1 subunits. Recording electrodes were fabricated from borosilicate glass capillaries, which, when filled with intracellular solution, had resistances of 1.3 – 2.3 MΩ for whole‐cell recordings. The electrode solution contained (in mm): 140 CsCl, 2 MgCl2, 1.1 EGTA, 3 Mg‐ATP, 10 Hepes (pH 7.4 with CsOH). The extracellular solution contained (in mm): 140 NaCl, 4.7 KCl, 1.2 MgCl2, 2.5 CaCl2, 10 Hepes, 10 glucose (pH 7.4 with NaOH). Cells were voltage clamped at an electrode potential of –60 mV. Currents were evoked by rapid application of GABA using the three‐pipe Perfusion Fast Step system (Warner Instruments, San Francisco, CA, USA), as described previously (Baptista‐Hon et al. 2013).
All electrophysiological data were recorded using an Axopatch 200B amplifier (Axon Instruments, Burlingame, CA, USA). Data were low pass filtered at 2 kHz, digitized at 20 kHz using a Digidata 1320 A interface and acquired using pCLAMP8 software (all from Molecular Devices, Sunnyvale, CA, USA).
SCAM
The cysteine sulfhydryl‐specific modifying effect of a MTS reagent was determined using 2‐aminoethyl methanethiosulphonate (MTSEA) or phenylmethanethiosulphonate (PhMTS) (Toronto Research Chemicals, Toronto, Ontario, Canada). Unless otherwise indicated, HEK‐293 cells transfected with WT or Cys‐substituted subunits were exposed repeatedly to an EC50 concentration of GABA to evoke stable baseline currents (I control). Freshly diluted MTSEA (up to 10 mm) was then applied episodically to cells prior to their GABA EC50 exposure. Altered current amplitudes following MTSEA exposure reveals accessibility to sulfhyrdryl modification (I modified). The extent of modification was measured as a percentage change:
% change = × 100
Cells were subsequently exposed to the reducing agent dithiothreitol (DTT) (10 mm) to reverse modification by MTSEA.
Rates of MTSEA modification were measured using empirically determined concentrations of MTSEA that produced cumulative changes in the amplitude of GABA‐evoked currents reaching steady‐state within the duration of the recording. MTSEA was applied episodically for intervals between 0.1 and 2 s, prior to a challenge with EC50 GABA. In all cases, the steady‐state current amplitude reached that of I modified when a saturating concentration of MTSEA was used, indicating that the modification reaction had reached completion. To determine whether the rate of MTSEA modification of the substituted cysteine can be altered in the presence of orthosteric agonists or allosteric agonists, a maximal concentration of GABA (EC100), or an activating concentration of propofol (10 μm) was simultaneously applied with MTSEA.
MD simulations
MD simulations were carried out with the simulation software Gromacs, version 4.67 (Pronk et al. 2013). The ambersb99_ildn force field (Hornak et al. 2006; Lindorff‐Larsen et al. 2010) for amino acids and the Berger force field for POPC lipids (Berger et al. 1997) were applied. Temperature and pressure were kept constant at 298 K and 1 bar, using the v‐rescale thermostat (Bussi et al. 2007) and the Parrinello‐Rahman barostat (Parrinello & Rahman, 1981) with coupling constants of 0.5 and 2 ps, respectively. Electrostatic interactions were computed using the Particle mesh Ewald method with a real space cut‐off of 12 Å. Van der Waals interactions were calculated using a cut‐off of 12 Å. The LINCS algorithm (Hess et al. 1997) was applied to constrain all bonds. Virtual sites (Feenstra et al. 1999) were employed for hydrogen atoms, permitting the use of a simulation timestep of 5 fs. The total sampling time was 1.64 μs.
pK a values of all titrable groups of the GABAA β3 homopentamer (Protein Data Bank code: 4COF) (Miller & Aricescu, 2014) were calculated using the H++ webserver (Anandakrishnan et al. 2012). The transmembrane domain of the protein was first aligned to a POPC bilayer, using LAMBADA (Schmidt & Kandt, 2012) and slightly moved manually to match protein–lipid interactions (Contreras et al. 2011). The protein was inserted into the membrane, employing the software InflateGRO2 (Schmidt & Kandt, 2012). A NaCl concentration of ∼180 mm was added and additional counter ions were introduced to neutralize the simulation system.
Data analysis
The peak amplitudes of agonist‐evoked currents were measured using Clampfit, version 10 (Molecular Devices). Individual relationships of current amplitude to GABA concentration were fitted with a logistics equation:
from which GABA EC50 and Hill slope (n H) values were determined.
The rate of MTSEA modification of Cys‐substituted receptors was measured by fitting single or double exponential functions to peak current amplitude data following cumulative MTSEA applications. The double exponential function is defined by:
where τf and τs represent the fast and slow time constants, respectively. A f and A s represent the proportion of the fast and slow components, respectively, such that A f and A s sum to 1 – Plateau. Rates of MTSEA modification are provided as weighted τ (τw) values, calculated using:
Pseudo first‐order rate constants were derived from a modified single or double exponential fit to peak current amplitude data following MTSEA applications. The double exponential function is defined by:
where K f and K s represent the fast and slow first‐order rates, respectively. A f and A s represent the proportion of the fast and slow components. Because all amplitude data are normalized to I GABA at t = 0, A f and A s sum to 1 – Plateau. Second‐order rate constants were obtained by dividing K by the concentration of MTSEA used (Holden & Czajkowski, 2002).
The solvent accessible surface area (SASA) was calculated as described previously (Eisenhaber et al. 1995) using data from the MD simulations. The solvent accessible area was graphically represented using trj_cavity (Paramo et al. 2014). All figures containing molecular information were produced using VMD (Humphrey et al. 1996).
Statistical analysis
Data are presented as the mean ± SEM or SD as indicated. Differences in means of three or more groups were compared using one‐way ANOVA, with a post hoc Tukey or Dunnet's test, as appropriate. Pairwise comparisons were performed using Student's t test. P < 0.05 was considered statistically significant. Statistical analyses were performed using Prism, version 5 (GraphPad Software, San Diego, CA, U.S.A.).
Results
Residues in the β1 strand of the β3 homopentamer are solvent accessible
Changes in the accessibility of residues during GABAA receptor activation have been determined using SCAM (Boileau et al. 1999). Two criteria are required for successful SCAM. First, the substituted Cys must be solvent accessible and, second, it must participate in a receptor function that is affected by modification. Both of these criteria apply to the GABAA receptor α1 subunit β2 strand loop D residue at position 64. This residue is solvent accessible and its modification by MTS reagents led to a reduction in GABA‐evoked current amplitudes in receptors harbouring the α1(F64C) substitution (Boileau et al. 1999). Phe64 participates in GABA binding and efficacy (Boileau et al. 1999; Szczot et al. 2014). Although residues in the β1 strand do not participate in agonist binding to the GABAA β3 homopentamer structure, we examined whether they are accessible through the binding pocket (Miller and Aricescu, 2014). We performed MD simulations of the whole protein but focused our analysis on residues located within the β1 strand of the β3 homopentamer (Fig. 1 A).
Figure 1. Amino acids implicated in agonist binding to GABAA and GluCl receptors in β1 and β2 strands, respectively .
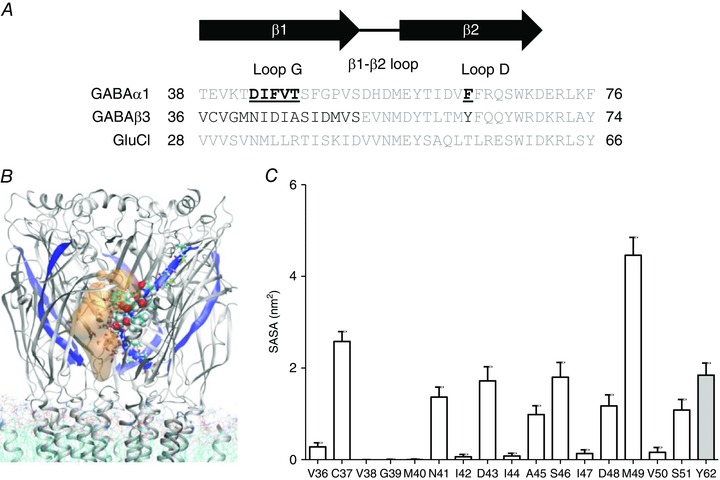
A, amino acid sequence alignment of the mouse GABAA α1 and β3 subunits, and the C. elegans GluCl α subunit. The region shown in the sequence alignment contains the residues relevant to the present study. Residues in the GABAA β3 subunit evaluated using MD simulations are shown in black. The residues in the β1 strand of the α1 subunit mutated to Cys are bold and underlined. B, graphical representation of the solvent accessible area of the GABAA β3 homopentamer from a representative snapshot. The β1 strand is shown in blue in all five subunits, residues of particular interest in one subunit are displayed as van der Waals spheres, and water molecules within a distance of 4.5 Å of these residues are shown as sticks. The solvent accessible crevice near the β1 strand is displayed in orange (transparent). C, SASA of β1 strand residues and Tyr62 (equivalent to α1 subunit Phe64) in the GABAA β3 homopentamer. The SASA of the homologous residues chosen for functional analysis are (mean ± SD): 1.34 ± 0.2 nm2 for Asn41, 0.05 ± 0.04 nm2 for Ile42, 1.85 ± 0.26 nm2 for Asp43, 0.07 ± 0.06 nm2 for Ile44 and 1.07 ± 0.18 nm2 for Ala45 in the β1 strand. Tyr62 in loop D of the β2 strand had a SASA of 1.93 ± 0.2 nm2.
Our simulations reveal that a number of residues in the β1 strand of the GABAA β3 homopentamer are solvent accessible. A representative snapshot of the access pathways of water to the agonist binding site observed in the simulations is shown in Fig. 1 B. Consistent with previous reports, the critical β2 strand loop D residue, equivalent to 64 in the α1 subunit, which is Tyr62 in the β3 homopentamer, is also accessible. We quantified the SASA for these residues (Fig. 1 C). The SASA values for residues from 41 to 45 are characteristic of a β‐strand structure with an alternating pattern of accessibility. While residues Asn41, Asp43 and Ala45 display significant solvent accessibility, Ile42 and Ile44 are inaccessible. The number of water molecules observed to be in contact with the residues throughout our simulations agrees well with their SASA (data not shown). Other residues (e.g. Cys37, Ser46, Asp48, Met49 and Ser51) also have significant SASA values and may therefore be accessible. However, inspection of the model reveals that the highly accessible Cys37 and Met49 are located outside the binding pocket at the surface of the receptor and within the outer vestibule, respectively. Furthermore, in the β3 homopentamer model, the β1 strand has a sharp kink after residue 45, dipping towards the vestibule.
β1 strand residues influence the apparent potency of GABA
Our MD simulations (Fig. 1) suggest that several residues within the β1 strand of GABAA receptors are part of a solvent accessible pocket contiguous with the ligand‐binding domain. We investigated the functional consequences of individual Cys substitutions from Asp43 to Thr47 in the α1 subunit. These residues are homologous to those between Asn41 and Ala45 in the GABAA β3 homopentamer according to our sequence alignment (Fig. 1 A). For comparison, we also included Cys‐substituted Phe64 in loop D of the β2 strand in our analysis. WT or Cys‐substituted α1 subunits were transiently expressed in HEK‐293 cells with β2 and γ2 subunits. Representative examples of GABA‐evoked currents mediated by Cys‐substituted GABAA receptors are shown in Fig. 2 A. GABA concentration–response relationships are plotted in Fig. 2 B. Logistic functions fitted to the data reveal rightward shifts in the GABA concentration–response relationship for some Cys‐substituted receptors. The average best‐fit parameters derived from logistic functions fitted to data recorded from several different cells are summarized in Table 1. Consistent with previous studies, α1(F64C)β2γ2 receptors displayed a large increase in EC50 (P < 0.0001; t test vs. WT α1β2γ2 receptors), confirming an important role for this β2 strand residue in GABAA receptor function. For β1 strand Cys‐substituted α1β2γ2 receptors, one‐way ANOVA with a Dunnet's comparison revealed a significant difference in GABA EC50 between α1(D43C)β2γ2 and WT α1β2γ2 receptors (P < 0.0001) (Table 1). α1(I44C)β2γ2 and α1(T47C)β2γ2 receptors showed tendencies towards increased EC50 values, although these were not significantly different from WT. α1(V46C)β2γ2 receptors had EC50 values indistinguishable from WT α1β2γ2 receptors. The Hill slope values for these Cys‐substituted GABAA receptors were similar to that of WT α1β2γ2 receptors (Table 1). Similar to α1(F64C)β2γ2, both α1(D43C)β2γ2 and α1(T47C)β2γ2 receptors had reduced GABA‐evoked peak current densities compared to WT, which were statistically significant (P < 0.0001 and P < 0.05, respectively; one‐way ANOVA with post hoc Dunnet's comparison with α1β2γ2 receptors) (Table 1). The Cys substitution in α1(F45C)β2γ2 receptors produced more complicated changes in function, which are described below.
Figure 2. β1 strand residues influence the apparent potency of GABA .
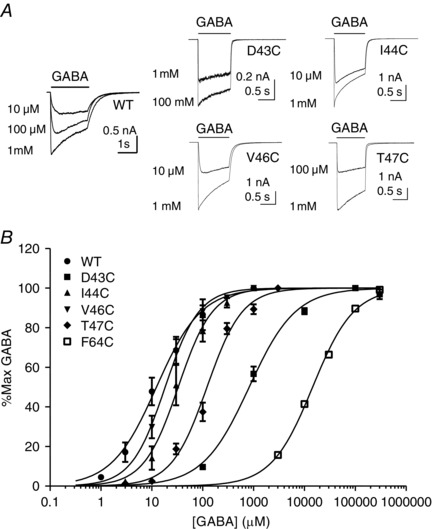
A, representative examples of whole‐cell currents evoked by different concentrations of GABA (indicated) mediated by WT or Cys‐substituted α1β2γ2 receptors. The bar indicates GABA application. B, concentration–response relationships for WT or Cys‐substituted α1β2γ2 GABAA receptors. Current amplitudes are expressed as a percentage of the maximum current amplitude recorded from each cell. Each point represents the mean ± SEM of at least four recordings. The sigmoidal curve represents the logistic function fitted to the data points. Mean parameters and statistical comparisons from the logistic function are summarized in Table 1.
Table 1.
Summary of logistic function fit parameters and current density values
| GABA EC50 (μm) | Hill slope | Current density (pA pF−1) | |
|---|---|---|---|
| α1β2γ2 | 17.8 ± 5.1 (6) | 1.12 ± 0.17 (6) | 1400 ± 250 (20) |
| α1(D43C)β2γ2 | 857 ± 161 (5)* | 1.01 ± 0.059 (5) | 210 ± 69 (8)* |
| α1(I44C)β2γ2 | 38.4 ± 11.7 (5) | 1.68 ± 0.15 (5) | 1200 ± 330 (7) |
| α1(F45C)β2γ2 | HP 0.52 (16) | HP 1.91 (16) | 880 ± 130 (27) |
| LP 150 (16) | LP 0.51 (16) | ||
| α1(V46C)β2γ2 | 19.0 ± 4.1 (4) | 1.48 ± 0.13 (4) | 890 ± 230 (7) |
| α1(T47C)β2γ2 | 135 ± 13.7 (9) | 1.42 ± 0.20 (9) | 610 ± 110 (12)* |
| α1(F64C)β2γ2 | 19000 ± 1600 (3)* | 1.00 ± 0.29 (3) | 77.9 ± 33.3 (13)* |
Data are provided as the mean ± SEM of (n) number of experiments, except EC50 and Hill slope values of α1(F45C)β2γ2 receptors, where the average fit parameters of the high apparent potency (HP) and low apparent potency (LP) components are reported. *Significant difference (P < 0.05) from one‐way ANOVA with Dunnet's post hoc comparison with α1β2γ2 values. α1(F45C)β2γ2 receptors were not statistically compared for EC50 and Hill slope values because of the presence of two components in the GABA concentration–response relationship. †Significant difference (P < 0.05) between α1(F64C)β2γ2 receptors and α1β2γ2 receptors from t test.
The α1 F45C substitution causes a biphasic GABA concentration–response relationship and spontaneous gating
The GABA concentration–response relationship for α1(F45C)β2γ2 receptors is shown in Fig. 3 A, along with representative examples for GABA‐evoked currents. A single component logistic function provided an inadequate representation of the concentration–response relationship for GABA‐evoked currents mediated by α1(F45C)β2γ2 receptors. By contrast, a two‐component logistic function provided a good fit across the entire GABA concentration range (Fig. 3 A, solid black line). An F test applied to statistically discriminate between the two approaches revealed that the two‐component logistic function provided a significantly improved fit to the data (P = 0.025). The solid grey lines in Fig. 3 A illustrate the two components of the GABA concentration–response relationship. The logistic fit for WT α1β2γ2 receptors is also reproduced here (dashed grey line) for comparison. Table 1 contains the values derived from the two‐component logistic function consistent with high and low apparent potency components for GABA‐evoked activation of α1(F45C)β2γ2 receptors.
Figure 3. α1(F45C)β2γ2 GABAA receptors display a biphasic GABA concentration–response relationship and are more spontaneously active .
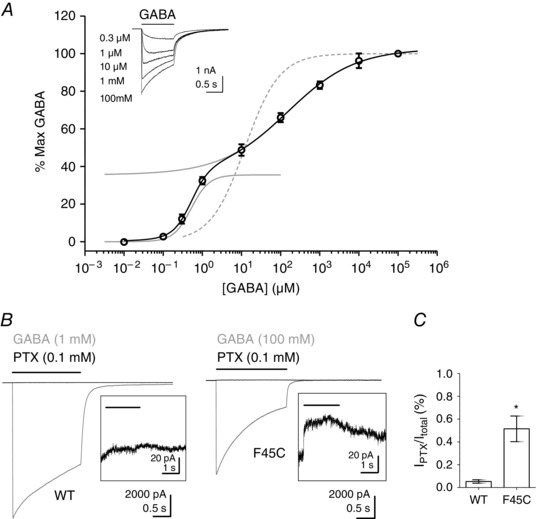
A, concentration–response relationship for α1(F45C)β2γ2 GABAA receptors. Current amplitudes are expressed as a percentage of maximum. Each point represents the mean ± SEM of at least four recordings. The solid black line shows the two component logistic function fit to the data points. The two solid grey lines show the separate components of the logistic function fit. The fit parameters are summarized in Table 1. The logistic function fit for WT α1β2γ2 receptors is also reproduced for comparison (grey dotted line). Inset shows a representative example of whole‐cell currents evoked by different concentrations of GABA (indicated) mediated by α1(F45C)β2γ2 GABAA receptors. The bar indicates GABA application. B, representative examples of whole‐cell currents evoked by a maximally efficacious concentration of GABA (grey traces) or inhibition of standing current by PTX (black trace) at α1β2γ2 or α1(F45C)β2γ2 receptors. The black bar indicates GABA or PTX application. The inset shows, on an expanded scale, PTX inhibition of standing current. C, graph showing mean ± SEM PTX inhibited standing current (I PTX) expressed as a percentage of the total current (I total) for α1β2γ2 (n = 5) and α1(F45C)β2γ2 (n = 10) GABAA receptors. The difference in mean PTX component was significantly higher at α1(F45C)β2γ2 receptors (* P = 0.014; t test).
GABAA receptors with high levels of spontaneous gating generally have a higher sensitivity to GABA (Mortensen et al. 2003; Hadley & Amin, 2007). The high apparent potency component of GABA activation of α1(F45C)β2γ2 receptors may therefore be associated with spontaneous activity. We investigated the possibility of spontaneous gating by recording the inhibition of basal currents by picrotoxin (PTX) in WT α1β2γ2 and α1(F45C)β2γ2 receptors. Figure 3 B shows representative examples of maximally effective GABA‐evoked currents (grey trace) and inhibition of basal currents by PTX (black trace). WT α1β2γ2 receptors display very little PTX inhibited basal current (Fig. 3 B inset). By comparison, the magnitude of PTX inhibited basal current in α1(F45C)β2γ2 receptors was large (Fig. 3 B, inset). We quantified spontaneous current as the magnitude of the PTX component (I PTX) expressed as a percentage of the total amount of current (I GABA + I PTX). The maximum GABA‐evoked current densities in WT α1β2γ2 and α1(F45C)β2γ2 receptors did not differ significantly (Table 1). Therefore, the maximum efficacy of GABA is probably not altered by the F45C substitution. The mean I PTX/I total values are shown in Fig. 3 C. α1(F45C)β2γ2 receptors had an increased proportion of spontaneous current from 0.05 ± 0.02% (n = 5) in WT α1β2γ2 receptors to 0.52 ± 0.11% (n = 10). The difference was statistically significant (P = 0.014; t test). These data suggest that F45C substitution increased spontaneous gating and therefore Phe45 in the β1 strand plays critical and complex roles in GABAA receptor function.
Several residues in the β1 strand of the GABAA α1 subunit are accessible to MTSEA modification
β1 strand residues in the GluCl α subunit (Hibbs & Gouaux, 2011) and GABAA β3 homopentamer (Miller & Aricescu, 2014) models have side chains facing into the orthosteric binding pocket. Our MD simulation data demonstrate that residues in the GABAA β3 homopentamer, equivalent to amino acids 43–47 in the α1 subunit, are solvent accessible (Fig. 1 C). Furthermore, receptors containing α1 subunit D43C, F45C and T47C substituents all exhibit functions distinguishable from WT receptors (Figs 2 and 3 and Table 1). Therefore, α1 subunit β1 strand residues may be accessible to MTS reagents and their modification may alter function.
We used SCAM to investigate whether the substituted cysteine residues in the β1 strand of the GABAA receptor are accessible to the sulfhydryl reagent, MTSEA. The use of a similar approach previously in Xenopus oocytes demonstrated the accessibility of substituted cysteines in α1(F64C) containing GABAA receptors (Boileau et al. 1999; Holden & Czajkowski, 2002). We adapted the technique for use in whole‐cell patch clamp recordings from HEK‐293 cells using rapid application of GABA and MTSEA (see Methods), and used α1(F64C)β2γ2 receptors for comparison.
We first tested whether MTSEA (10 mm) modulates GABAA receptors containing WT α1 subunits (Fig. 4 A). Control currents were evoked by an EC50 concentration of GABA (10 μm) in HEK‐293 cells expressing α1β2γ2 GABAA receptors. Modification by MTSEA (10 mm) was tested by pre‐application for 2 s prior to GABA. Five applications were tested, such that cells were exposed to MTSEA cumulatively for 10 s. The amplitudes of GABA‐evoked currents appeared modestly enhanced following MTSEA treatment in some cells, although the mean did not significantly differ from that of controls (n = 6). We next confirmed that MTSEA (2 mm) modifies the function of α1(F64C)β2γ2 receptors. MTSEA substantially reduced the current amplitude evoked by an EC50 concentration of GABA (10 mm) (Fig. 4 A). The application of the reducing agent DTT (10 mm) reversed the modification by MTSEA. This result is consistent with the results of a previous study of the effects of SCAM on GABAA receptors containing the α1(F64C) subunit (Boileau et al. 1999).
Figure 4. Accessibility of Cys‐substituted β1 strand residues to sulfhydryl modification .
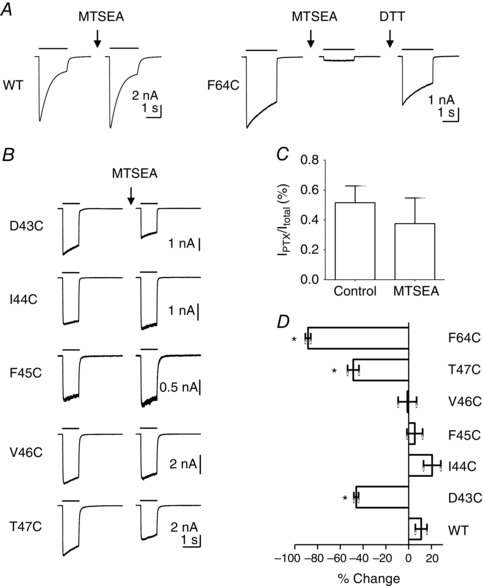
A, representative examples of the effect of MTSEA on WT α1β2γ2 and α1(F64C)β2γ2 receptors. MTSEA (10 mm) was applied to a cell expressing WT α1β2γ2 receptors prior to recording a current evoked by the EC50 concentration (10 μm, black bar) of GABA. Also shown is the effect of MTSEA (2 mm) on α1(F64C)β2γ2 receptors. MTSEA substantially reduced the amplitude of the EC50 (10 mm) GABA‐evoked (black bar) current through α1(F64C)β2γ2 receptors. Modification was reversed following application of DTT (10 mm). B, representative examples of MTSEA (2 mm) on β1 strand Cys‐substituted α1β2γ2 receptors. MTSEA (arrow) reduced the current amplitude of α1(D43C)β2γ2 and α1(T47C)β2γ2 receptors. C, effect of MTSEA (2 mm) on spontaneous activity of α1(F45C)β2γ2 receptors. Spontaneous activity was quantified as the I PTX/I total. The level of spontaneous activity under control conditions was 0.51 ± 0.11% (n = 10). In the presence of MTSEA (2 mm), the level of spontaneous activity was 0.38 ± 0.17% (n = 5). The difference was not statistically significant. (P > 0.05; t test). D, graph showing mean ± SEM percentage change in current amplitude following MTSEA treatment in WT and mutant α1β2γ2 receptors. The change in GABA‐evoked current amplitude was 11 ± 5.1% (n = 6) for WT α1β2γ2, –46 ± 2.1% (n = 8) for α1(D43C)β2γ2, 21 ± 7.5% (n = 3) for α1(I44C)β2γ2, 5.4 ± 6.9% (n = 4) for α1(F45C)β2γ2, –1.1 ± 8.1% (n = 3) for α1(V46C)β2γ2, –49 ± 5.0% (n = 15) for α1(T47C)β2γ2 and –88 ± 2.4% (n = 4) for α1(F64C)β2γ2 receptors. There was a statistically significant difference in the percentage change between α1(D43C)β2γ2, α1(T47C)β2γ2 and α1(F64C)β2γ2 receptors compared to WT α1β2γ2, receptors (* P < 0.0001; one‐way ANOVA; post hoc Dunnet's comparison vs. WT).
Using the same approach, we applied MTSEA (10 mm) to GABAA receptors containing Cys‐substituted β1 strand residues. Exemplar currents evoked by an EC50 concentration of GABA before and after MTSEA application are shown in Fig. 4 B. MTSEA application caused a reduction in GABA‐evoked current amplitude in α1(D43C)β2γ2 and α1(T47C)β2γ2 receptors. MTSEA did not affect the amplitude of α1(I44C)β2γ2, α1(F45C)β2γ2 or α1(V46C)β2γ2 receptors. The equivalent residues to Cys44 and Cys46 in the GABAA β3 homopentamer showed low SASA values and the failure of MTSEA to affect function is consistent with their lack of solvent accessibility (Fig. 1 C).
It is interesting that α1(F45C)β2γ2 receptors were unaffected by MTSEA despite a high SASA (Fig. 1 C) and the importance of the identity of this residue in receptor function (Fig. 3). It is possible that modification by MTSEA, which would add a positive charge to the Cys residue, was functionally silent in these receptors. We therefore used a different sulfhydryl modifying reagent, PhMTS, which adds an aromatic group to an accessible Cys, a modification that would mimic the native Phe. However, PhMTS (200 μm) applied for more than 2 min also had no effect on the function of α1(F45C)β2γ2 receptors (data not shown). In addition, we repeated the MTSEA experiment on α1(F45C)β2γ2 receptors using 1 mm GABA as the test concentration. The effect of 1 mm GABA was maximal at WT α1β2γ2 receptors (Fig. 2 C), whereas this concentration was only 80% effective at α1(F45C)β2γ2 receptors (Fig. 3 A). However, MTSEA had no effect on the amplitude of GABA (1 mm)‐evoked currents (data not shown). In addition to an altered GABA concentration–response relationship, α1(F45C)β2γ2 receptors displayed higher spontaneous activity compared to WT α1β2γ2 receptors. We therefore examined whether MTSEA could influence the extent of spontaneous activity mediated by α1(F45C)β2γ2 receptors. Cells were treated with MTSEA (2 mm) for 2 min and spontaneous activity was measured as the percentage of I PTX to I total. Figure 4 C shows I PTX/I total under control conditions and in the presence of MTSEA (2 mm). There was no significant difference in the level of spontaneous activity in the presence of MTSEA. Taken together, these data suggest that, although Cys43 and Cys47 are accessible to MTSEA, Cys45 is inaccessible.
We quantified the extent of MTSEA modification determining the percentage changes in EC50 GABA‐evoked current amplitude for each Cys‐substituted receptor before and after MTSEA application. These values are plotted in Fig. 4 D. Application of MTSEA caused a significant reduction in the amplitudes of GABA‐evoked currents mediated by α1(D43C)β2γ2, α1(T47C)β2γ2 and α1(F64C)β2γ2 receptors relative to those mediated by WT receptors (P < 0.0001; one‐way ANOVA; post hoc Dunnet's comparison vs. α1β2γ2).
Receptor activation influences MTSEA modification of D43C and T47C containing receptors
Receptor activation by an agonist might influence the accessibility of a substituted Cys. This could occur as a result of the bound agonist directly protecting the residue from modification. Alternatively, activation may cause a conformational rearrangement of the receptor in which the Cys becomes less accessible. The α1(F64C) was modified more slowly by an MTS reagent applied with GABA to Xenopus oocytes expressing α1(F64C)β2γ2 receptors (Boileau et al. 1999). We investigated whether GABA also affects the modification of α1(D43C)β2γ2 and α1(T47C)β2γ2 receptors by applying MTSEA in its presence and absence. Once again, we modified the approach for use in voltage clamped HEK‐293 cells (see Methods).
First, using this approach, we compared the rate of MTSEA modification of α1(D43C)β2γ2, α1(T47C)β2γ2 and α1(F64C)β2γ2 receptors. Figure 5 A shows representative examples of the effect of rapid and short applications of MTSEA on GABA‐evoked currents mediated by α1(D43C)β2γ2, α1(T47C)β2γ2 and α1(F64C)β2γ2 receptors. The concentration of MTSEA used for each mutant was chosen empirically on the basis of a measurable reduction in the amplitude of the EC50 GABA‐evoked current during the time course of the experiment. For α1(D43C)β2γ2 and α1(F64C)β2γ2 receptors, MTSEA (100 μm and 10 μm, respectively) was applied episodically for 100 ms, prior to application of a test concentration of GABA (EC50). For both receptors, the modification was complete within 500 ms. The rate of MTSEA (100 μm) modification of α1(T47C)β2γ2 receptors was slower and therefore MTSEA was applied episodically for 500 ms prior to the application of GABA (EC50). We analysed the change in EC50 GABA‐evoked current amplitude and expressed these as a percentage of the control current. Figure 5 B shows the rate of MTSEA evoked reduction of GABA‐evoked currents mediated by α1(D43C)β2γ2, α1(T47C)β2γ2 and α1(F64C)β2γ2 receptors. In all cases, the time course of MTSEA modification was well fitted with a single exponential. The mean ± SEM time constants of modification are plotted in Figure 5 C. One‐way ANOVA with a post hoc Tukey's comparison of the time constants revealed that both α1(D43C)β2γ2 and α1(F64C)β2γ2 receptors exhibited significantly smaller modification time constants compared to that of α1(T47C)β2γ2 receptors (P < 0.0001). We also determined the second‐order rate constants for MTSEA modification (see Methods). Second‐order rate constants are summarized in Table 2. The second‐order rate constants for α1(F64C)β2γ2 receptors were significantly faster than those of α1(D43C)β2γ2 and α1(T47C)β2γ2 receptors (P < 0.0001; one‐way ANOVA; post hoc Tukey's comparison) (Table 2). Our second‐order rate constant for MTSEA modification, derived from HEK cells expressing α1(F64C)β2γ2 receptors, is similar to that reported by Holden and Czajkowski (2002) in oocytes. The plateau of modification for α1(D43C)β2γ2, α1(T47C)β2γ2 and α1(F64C)β2γ2 receptors, which indicates the maximal extent of functional change for the concentration of MTSEA tested, did not differ from those shown in Figure 4 D, which used a high concentration of MTSEA (2 mm). This demonstrates that modification was complete. Our data suggest that Cys64 and Cys43 are more accessible to modification than Cys47.
Figure 5. Rate of MTSEA modification of Cys64, Cys43 and Cys47 .
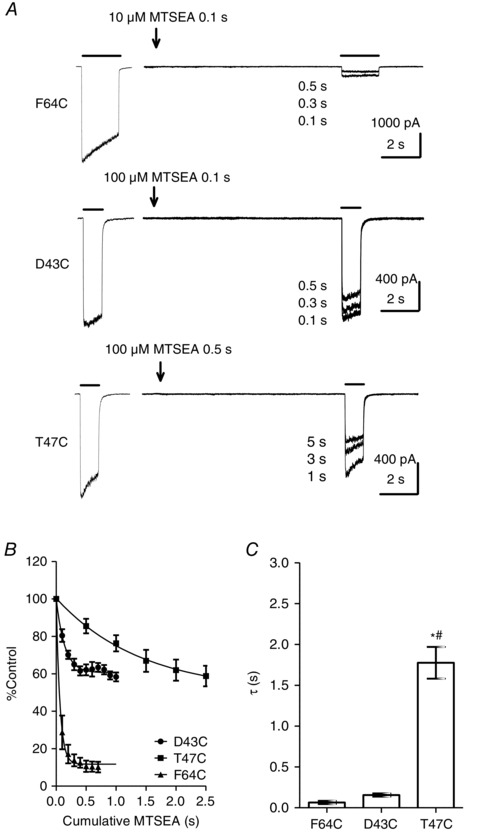
A, representative examples of EC50 GABA‐evoked currents before and after cumulative MTSEA application at α1(F64C)β2γ2, α1(D43C)β2γ2 and α1(T47C)β2γ2 receptors. The first current in each trace indicates the steady‐state EC50 GABA‐evoked current amplitude under control conditions. MTSEA was applied for the indicated time (arrow) and a second EC50 GABA‐evoked current was recorded. Cumulative application of MTSEA led to a progressive reduction in EC50 GABA‐evoked current amplitudes. B, rate of MTSEA modification of α1(F64C)β2γ2 (n = 4), α1(D43C)β2γ2 (n = 6) and α1(T47C)β2γ2 (n = 8) receptors. The line shows the exponential function fitted to the data points. C, graph showing mean ± SEM τ of MTSEA modification of α1(F64C)β2γ2 (0.065 ± 0.020 s; n = 4), α1(D43C)β2γ2 (0.16 ± 0.020 s; n = 6) and α1(T47C)β2γ2 (1.8 ± 0.19 s; n = 8) receptors. The τ of MTSEA modification was significantly larger for α1(T47C)β2γ2 receptors compared to α1(D43C)β2γ2 and α1(F64C)β2γ2 receptors (* # P < 0.0001; one‐way ANOVA; post hoc Tukey's comparison).
Table 2.
Summary of second‐order rate constants for MTSEA modification
| MTSEA k2 (m −1 s−1) | MTSEA + GABA k2 (m −1 s−1) | MTSEA + propofol k2 (m −1 s−1) | |
|---|---|---|---|
| α1(D43C)β2γ2 | 70,030 ± 9,629 (6) | 34,530 ± 10,420 (6)* | 38,860 ± 8,437 (7) |
| α1(T47C)β2γ2 | 6,216 ± 784.8 (8) | 3,892 ± 873.6 (5) | 6,049 ± 1,271 (6) |
| α1(F64C)β2γ2 | 1,944,000 ± 460,500 (4)† | 24,660 ± 9,129 (4)* | 1,832,000 ± 205,400 (5) |
Data are provided as the mean ± SEM of (n) number of experiments. The second‐order rate constants are calculated by dividing the rate of decay, obtained from the exponential fits to data that generated the rate of MTSEA modification data, by the concentration of MTSEA used. The concentration of MTSEA used was 100 μm for α1(D43C)β2γ2 and α1(T47C)β2γ2 receptors, and 10 μm for α1(F64C)β2γ2 receptors. *Significant difference (P < 0.05) from one‐way ANOVA with post hoc Tukey's comparison between the presence or absence of saturating concentrations of GABA or activating concentrations of propofol. †Significant difference (P < 0.05) from one‐way ANOVA with post hoc Tukey's comparison between MTSEA k2 of different mutants.
We next repeated the experiment in the presence of either a maximal concentration of GABA or an activating concentration of propofol (10 μm), applied simultaneously with MTSEA. In all cases [α1(F64C)β2γ2, α1(D43C)β2γ2 and α1(T47C)β2γ2 receptors], the co‐application of either GABA or propofol with MTSEA induced inward currents. Figure 6 A shows the rate of MTSEA (10 μm) modification of α1(F64C)β2γ2 receptors in the presence of either GABA (300 mm) or propofol (10 μm). The rate of modification was again measured by episodic (100 ms) application of MTSEA (10 μm) with GABA (300 mm) or propofol (10 μm) for the first 500 ms. Propofol did not affect the rate of MTSEA modification (Fig. 6 A, inset). In the presence of GABA (300 mm), however, the extent of modification following 500 ms of MTSEA exposure was less than in its absence (Fig. 6 A, inset). We therefore increased the duration of each episodic application to 2 s. The rate of modification in the presence of GABA was best fitted with a double exponential (Fig. 6 A). The τw was calculated for modification when MTSEA was applied simultaneously with GABA and compared with the τ for modification in the absence of GABA. The mean ± SEM time constants are plotted in Fig. 6 B. The second‐order rate constants derived from the exponential fits are summarized in Table 2. GABA (300 mm), but not propofol (10 μm), significantly slowed the rate of MTSEA (10 μm) modification of α1(F64C)β2γ2 receptors (P = 0.0004; one‐way ANOVA; post hoc Tukey's comparison). Our data are consistent with previous observations indicating that GABA, but not the allosteric agonist pentobarbital, influences the modification of α1(F64C) containing GABAA receptors (Boileau et al. 1999).
Figure 6. Effect of GABA and propofol on MTSEA modification of Cys‐substituted receptors .
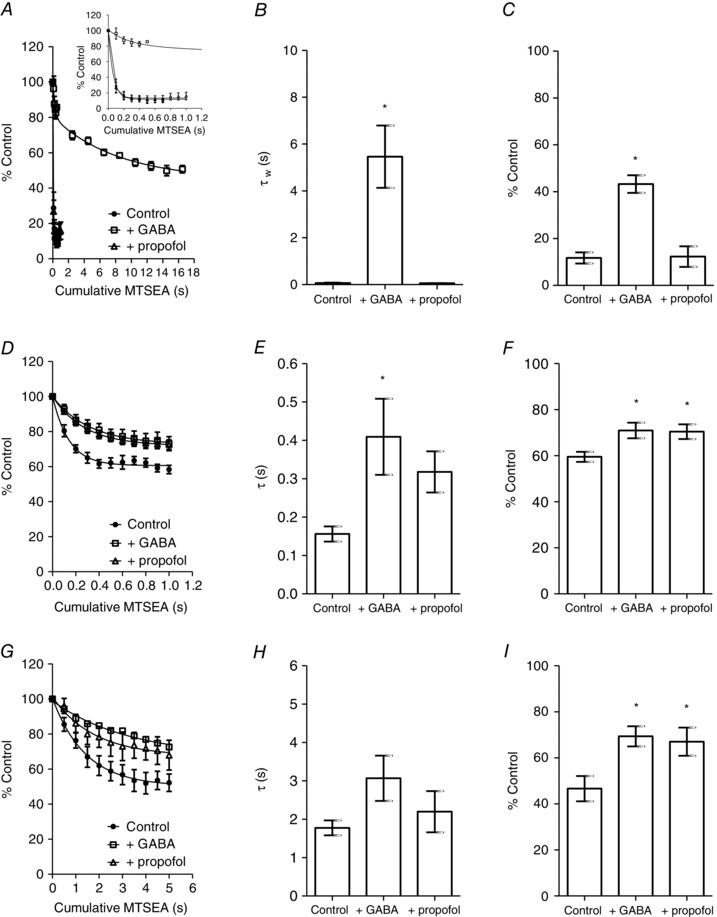
A, rate of MTSEA (10 μm) modification of α1(F64C)β2γ2 receptors in the control (solid circles) and in the presence of GABA (300 mm; open squares) or propofol (10 μm; open triangles). The line shows the exponential function fitted to the data points. Inset in (A) shows, on an expanded time scale, the difference in rate of MTSEA modification in the presence or absence of agonists. B, graph showing mean ± SEM τ MTSEA modification of α1(F64C)β2γ2 receptors. The τ of MTSEA modification in control, GABA and propofol was 0.065 ± 0.02 s (n = 4), 5.5 ± 1.3 s (n = 4) and 0.057 ± 0.001 s (n = 5). The presence of GABA significantly increased the τ of MTSEA modification (* P < 0.001; one‐way ANOVA; post hoc Tukey's comparison). C, graph showing mean ± SEM extent of MTSEA modification of α1(F64C)β2γ2 receptors. The extent of MTSEA modification in control, GABA and propofol was 12 ± 2.4% (n = 4), 43 ± 3.8% (n = 4) and 12 ± 4.4 s (n = 5). The presence of GABA significantly reduced the extent of MTSEA modification (* P < 0.0001; one‐way ANOVA; post hoc Tukey's comparison). D, as in (A), the rate of MTSEA (100 μm) modification of α1(D43C)β2γ2 receptors. E, mean ± SEM τ MTSEA modification of α1(D43C)β2γ2 receptors. The τ of MTSEA modification in control, GABA (300 mm) and propofol (10 μm) was 0.16 ± 0.02 s (n = 6), 0.41 ± 0.10 s (n = 6) and 0.32 ± 0.054 s (n = 7). The presence of GABA significantly increased the τ of MTSEA modification (* P < 0.05; one‐way ANOVA; post hoc Tukey's comparison). F, mean ± SEM extent of MTSEA modification of α1(D43C)β2γ2 receptors. The extent of MTSEA modification in control, GABA and propofol was 60 ± 2.2% (n = 6), 71 ± 3.4% (n = 6) and 70 ± 3.2% (n = 7). The presence of GABA and propofol significantly reduced the extent of MTSEA modification (* P < 0.05; one‐way ANOVA; post hoc Tukey's comparison). G, as in (A), the rate of MTSEA (100 μm) modification of α1(T47C)β2γ2 receptors. H, mean ± SEM τ MTSEA modification of α1(T47C)β2γ2 receptors. The τ of MTSEA modification in control, GABA (30 mm) and propofol (10 μm) was 1.8 ± 0.19 s (n = 8), 3.1 ± 0.59 s (n = 5) and 2.2 ± 0.54 s (n = 6). There was no significant difference in the τ of modification (P = 0.13; one‐way ANOVA). I, mean ± SEM extent of MTSEA modification of α1(T47C)β2γ2 receptors. The extent of MTSEA modification in control, GABA and propofol was 47 ± 5.5% (n = 8), 69 ± 4.4% (n = 5) and 67 ± 6.1% (n = 6). The presence of GABA and propofol significantly reduced the extent of MTSEA modification (* P < 0.05; one‐way ANOVA; post hoc Tukey's comparison).
Using the same approach, we determined the effect of GABA and propofol on MTSEA (100 μm) modification of α1(D43C)β2γ2 and α1(T47C)β2γ2 receptors. The concentrations of GABA used were 300 mm and 30 mm, respectively, and propofol at a concentration of 10 μm was used for both receptors. The time course for MTSEA modification in the control, in the presence of GABA and in the presence of propofol for α1(D43C)β2γ2 receptors is shown in Fig. 6 D. MTSEA in the presence or absence of agonists was applied cumulatively for 100 ms. The rates of modification in all three conditions were well fitted with single exponential functions (Fig. 6 D). The mean ± SEM time constants and the extent of functional change are plotted in Fig. 6 E and 6 F, respectively. Second‐order rate constants are summarized in Table 2. In the presence of GABA (300 mm), there was a significant increase in the τ of MTSEA modification (P < 0.05; one‐way ANOVA; post hoc Tukey's comparison vs. control). There was a tendency for the τ of modification to increase in the presence of propofol, although this was not statistically significant. However, there was also no significant difference in the τ of modification in the presence of GABA and in the presence of propofol. Furthermore, comparison of the extent of functional change revealed a statistically significant reduction between that in the presence of GABA and control (P < 0.05; one‐way ANOVA; post hoc Tukey's comparison vs. control), as well as that in the presence of propofol and control (P < 0.05; one‐way ANOVA; post hoc Tukey's comparison vs. control) (Fig. 6 F). These data therefore suggest that both GABA and propofol influence MTSEA modification of Cys43. This is in contrast to Cys64 in loop D where the allosteric agonist propofol did not influence MTSEA modification (Fig. 6 A–C).
Figure 6 G shows the time course of MTSEA (100 μm) modification of α1(T47C)β2γ2 receptors in the control, in the presence of GABA (30 mm) and in the presence of propofol (10 μm). MTSEA in the presence or absence of agonists was applied cumulatively for 500 ms. The time course for modification in each case was well fitted with a single exponential function. The mean ± SEM τ of modification and the extent of functional change are plotted in Figure 6 H and I, respectively. Second‐order rate constants are summarized in Table 2. GABA (30 mm) and propofol (10 μm) did not significantly alter the τ of MTSEA modification of α1(T47C)β2γ2 receptors. However, both GABA and propofol significantly reduced the extent of MTSEA‐mediated inhibition (P < 0.05; one‐way ANOVA; post hoc Tukey's comparison vs. control) (Fig. 6 I).
Discussion
In the present study, we investigated the role of the α1 subunit β1 strand in GABAA receptor function. Guided by MD simulation data on the SASA of equivalent residues of the GABAA β3 homopentamer, we mutated five continuous residues on the β1 strand of the GABAA α1 subunit to Cys. Whole‐cell voltage clamp electrophysiology revealed that D43C, F45C and T47C substitutions caused changes in the GABA concentration–response relationship, demonstrating for the first time that the β1 strand residues play a role in mammalian pLGIC function.
The β1 strand runs adjacent and parallel to the critical β2 strand, which contains loop D. A crucial Phe (Phe64) in the β2 strand of the GABAA receptor α1 subunit plays a critical role in both GABA binding and the conformational transition that leads to orthosteric gating of the channel (Boileau et al. 1999; Szczot et al. 2014). These effects also manifest as a large rightward shift in the apparent potency for GABA. The agonist concentration–response relationship (and therefore apparent potencies) are composites of binding and gating events, and both can influence the concentration–response relationship (Colquhoun, 1998). Indeed, α1 subunit Phe64 in the β2 strand, which causes large rightward shifts in the apparent potency of GABA, when Cys‐substituted, is an example of a residue with a dual action in binding and gating efficacy (Boileau et al. 1999; Szczot et al. 2014). Substituted Cys64 in the GABAA receptor α1 subunit was accessible to sulfydryl modification when expressed in Xenopus oocytes (Boileau et al. 1999; Holden & Czajkowski, 2002). Using fast applications of MTSEA, we showed that the same substitution in α1β2γ2 receptors expressed in HEK‐293 cells was modified with a similar second‐order rate constant. Furthermore, as noted previously, the presence of GABA slows the rate of Cys64 modification, whereas the allosteric agonist propofol had no effect. This is in contrast to α1(D43C)β2γ2 and α1(T47C)β2γ2 receptors, where both GABA and propofol affect MTSEA‐mediated modification of the substituted Cys, suggesting that β1 strand residues play a role different from that of Phe64 in the β2 strand in terms of GABAA receptor activation.
Changes in Cys accessibility during receptor activation can be a result of either direct or indirect effects of agonists. Direct agonist effects probably arise from hindrance of the substituted Cys by the bound agonist (protection), whereas indirect effects probably arise from conformational changes induced by the agonist that alter the position of the substituted Cys, making it less accessible. Our data show that an activating concentration of propofol impairs the accessibility of both Cys43 and Cys47 in a manner similar to a maximally effective concentration of GABA. This is consistent with a conformational change associated with gating resulting in reduced accessibility of positions 43 and 47. This also suggests that the structural rearrangements in the β1 strand of the GABAA receptor α1 subunit are similar, irrespective of agonist binding in the vicinity or far away from the orthosteric binding site. A conformational change in this region makes sense given the anti‐parallel location of the β1 relative to the β2 strand, together constituting a hairpin structure that participates in the transduction of binding to GABAA channel opening. Indeed, Kash et al. (2003) implicated an interaction between the loop connecting the β1 and β2 strands of the α1 subunit, and the TM2–3 loop during GABAA receptor activation. Furthermore, we have previously demonstrated that substitution of the conserved α1 subunit TM2‐3 Lys278 with methionine reduces the efficacy of activation by GABA and propofol (Hales et al. 2006). The same mutation also reduced the level of spontaneous gating of α1(K278M)β2γ2 GABAA receptors consistent with a global reduction in gating (Othman et al. 2012). Comparison of the open (ivermectin and glutamate bound) and closed (apo) structures of GluCl also reveals movement in this region associated with channel opening (Althoff et al. 2014). During activation, there is a downward displacement of the β1–β2 loop, increasing its proximity to the TM2–3 loop. The agonist bound and apo‐GluCl models also reveal large movements of residues within the β1 and β2 strands, particularly Arg37 in the β1 strand, homologous to Thr47 in the present study. Comparison of the open (glycine bound) and closed (strychnine bound) structures of the zebrafish glycine receptor reveal similar movements of the β1–β2 loop (Du et al. 2015). It is therefore becoming evident that residues along the β1 and β2 strands are well positioned to participate in the transduction of agonist binding to gating.
Although α1(D43C)β2γ2 and α1(T47C)β2γ2 receptors were sensitive to MTSEA modification, α1(I42C)β2γ2, α1(F45C)β2γ2 and α1(V46C)β2γ2 receptors were not. The low SASA values of the residues homologous to those in position 42 and 46 of the GABAA β3 homopentamer (Ile42 and Ile44, respectively) (Fig. 1) suggest that these residues are not solvent exposed but, instead, have their side chains orientated within a hydrophobic environment. However, the homologous position to Cys45 in the GABAA β3 homopentamer (Asp43) is solvent accessible; this contrasts with our SCAM data, which demonstrate that Cys45 is not modifiable by MTSEA or PhMTS. It is possible that the carboxylate group of Asp43 (equivalent to Phe45 in α1) causes an increased hydration of the agonist binding site in the case of the β3 homopentamer, as it has previously been shown that negatively‐charged side chains can promote solvation even in highly hydrophobic regions (Krah et al. 2010; Villinger et al. 2010). The structure of the α1 subunit within the heteromeric α1β2γ2 receptor may also differ significantly from the β3 subunit, altering the environment of α1 residue 45 relative to the equivalent β3 residue 43. The most obvious structural difference between these two subunits in the vicinity of this residue is in loop F, which, together with loop D, sandwich the β1 strand. Loop F has two fewer residues in the α1 subunit compared to the β3 subunit.
Our data also demonstrate that α1(F45C)β2γ2 receptors have a biphasic GABA concentration–response relationship with increased spontaneous gating. The component of the biphasic GABA concentration–response relationship, with the higher apparent potency, may occur as a consequence of increased spontaneous gating observed for α1(F45C)β2γ2 relative to WT receptors. Our finding that α1(F45C)β2γ2 exhibits increased spontaneous gating suggests that residue Phe45 stabilizes the receptor in the inactive conformation.
Mutations that increase spontaneous gating typically cause a left‐shifted concentration–response relationship, where receptors have a higher sensitivity to agonists (Mortensen et al. 2003; Hadley & Amin, 2007). The component of the α1(F45C)β2γ2 concentration–response relationship, with a lower apparent potency, has a shallow Hill coefficient (∼0.5) (Fig. 3 A and Table 1), suggesting that co‐operativity between adjacent GABA binding sites is impaired. It is worth considering how residue 45 in the first occupied orthosteric site may participate in communication with the subsequently occupied binding site. As noted previously, the β1 strand lies between loop D on the β2 strand and loop F. Loop F in GABAA ρ1 homopentamers and α1β2γ2 heteromers has been implicated in co‐operativity of binding and gating, respectively. Site‐directed fluorescence spectroscopy of GABAA ρ1 receptors, implicated structural rearrangements in loop F during orthosteric ligand binding, which were unrelated to gating (Khatri et al. 2009). Interestingly, for GABAA ρ1 receptors, a number of loop F Cys substitutions appeared to reduce the Hill coefficient. Loop F residues in the γ2 subunit are also implicated in transducing benzodiazepine binding events to altered gating (Hanson & Czajkowski, 2008) and it has been postulated that loop F residues play a role in the cooperativity of agonist binding (Khatri & Weiss, 2010). Phe45 in the β1 strand of the GABAA α1 subunit may transduce structural rearrangements within the binding pocket to the adjacent binding domain, via loop F. Therefore, the lack of Phe45 in the α1(F45C)β2γ2 receptor may lead to an uncoupling of binding between the two orthosteric binding sites. In addition, the existence of receptors in either the spontaneously open conformation or in the closed state may provide an explanation for the high and low sensitivity components of the concentration–response relationship.
Residues in the β1 and β2 strands of the Erwinia chrysanthemi ligand‐gated ion channel (Pan et al. 2012) and neuronal α7 ACh receptors (Quiram et al. 2000) have been implicated in the recognition of antagonists. The C. elegans GluCl α subunit homopentamer is the only structure that implicates β1 strand residues in agonist binding and, consequently, this region was postulated to be a seventh ligand‐binding loop, termed loop G (Hibbs & Gouaux, 2011). Arg45 in the C. elegans GluCl α subunit appears to participate in glutamate binding. By contrast, β1 strand residues of the GABAA β3 (Miller & Aricescu, 2014), as well as the 5‐HT3A receptors (Hassaine et al. 2014), lie outside the ligand‐binding site. Emerging structural data suggest that a direct role for β1 strand residues in agonist binding may be restricted to the C. elegans GluCl (Blarre et al. 2014).
Our findings suggest that naturally occurring mutations and polymorphisms affecting the β1 strand may result in functional deficits that could be pathological. We are unaware of any such mutations that have been identified to date, although knowledge of the importance of this region of the receptor may prove to be important in future studies.
Refinement of the SCAM for use with rapid solution exchange should enable an investigation of altered accessibility caused by specific states of the receptor induced by agonist activation. We anticipate that this approach will help to bridge the gap between static structural models and real time recordings of electrophysiology. This will help to discriminate between the conformational changes caused by activation and desensitization.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
DTB‐H, UZ and TGH designed the work. DTB‐H and AK acquired the data. DTB‐H, AK, UZ and TGH analysed and interpreted the data. DTB‐H, AK, UZ and TGH drafted the work and revised it critically for intellectual content. All authors approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions relating to the accuracy and integrity of the work are appropriately investigated and resolved. All authors qualify for authorship and only those who qualify for authorship are listed.
Funding
The authors would like to thank Tenovus Scotland and Scottish Universities’ Physics Alliance (SUPA) for funding.
References
- Althoff T, Hibbs RE, Banerjee S & Gouaux E (2014). X‐ray structures of GluCl in apo states reveal a gating mechanism of Cys‐loop receptors. Nature 512, 333–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandakrishnan R, Aguilar B & Onufriev AV (2012). H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modelling and simulations. Nucleic Acids Res 40, W537–W541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista‐Hon DT, Deeb TZ, Lambert JJ, Peters JA & Hales TG (2013). The minimum M3‐M4 loop length of neurotransmitter‐activated pentameric receptors is critical for the structural integrity of cytoplasmic portals. J Biol Chem 288, 21558–21568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger O, Edholm O & Jahnig F (1997). Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys J 72, 2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blarre T, Bertrand HO, Acher FC & Kehoe J (2014). Molecular determinants of agonist selectivity in glutamate‐gated chloride channels which likely explain the agonist selectivity of the vertebrate glycine and GABAA‐rho receptors. PLoS ONE 9, e108458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau AJ, Evers AR, Davis AF & Czajkowski C (1999). Mapping the agonist binding site of the GABAA receptor: evidence for a beta‐strand. J Neurosci 19, 4847–4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau AJ, Newell JG & Czajkowski C (2002). GABA(A) receptor beta 2 Tyr97 and Leu99 line the GABA‐binding site. Insights into mechanisms of agonist and antagonist actions. J Biol Chem 277, 2931–2937. [DOI] [PubMed] [Google Scholar]
- Bussi G, Donadio D & Parrinello M (2007). Canonical sampling through velocity rescaling. J Chem Phys 126, 014101. [DOI] [PubMed] [Google Scholar]
- Calimet N, Simoes M, Changeux JP, Karplus M, Taly A & Cecchini M (2013). A gating mechanism of pentameric ligand‐gated ion channels. Proc Natl Acad Sci USA 110, E3987–E3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D (1998). Binding, gating, affinity and efficacy: the interpretation of structure‐activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol 125, 924–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras FX, Ernst AM, Wieland F & Brugger B (2011). Specificity of intramembrane protein‐lipid interactions. Cold Spring Harb Perspect Biol 3, pii: a004705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromer BA, Morton CJ & Parker MW (2002). Anxiety over GABA(A) receptor structure relieved by AChBP. Trends Biochem Sci 27, 280–287. [DOI] [PubMed] [Google Scholar]
- Du J, Lu W, Wu S, Cheng Y & Gouaux E (2015). Glycine receptor mechanism elucidated by electron cryo‐microscopy. Nature 526, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhaber F, Lijnzaad P, Argos P, Sander C & Scharf M (1995). The double cubic lattice method: efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. J Comput Chem 16, 12. [Google Scholar]
- Feenstra AK, Hess B & Berendsen Herman JC (1999). Improving efficiency of large time‐scale molecular dynamics simulations of hydrogen‐rich systems. J Comput Chem 20, 13. [DOI] [PubMed] [Google Scholar]
- Goldschen‐Ohm MP, Wagner DA & Jones MV (2011). Three arginines in the GABAA receptor binding pocket have distinct roles in the formation and stability of agonist‐ versus antagonist‐bound complexes. Mol Pharmacol 80, 647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley SH & Amin J (2007). Rat alpha6beta2delta GABAA receptors exhibit two distinct and separable agonist affinities. J Physiol 581, 1001–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales TG, Deeb TZ, Tang H, Bollan KA, King DP, Johnson SJ & Connolly CN (2006). An asymmetric contribution to gamma‐aminobutyric type A receptor function of a conserved lysine within TM2‐3 of alpha1, beta2, and gamma2 subunits. J Biol Chem 281, 17034–17043. [DOI] [PubMed] [Google Scholar]
- Hanson SM & Czajkowski C (2008). Structural mechanisms underlying benzodiazepine modulation of the GABA(A) receptor. J Neurosci 28, 3490–3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassaine G, Deluz C, Grasso L, Wyss R, Tol MB, Hovius R, Graff A, Stahlberg H, Tomizaki T, Desmyter A, Moreau C, Li XD, Poitevin F, Vogel H & Nury H (2014). X‐ray structure of the mouse serotonin 5‐HT3 receptor. Nature 512, 276–281. [DOI] [PubMed] [Google Scholar]
- Heckman KL & Pease LR (2007). Gene splicing and mutagenesis by PCR‐driven overlap extension. Nat Protoc 2, 924–932. [DOI] [PubMed] [Google Scholar]
- Hess B, Bekker H, Berendsen HJC & Fraaije JGEM (1997). LINCS: A linear constraint solver for molecular simulations. J Comput Chem 18, 10. [Google Scholar]
- Hibbs RE & Gouaux E (2011). Principles of activation and permeation in an anion‐selective Cys‐loop receptor. Nature 474, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden JH & Czajkowski C (2002). Different residues in the GABA(A) receptor alpha 1T60‐alpha 1K70 region mediate GABA and SR‐95531 actions. J Biol Chem 277, 18785–18792. [DOI] [PubMed] [Google Scholar]
- Hornak V, Abel R, Okur A, Strockbine B, Roitberg A & Simmerling C (2006). Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A & Schulten K (1996). VMD: visual molecular dynamics. J Mol Grap 14, 33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- Kash TL, Jenkins A, Kelley JC, Trudell JR & Harrison NL (2003). Coupling of agonist binding to channel gating in the GABA(A) receptor. Nature 421, 272–275. [DOI] [PubMed] [Google Scholar]
- Khatri A, Sedelnikova A & Weiss DS (2009). Structural rearrangements in loop F of the GABA receptor signal ligand binding, not channel activation. Biophys J 96, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri A & Weiss DS (2010). The role of Loop F in the activation of the GABA receptor. J Physiol 588, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krah A, Pogoryelov D, Meier T & Faraldo‐Gomez JD (2010). On the structure of the proton‐binding site in the F(o) rotor of chloroplast ATP synthases. J Mol Biol 395, 20–27. [DOI] [PubMed] [Google Scholar]
- Lindorff‐Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO & Shaw DE (2010). Improved side‐chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller PS & Aricescu AR (2014). Crystal structure of a human GABA receptor. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen M, Wafford KA, Wingrove P & Ebert B (2003). Pharmacology of GABA(A) receptors exhibiting different levels of spontaneous activity. Eur J Pharmacol 476, 17–24. [DOI] [PubMed] [Google Scholar]
- Newell JG & Czajkowski C (2003). The GABAA receptor alpha 1 subunit Pro174‐Asp191 segment is involved in GABA binding and channel gating. J Biol Chem 278, 13166–13172. [DOI] [PubMed] [Google Scholar]
- Othman NA, Gallacher M, Deeb TZ, Baptista‐Hon DT, Perry DC & Hales TG (2012). Influences on blockade by t‐butylbicyclo‐phosphoro‐thionate of GABAA receptor spontaneous gating, agonist activation and desensitization. J Physiol 590, 163–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Chen Q, Willenbring D, Yoshida K, Tillman T, Kashlan OB, Cohen A, Kong XP, Xu Y & Tang P (2012). Structure of the pentameric ligand‐gated ion channel ELIC cocrystallized with its competitive antagonist acetylcholine. Nat Commun 3, 714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paramo T, East A, Garzon D, Ulmschneider MB & Bond PJ (2014). Efficient characterization of protein cavities within molecular simulation trajectories: trj_cavity. J Chem Theory Comp 10, 2151–2164. [DOI] [PubMed] [Google Scholar]
- Parrinello M & Rahman A (1981). Polymorphic transitions in single‐crystals – a new molecular‐dynamics method. J Appl Phys 52, 9. [Google Scholar]
- Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B & Lindahl E (2013). GROMACS 4.5: a high‐throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiram PA, McIntosh JM & Sine SM (2000). Pairwise interactions between neuronal alpha(7) acetylcholine receptors and alpha‐conotoxin PnIB. J Biol Chem 275, 4889–4896. [DOI] [PubMed] [Google Scholar]
- Schmidt TH & Kandt C (2012). LAMBADA and InflateGRO2: efficient membrane alignment and insertion of membrane proteins for molecular dynamics simulations. J Chem Inf Model 52, 2657–2669. [DOI] [PubMed] [Google Scholar]
- Smith GB & Olsen RW (1995). Functional domains of GABAA receptors. Trends Pharmacol Sci 16, 162–168. [DOI] [PubMed] [Google Scholar]
- Szczot M, Kisiel M, Czyzewska MM & Mozrzymas JW (2014). alpha1F64 Residue at GABA(A) receptor binding site is involved in gating by influencing the receptor flipping transitions. J Neurosci 34, 3193–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PN, Laha KT & Wagner DA (2011). A tight coupling between beta(2)Y97 and beta(2)F200 of the GABA(A) receptor mediates GABA binding. J Neurochem 119, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalan SP & Czajkowski C (2008). A conserved salt bridge critical for GABA(A) receptor function and loop C dynamics. Proc Natl Acad Sci USA 105, 13604–13609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villinger S, Briones R, Giller K, Zachariae U, Lange A, de Groot BL, Griesinger C, Becker S & Zweckstetter M (2010). Functional dynamics in the voltage‐dependent anion channel. Proc Natl Acad Sci USA 107, 22546–22551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner DA, Czajkowski C & Jones MV (2004). An arginine involved in GABA binding and unbinding but not gating of the GABA(A) receptor. J Neurosci 24, 2733–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting PJ, McKernan RM & Wafford KA (1995). Structure and pharmacology of vertebrate GABAA receptor subtypes. Int Rev Neurobiol 38, 95–138. [DOI] [PubMed] [Google Scholar]